
Abstract
Background
Septic shock is the most severe phase of sepsis and is associated with high rates of mortality. However, early stage prediction of septic shock outcomes remains difficult. Metabolomic techniques have emerged as a promising tool for improving prognosis.
Methods
Orthogonal projections to latent structures-discriminant analysis (OPLS-DA) models separating the serum metabolomes of survivors from those of non-survivors were established with samples obtained at the intensive care unit (ICU) admission (H0) and 24 h later (H24). For 51 patients with available H0 and H24 samples, multi-level modeling was performed to provide insight into different metabolic evolutions that occurred between H0 and H24 in the surviving and non-surviving patients. Relative quantification and receiver operational characteristic curves (ROC) were applied to estimate the predictability of key discriminatory metabolites for septic shock mortality.
Results
Metabolites that were involved in energy supply and protein breakdown were primarily responsible for differentiating survivors from non-survivors. This was not only seen in the H0 and H24 discriminatory models, but also in the H0-H24 paired models. Reanalysis of extra H0-H24 paired samples in the established multi-level model demonstrated good performance of the model for the classification of samplings. According to the ROC results, nine discriminatory metabolites defined consistently from the unpaired model and the H0-H24 time-trend change (ΔH24-H0) show good prediction of mortality. These results suggest that NMR-based metabolomic analysis is useful for a better overall assessment of septic shock patients.
Conclusions
Dysregulation of the metabolites identified by this study is associated with poor outcomes for septic shock. Evaluation of these compounds during the first 24 h after ICU admission in the septic shock patient may be helpful for estimating the severity of cases and for predicting outcomes.
Trial registration
All human serum samples were collected and stored, provided by the “center of biologic resources for liver disease”, in Jean Verdier Hospital, Bondy, France (BB-0033-00027).
Similar content being viewed by others


Background
Septic shock is the most severe phase of sepsis [1, 2]. It is defined as sepsis complicated either by hypotension that is refractory to fluid resuscitation or by hyperlactacidemia and is often accompanied by acute organ failure. Mortality rates associated with septic shock are 20 to 30% in many series, principally due to multiple organ dysfunction syndrome (MODS) [3]. Common strategies for the treatment of septic shock include prompt initiation of therapy to treat the underlying infection with antibiotics, vasopressor therapy, and support for failing organs. In recent years, early goal-directed therapy (EGDT), which improves curative effect, has been extensively applied to improve rescue outcomes [4, 5]. However, early personalized prognosis and diagnosis remain challenging due to the complicated etiology and pathogenesis of septic shock. Determination of an acute prognosis in the early stage of sepsis is of great importance to improve therapeutic efficacy and will aid in the development of adapted strategies for different cases. In fact, evaluation of existing biomarkers (e.g., TNF-α, IL-6, and PCT) and clinical scores such as the sequential organ failure assessment (SOFA) [6] have been applied prognostically but their performance (sensitivity, specificity) has not proven adequate for all cases [7]. Thus, new methods for reliable early prognosis are still urgently needed.
Metabolomics has been proven to be a promising tool that aid in the prognosis of sepsis. This is because metabolomics allows to provide comprehensive information of personalized metabolome and therefore to enable the prediction of personalized outcome for septic patients. Previous studies have shown that there are considerable differences in the metabolome fingerprints between septic shock survivors and non-survivors. However, notably, most of the previous studies in human septic patients were designed to be performed by analysis of one unique sampling, and no studies have derived dynamic alterations of patient metabolomes during clinical therapy. However, good outcomes for septic shock are associated with a less severe disease course and a positive therapeutic response to treatment. In a previous study, we reported comprehensive differences in the metabolic profiles between septic shock survivors and non-survivors at the admission to the intensive care unit (ICU), based on a liquid chromatography-mass spectrometry (LC-MS) approach [8]. In this current study, samples from the septic shock patients which were obtained 24 h after ICU admission were also included. The aim of the present study was to analyze the discriminatory ability of metabolic profiles between septic shock survivors and non-survivors at the beginning and 24 h after ICU admission and also to describe the evolution of metabolic profiles for septic shock patients during this period by using 1H NMR spectroscopy-based metabolomics.
Materials and methods
Patient inclusion
Between January 2009 and December 2011, all consecutive adults admitted to our intensive care unit were enrolled in this study if they had an indisputable or probable septic shock in the first 24 h after ICU admission [9]. Septic shock was defined as the presence of a clinically or microbiologically documented infection and on-going treatment with vasopressor therapy (norepinephrine or epinephrine at a dose ≥ 0.25 μg per kilogram of body weight per minute or at least equal to 1 mg per hour) for at least 6 h to maintain a systolic blood pressure of at least 90 mmHg or a mean blood pressure of at least 65 mmHg. Non-inclusion criteria were (i) patient younger than 18 years, (ii) patient with solid cancer or blood cancer, and (iii) patient with liver cirrhosis or chronic kidney disease. Patients were treated according to the international guidelines for the management of sepsis and shock septic [5].
Biological parameters, hemodynamic parameters, and the use of catecholamine and mechanical ventilation were recorded at inclusion. Cause of septic shock was recorded. To evaluate the severity of the disease, the Sequential Organ Failure Assessment (SOFA) score was calculated during the first day of admission [10]. ICU and hospital length of stay and mortality were recorded. The survival status of each patient was noted 7 days after the first sample.
Sample collection
All the first samplings (H0) were obtained withdrawn just before or immediately after clinical vasopressor therapy initiation on the patients. The second samples were withdrawn 24 h after the beginning of the vasopressor introduction. Blood samples were collected in serum separator tubes (SST). SST were stored for at least 30 min and not more than 1 h and 30 min. After centrifugation (1000×g, 25 °C, 10 min), the serum was stored at −80 °C. All human serum samples were collected and stored, provided by the “center of biologic resources for liver disease”, in Jean Verdier Hospital, Bondy, France (BB-0033-00027). Written informed consent was obtained from all subjects or their surrogate decision-maker. The local ethics committee approved the protocol.
Regrouping and matching of samples
As shown in Fig. 1, 122 samples from 70 patients were obtained. Seventy samples were drawn at ICU admission and are noted as H0 samples. 52 samples were obtained 24 h after the first sampling and are noted as H24 samples. During analysis, one H0 sample from a non-survivor who did not have a matching H24 sample was excluded as a spectral outlier. One H24 sample from a survivor was excluded while the H0 sample that belonged to the same patient was retained. The exclusion of the sample was due to the drastically affected NMR gain parameter. The spectrum of this sample was therefore found to be clearly different from the others. Among the non-survivors, 11 patients died prior to the H24 sampling and their H24 samples were therefore not available. For the other samples, each H24 sample was matched with the H0 sample which was collected from the same patient. In this case, 32 pairs for survivors and 19 pairs for non-survivors were obtained. For both septic shock survivor (SSS) and non-survivor (SSN) H0-H24 pairs, two thirds were randomly taken into the training set while the remaining were put into the test set.

Regrouping and matching of samples. One H24 sample from a survivor was excluded due to the problem of NMR gain parameter; one H0 sample from a non-survivor was excluded as it was found to be an outlier in PCA. For the paired H0-H24 samples obtained from the same patients, the pairs have been divided into training set and test set. The pairs of survivors and non-survivors were analyzed separately. Samples in the training set were analyzed for establishing discriminatory models between H0 and H24 samples. The pairs in the test set were reanalyzed in the established models
Sample preparation and NMR data acquisition
Samples were defrosted at room temperature. A volume of 450 μL of each sample was diluted with 50 μL of D2O in an NMR tube of 5 mm diameter. All the samples were then analyzed with a 500-MHz NMR spectrometer (Advance III, Bruker, Germany) at 297 K. The free induction decay (FID) signals were collected onto 64k data points, with a spectral width of 6000 Hz. The 1D 1H NMR spectra were recorded by the Carr-Purcell-Meiboom-Gill (CPMG) sequence [11] with 128 transients for each spectrum. For several samples, 2D NMR experiments (TOCSY and JRES sequences) were achieved to confirm spectral assignments. The mixing time of the TOCSY spectra was 80 ms with 32 transients.
Data processing
After the FIDS were acquired for all the samples, they were processed using the NMRPipe software [12]. All FIDs were multiplied by a 0.3-Hz exponential line broadening factor prior to Fourier transformation. Phasing of each spectrum was manually adjusted, and baselines were corrected using a linear method. All the spectra were divided into 0.001 ppm buckets between − 1 and 10 ppm. The residual water signal (4.6 to 5.5 ppm) was excluded, and the spectral region from 3.16 to 4 ppm was also removed since signals observed in this section represented the infusion of hydroxyethyl starch (HES), which was applied in the ICU to heighten blood tension for the patients who suffered from hypotension. The spectra were then normalized using the probabilistic quotient method [13]. All the buckets were centered by the method of auto-scaling. The peaks were adequately assigned using the Human Metabolome Database (HMDB, www.hmdb.ca) NMR library, the Chenomx software (Chenomx Inc., Canada), and the 2D experiments. As to the annotation of NMR peaks, an exemplar NMR spectrum has been shown in Additional file 1: Figure S1 with some of the assignments.
Statistical analyses
All the multivariate analyses were achieved using an in-house code which is based on the code of Trygg and Wold [14], developed using Matlab software (version 2012b, MA, USA). Prior to the establishment of discriminatory analyses, a principal component analysis (PCA) with H0 samples from all the included non-survivors shows that the main variability among these samples does not correspond to the time of the death, as shown in Additional file 1: Figure S2. Another PCA for all the acquired spectra was performed to ensure that there were no outliers (Additional file 1: Figure S3). Orthogonal projections to latent structures-discriminant analyses (OPLS-DA) were performed for differentiating survivors from non-survivors with H0 and H24 samples, respectively. Samples obtained at H0 and H24 from the same patients were paired and analyzed in multilevel models to study the interindividual variability [15]. The paired samples were divided into survivor and non-survivor groups. Two OPLS-DA multilevel models were applied with the survivors and non-survivors, respectively, separating H0 from H24 samples. The models were all validated by cross-validation with 500 permutations of variable X and Y, where X represents the data matrix and Y represents the discriminatory variable for each model [15, 16]. For the univariate analyses analyzing the significant differences between two groups, the P values were calculated with Student’s T test. The false discovery rate (FDR) was calculated by the Multiple Experiment Viewer Toll (version 4.9.0, OriginLab, Northampton, USA); the correction of P value is performed with “adjusted Bonferroni correction” [17]. The threshold of FDR was set at 0.1 for the screening of the discriminatory metabolites, that is, the variables with FDRs superior to 0.1 were not considered as important discriminants. A significant difference between compared groups was defined with an adjusted P value inferior to 0.05.
Results
Baseline characteristics of patients
The baseline biological characteristics of all the included patients are shown in Table 1. Partial pressure of arterial oxygen (PaO2) and the ratio of PaO2 to the percentage of inspired oxygen (FiO2) were significantly different between the survivors and non-survivors at H0. Lactate level in non-survivors was also found to be significantly increased than those in survivors. For the clinical scores, SAPSII and SOFA, SAPSII was able to discriminate between survivors and non-survivors. However, due to the sample size, it was not satisfying to predict mortality with SOFA in this study, according to the results both at H0 and H24.
Discriminatory analyses separating septic survivors from non-survivors with samples drawn before treatment (H0)
For the H0 samples, a total of 69 samples were analyzed using an OPLS-DA model (PCA models separating the survivors from the non-survivors prior to the exclusion of the outlier have been illustrated in Additional file 1: Figure S3). Among these, 40 samples were obtained from survivors and 29 were from non-survivors. As is shown in Fig. 2a for H0, a clear separation between the two groups of patients is demonstrated by the score plot. The Q2Y, which indicates the predictability of the model, was equal to 0.60 with three components, and the R2Y, which indicates the fraction of explained variance of the Y variable, was 0.75, where Y corresponded to the survival condition in the model for the SSS vs. SSN comparison. Cross-validation showed that the model was not over-fitted (Additional file 1: Figure S4). In the loading plot (Fig. 2c), the peaks are colored according to the correlation coefficients, which relate to their contribution to the discriminatory model. Corresponding discriminatory metabolites have been listed in Table 2 with their chemical shifts, multiplicity, correlations, variance importance projections (VIPs), and P values. The concentrations of various amino acids such as alanine, glutamate, glutamine, methionine, and aromatic amino acids were increased in the non-survivors as compared to the survivors. Significant variations between the two groups were also found in energy-associated metabolites including two tricarboxylic acid (TCA) cycle intermediates, citrate and fumarate, and lactate and pyruvate. Ketone bodies, 3-hydroxybutyrate, and acetate were also elevated in the non-surviving patients. The only decreased signal was observed for the N-acetyl moieties of glycoproteins. Together, the results showed considerable differences in the metabolic profiles between the survivors and the non-survivors at H0.

OPLS-DA between septic shock survivors and non-survivors at H0 and H24. a, b Score plots for the H0 and H24 models, respectively. Blue dots represent the survivors and yellow dots represent the non-survivors. Tpred: The components that predict the differences between the groups; Torth: components that do not predict the differences between the groups; c, d Loading plot for the H0 and H24 models, respectively. The color of the peaks indicates the correlation between the marked peak and the classification of the sample. Colors that are close to red correspond to a higher correlation. Positive peaks in the loading plot correspond to metabolites which increased in non-survivors; negative peaks correspond to metabolites that decreased in non-survivors
Discriminatory analyses separating septic survivors from non-survivors with samples drawn 24 h after ICU admission (H24)
A second OPLS-DA model differentiating metabolic profiles of SSS from those of SSN were performed with 51 H24 samples. Of these samples, 19 non-survivors were compared with 32 survivors. As shown in Fig. 2b, a separation between SSS and SSN was observed. The R2Y and Q2Y values in the model were equal to 0.86 and 0.46, respectively, and were calculated with three components. The validation by permutations is shown in Additional file 1: Figure S5. Significant discriminant metabolites were identified referring to the loading plot (Fig. 2d) and are listed in Table 3. Interestingly, increasing levels of most amino acids and energy-related metabolites, as well as the decreases of N-acetyl moieties of glycoproteins, were still detected in SSN, compared with SSS, in line with the findings of the H0 model. Besides, an increase in ketone bodies and diminishing lipid-related signals were only present at H24, but not at H0, in the non-survivors when compared to survivors. Both H0 and H24 unpaired models revealed extensive variations in the metabolic profiles between SSS and SSN at the admission and 24 h after ICU admission.
Discriminatory analysis of the evolution of septic shock from H0 to H24 for septic shock survivors and non-survivors
On the basis of the separation found between SSS and SSN within the above two discriminant models, we hypothesized that the therapeutic response between SSS and SSN could be different. To verify this hypothesis, SSS and SSN groups were compartmentalized and studied by two multi-level OPLS-DA models which focused on the intraindividual variability of the metabolome between H0 and H24. The pairwise distance of metabolome variations between H0 and H24 samples were analyzed for the patients for whom both H0 and H24 samples were collected.
For the SSS group, 21 pairs were randomly included to establish a model separating the H0 sample from the H24 sample, as shown in Fig. 3a. The Q2Y was 0.78 with 2 components, and R2Y was 0.94. This model was subsequently applied to 11 other pairs. The predictions for these pairs are shown in Fig. 3b. R2Y for the prediction was 0.76, showing a prominent prediction of the classification among H0 and H24 samples. For the SSN model, separation between H0 and H24 samples was also observed, as shown in Fig. 3c. Thirteen pairs were used to set up a training model, and 6 pairs were included in the test set. Consequently, R2Y and Q2Y were 0.57 and 0.91, respectively, and R2Y for the reanalysis was 0.33 (as shown Fig. 3d). The loading plots for the two paired models are shown in Additional file 1: Figure S6. Metabolites which are listed in Table 4 exhibited opposite H0-H24 metabolome evolutions between SSN and SSS. Accordingly, increases of amino acids, energy-related metabolites, and creatinine and a decline of glycoprotein could be observed during the evolution from H0 to H24 for the non-survivors. However, this was not the case for survivors.

Score plots of OPLS-DA separating H0 from H24. For the patients whose H0 and H24 are both available, their H0 and H24 samples are matched in the discriminatory models. The pairs from the survivors and non-survivors are analyzed in two separated paired models. Blue dots represent the H24 samples and yellow dots represent the H0 samples. a Score plot for the training set separating H0 from H24 for the survivors. b Reanalysis of test set samples of survivors in the established H0-H24 multi-level model. c Score plot for the training set separating H0 from H24 for the non-survivors. d Reanalysis of test set samples of non-survivors in the established H0-H24 multi-level model
Discrimination between SSS and SSN based on the relative quantification of key discriminators
Spectral signals corresponding to the metabolites in Table 4 were integrated for the spectra of paired samples. As shown in Table 4, the molecules varied oppositely during the H0-H24 evolution between the survivors and non-survivors. The time-trend change of area, ΔSignal areaH24-H0, was calculated for each metabolite. Average values for ΔSignal areaH24-H0 of involved metabolites resulting from the SSN model were compared to those from the SSS model, as shown in Fig. 4a. Interestingly, the metabolites were also shown to be discriminant variables in the comparison between SSS and SSN in previously mentioned H0 and H24 unpaired models. We further calculated the area under the ROC curve for the metabolites in order to test their performance in the classification of surviving patients. ROCs for the discriminant metabolites in the H0 and H24 models, as well as for ΔH24-H0, were performed and are shown in Table 5. Accordingly, based on our data, most ROCs for the metabolites showed slightly better performance in the classification of survival than SAPSII and SOFA, not only within the H0 and H24 models, but also with the value of ΔH24-H0.

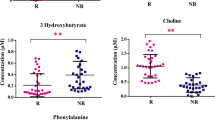
Levels of key discriminatory metabolites and their relevant metabolic pathway in the comparison between SSS and SSN patients during the H0-H24 evolution. a Levels of key discriminatory metabolites in the SSS and in SSN. The levels of the metabolites are calculated with the average of time-trend change (ΔH24-H0). Averages of ΔH24-H0 for the survivors and non-survivors have been respectively shown and calibrated by the standard deviation. *: p < 0.05. **: p < 0.01. a.u.: arbitrary unit; b relevant metabolic pathway for energy-related metabolites and amino acids that vary differentially between SSS and SSN in the H0-H24 evolution. Metabolites marked by red color are those that increase in the SSN compared to SSS in all the models. Solid flashes express a direct conversion between two metabolites and dotted lines represent undirect conversions between two metabolites, according to KEGG metabolic pathway database
Discussion
Effective prognosis can help to improve outcomes for septic shock patients. However, septic shock prognosis can be complicated by patient-specific factors that affect responsiveness to therapy. With the use of metabolomic techniques, we have determined the serum metabolome fingerprint of septic shock patients with both H0 and H24 samples. We also investigate the metabolic footprint along with the evolution from H0 to H24. To our knowledge, our study is the first to reveal time-trend metabolic differences using NMR-based metabolomics between septic shock survivors and non-survivors within 24 h after ICU admission.
Metabolic variations for H0 and H24 unpaired models separating SSS from SSN
The H0 and H24 unpaired models reveal the differences of metabolome fingerprint between SSS and SSN at the admission to ICU and at 24 h after the admission. Regarding the common discriminatory metabolites found in both models, consistent increases in energy-related metabolites, creatinine, 1-MH, and several amino acids, as well as decreases in glycoproteins are observed as important signals in the non-survivors. Such variations found in SSN at both H0 and H24 are likely to reflect more severe sepsis-induced inflammatory responses and organ dysfunctions that contribute to poor outcomes.
The deregulation of TCA cycle intermediates, such as more concentrated citrate found in the SSN, is one of the consequences of severe stress induced by sepsis [18]. Stress also results in an unregulated catabolism [19]. Enhanced degradation of glycoproteins indicates an aggravated stress in the non-survivors. Also, increases in various amino acids and ketone bodies at H24 in the non-survivors are known as effects of protein breakdown and enhanced lipid oxidation [20]. Notably, ketone bodies have recently been reported to be immune suppressors [21], and elevations in these metabolites may contribute to a negative response in critical illness [22].
Our results also provide evidence for the metabolic variations that are associated with severe organ dysfunction in non-survivors. As shown in Table 1, significantly lower oxygen pressures with higher blood lactate levels indicate the presence of more severe hypoxia in the non-survivors than in survivors. This may be due to mitochondrial disorder, defective TCA cycle [23], which results in dampened aerobic respiration and abnormal energy supply. Severe disorders in energy supply should be an import factor inducing organ failure [24,25,26]. Other variations involving organ dysfunction are found in creatinine and 1-MH. Their elevations in the comparison in SSN are also supported by some other previous studies [27, 28].
Different variations for some key discriminators between SSS and SSN H0-H24 multilevel models
As shown in Table 4, different pairwise alterations of relevant metabolites in the comparison between SSS and SSN groups indicate distinct trends in development along with clinical therapy. Interestingly, most of these metabolites related to septic shock evolution are in accordance with the discriminatory molecules found with the H0 and H24 unpaired models. Besides the deregulation of energy-related molecules, increases in four amino acids may be associated with severe protein breakdown and muscle wasting for the non-survivors. Notably, serum concentrations in some amino acids, such as alanine, glutamate, and phenylalanine, are otherwise documented to be involved with hemolysis associated with sepsis [29]. Moreover, as shown in Fig. 4b, glutamate is known to be a core amino acid for conversion into TCA cycle intermediates [30]. Its elevation, as well as the elevation of related amino acids such as glutamine and alanine, is associated with increases in citrate. Phenylalanine can be converted into fumarate. Increases in phenylalanine in patients with poor outcomes have been also reported in other studies [31,32,33]. The conversion from amino acids to TCA cycle intermediates is likely to provide supplementary energy during severe anoxic conditions, however, is detrimental for the outcomes [34]. Creatinine is known to be an important indicator for monitoring renal injury. Decreases in N-acetyl glycoproteins may correspond to a breakdown of proteins. Interestingly, it has been demonstrated by DeCoux et al. [35] that inflammation-induced enriched extracellular glycoproteins are associated with an optimal response during septic shock.
The present study not only provides support for the findings in our previous work, which investigated metabolic differences between SSS and SSN at H0 [8], but also reveals that different evolutions in the first 24 h after admission to ICU between septic shock survivors and non-survivors are linked to variations in metabolites identified by this study. The ROC results shown in Table 5 also show that the key metabolite discriminators are good classifiers for separating SSN from SSS during the first 24 h after ICU admission. We have reason to believe that sustained enrichment of energy-related metabolites and amino acids can provide early warning of a bad outcome.
Conclusion
In the present study, we have investigated metabolic differences between the survivors and non-survivors of septic shock with the samples obtained at ICU admission and with those obtained 24 h later. We have provided evidence that the sustained enrichment of energy-supply metabolites and amino acids is predictive of a bad outcome. We suggest that monitoring the relevant metabolites in the first 24 h may help to evaluate early therapeutic response.
Abbreviations
- AUROC:
-
Area under receiver operation curve
- FDR:
-
False discovery rate
- HB:
-
Hydroxybutyrate
- HES:
-
Hydroxyethyl starch
- ICU:
-
Intense care unit
- JRES:
-
J-resolved spectroscopy
- MH:
-
Methylhistidine
- MODS:
-
Multi-organ dysfunction syndrome
- NMR:
-
Nuclear magnetic resonance
- OPLS-DA:
-
Orthogonal projections to latent structures-discriminant analysis
- PCA:
-
Principal component analysis
- PCT:
-
Procalcitonin
- SAPSII:
-
New Simplified Acute Physiology Score
- SOFA:
-
Sequential organ failure assessment
- SSN:
-
Septic shock non-survivors
- SSS:
-
Septic shock survivors
- TCA:
-
Tricarboxylic acid
- TOCSY:
-
Total correlation spectroscopy
References
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest. 1992;101(6):1644–55.
Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 2003;29(4):530–8.
Balk RA. Systemic inflammatory response syndrome (SIRS): where did it come from and is it still relevant today? Virulence. 2014;5(1):20–6.
Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, Peterson E, Tomlanovich M. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345(19):1368–77.
Dellinger RL, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296–327.
Vincent JL, Moreno R, Takala J, Willatts S, DeMendonca A, Bruining H, Reinhart CK, Suter PM, Thijs LG. The SOFA (sepsis-related organ failure assessment) score to describe organ dysfunction/failure. Intensive Care Med. 1996;22(7):707–10.
Lee JW, Yamamoto T, Uchikata T, Matsubara A, Fukusaki E, Bamba T. Development of a polar lipid profiling method by supercritical fluid chromatography/mass spectrometry. J Sep Sci. 2011;34(24):3553–60.
Liu Z, Yin P, Amathieu R, Savarin P, Xu G. Application of LC-MS-based metabolomics method in differentiating septic survivors from non-survivors. Anal Bioanal Chem. 2016;408(27):7641–9.
Annane D, Bellissant E, Cavaillon J-M. Septic shock. Lancet. 2005;365(9453):63–78.
Vincent JL, de Mendonca A, Cantraine F, Moreno R, Takala J, Suter PM, Sprung CL, Colardyn F, Blecher S. Use of the SOFA score to assess the incidence of organ dysfunction/failure in intensive care units: results of a multicenter, prospective study. Crit Care Med. 1998;26(11):1793–800.
Carr HY, Purcell EM. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys Rev. 1954;94(3):630.
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–93.
Dieterle F, Ross A, Schlotterbeck G, Senn H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal Chem. 2006;78(13):4281–90.
Trygg J, Wold S. Orthogonal projections to latent structures (O-PLS). J Chemom. 2002;16(3):119–28.
Westerhuis JA, van Velzen EJ, Hoefsloot HC, Smilde AK. Multivariate paired data analysis: multilevel PLSDA versus OPLSDA. Metabolomics. 2010;6(1):119–28.
Westerhuis JA, Hoefsloot HCJ, Smit S, Vis DJ, Smilde AK, van Velzen EJJ, van Duijnhoven JPM, van Dorsten FA. Assessment of PLSDA cross validation. Metabolomics. 2008;4(1):81–9.
Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage. 2002;15(4):870–8.
Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, Tabita-Martinez J, Sellers KF, Rickels MR, Reilly MP. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes. 2010;59(1):172–81.
Preiser JC, Ichai C, Orban JC, Groeneveld AB. Metabolic response to the stress of critical illness. Br J Anaesth. 2014;113(6):945–54.
Pawlak M, Baugé E, Lalloyer F, Lefebvre P, Staels B. Ketone body therapy protects from lipotoxicity and acute liver failure upon Ppar α deficiency. Mol Endocrinol. 2015;29(8):1134–43.
Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25(1):42–52.
Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, Moore FA, Moldawer LL. Sepsis pathophysiology, chronic critical illness, and persistent inflammation-immunosuppression and catabolism syndrome. Crit Care Med. 2017;45(2):253–62.
Rodriguez MC, MacDonald JR, Mahoney DJ, Parise G, Beal MF, Tarnopolsky MA. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve. 2007;35(2):235–42.
Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(9328):219–23.
Strassburg CP. Shock liver. Best Pract Res Clin Gastroenterol. 2003;17(3):369–81.
Poole RC, Halestrap AP. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am J Phys Cell Phys. 1993;264(4):C761–82.
Lin ZY, Xu PB, Yan SK, Meng HB, Yang GJ, Dai WX, Liu XR, Li JB, Deng XM, Zhang WD. A metabonomic approach to early prognostic evaluation of experimental sepsis by (1) H NMR and pattern recognition. NMR Biomed. 2009;22(6):601–8.
Seymour CW, Yende S, Scott MJ, Pribis J, Mohney RP, Bell LN, Chen YF, Zuckerbraun BS, Bigbee WL, Yealy DM, et al. Metabolomics in pneumonia and sepsis: an analysis of the GenIMS cohort study. Intensive Care Med. 2013;39(8):1423–34.
Stringer KA, Younger JG, McHugh C, Yeomans L, Finkel MA, Puskarich MA, Jones AE, Trexel J, Karnovsky A. Whole blood reveals more metabolic detail of the human metabolome than serum as measured by 1H-NMR spectroscopy: implications for sepsis metabolomics. Shock. 2015;44(3):200–208.
Vaitheesvaran B, Xu J, Yee J, Q-Y L, Go VL, Xiao GG, Lee WN. The Warburg effect: a balance of flux analysis. Metabolomics. 2015;11(4):787–96.
Shapiro N, Howell MD, Bates DW, Angus DC, Ngo L, Talmor D. The association of sepsis syndrome and organ dysfunction with mortality in emergency department patients with suspected infection. Ann Emerg Med. 2006;48(5):583–90 590 e581.
Cooney RN, Kimball SR, Vary TC. Regulation of skeletal muscle protein turnover during sepsis: mechanisms and mediators. Shock. 1997;7(1):1–16.
Schakman O, Kalista S, Barbe C, Loumaye A, Thissen JP. Glucocorticoid-induced skeletal muscle atrophy. Int J Biochem Cell Biol. 2013;45(10):2163–72.
Hasselgren PO, Fischer JE. Sepsis: stimulation of energy-dependent protein breakdown resulting in protein loss in skeletal muscle. World J Surg. 1998;22(2):203–8.
DeCoux A, Tian Y, DeLeon-Pennell KY, Nguyen NT, de Castro Bras LE, Flynn ER, Cannon PL, Griswold ME, Jin YF, Puskarich MA, et al. Plasma Glycoproteomics reveals sepsis outcomes linked to distinct proteins in common pathways. Crit Care Med. 2015;43(10):2049–58.
Acknowledgements
We thank Dr. Pierre Nahon for his help with sample collection and for his constructive suggestions for the study design. We also would like to express our appreciation to Agnes Victor Bala for her help in the assignment of NMR spectra. We thank Sandy Field, PhD, for medical editing assistance. We acknowledge the NMR-PF facility (University Paris 13 France).
Funding
This study is supported by the funding of Anhui Medical University (XJ201729) and by the funding from the University of Paris 13.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
This study was designed by PS, RA, and LLM. ZL participated in conducting the NMR experiments, data analysis, and paper writing. MT provided the programming for the in-house program and for the data analysis. RA conducted sample collection and sample storage. XL, NB, and EH helped with the NMR experiments. All authors participated in the review and revision of the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all subjects or their surrogate decision-maker. The local ethics committee approved the protocol.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1
Table S1. Assignment of spectra recorded with one exemplar serum sample from a septic shock patient. Figure S1. Assignment of spectra recorded with an example of a representative 1H-NMR spectrum. The assigned peaks corresponding with the key metabolite discriminants have been marked in the figure. Figure S2. A PCA calculated with H0 samples from 11 nonsurvivors who died during the first 24 h (red dots) and those from the other non-survivors who died from the second day to the seventh day after the first sampling (blue dots). Figure S3. PCA model separating survivors from non-survivors with H0 samples before the exclusion of outlier. One sample of a non-survivor was observed as an outlier for the PCA. This outlier has been removed before statistical analyses. Blue dots: survivors, yellow dots: non-survivors. Figure S4 (respectively S5). Cross-validation by 200 times permutation between X and Y for the OPLS-DA model with H0 samples (respectively H24). The green dots stand for the obtained R2 value and the blue dots stand for the obtained Q2 value within the 200 permutations. The Y-axis represents R2 and Q2 calculated for every model while the X-axis represents the correlation coefficient between original and permuted response data. Figure S6. Loading plots for paired OPLS-DA models showing important discriminatory metabolites that contribute to the separation between H0 and H24 samples. The paired models for the survivors and non-survivors are shown separately. The peaks are assigned to corresponding discriminatory metabolites. The correlations between the assigned metabolites and the model have been shown with the colors. a: loading plot for the separation between H0 and H24 for the survivors; b: loading plot for the separation between H0 and H24 for the non-survivors. (DOCX 829 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, Z., Triba, M.N., Amathieu, R. et al. Nuclear magnetic resonance-based serum metabolomic analysis reveals different disease evolution profiles between septic shock survivors and non-survivors. Crit Care 23, 169 (2019). https://doi.org/10.1186/s13054-019-2456-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-019-2456-z