
Abstract
Acute respiratory distress syndrome (ARDS) and hospital-acquired pneumonia (HAP) are major problems of public health in intensive care units (ICUs), occurring in 15% of critically ill patients. Among the factors explaining ARDS development, sepsis is known as a frequent cause. Sepsis, ARDS, and HAP increase morbidity, mortality, length of stay in the ICU, and the overall costs of healthcare. The major challenge remains to identify accurately among critically ill patients those at risk of poor outcomes who could benefit from novel therapies. Endocan is released by the pulmonary endothelium in response to local or systemic injury. It inhibits mainly leukocyte diapedesis rather than leukocyte rolling or adhesion to the endothelial cells both in vitro and in vivo. Endocan was evaluated in 25 clinical reports, including 2454 critically ill patients and 452 healthy controls. The diagnostic value of endocan for sepsis or sepsis severity was equal to procalcitonin but its prognostic value was better. A predictive value for postoperative pneumonia was evidenced in two studies, and a predictive value for ARDS in four studies from three independent centers. This review presents an overview of the structure, expression, and functions of endocan. We also hereby summarize the potential applications of endocan in the prediction and prognosis of ARDS and HAP, as well as in the prognosis of sepsis.
Similar content being viewed by others

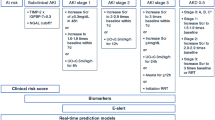
Background
Hospital-acquired pneumonia (HAP) and acute respiratory distress syndrome (ARDS) are major lung injuries that commonly occur in intensive care unit (ICU) patients. In ICU patients, HAP is mainly represented by postoperative pneumonia (POP) and ventilator-associated pneumonia (VAP), respectively defined as pneumonia occurring at least 48 h after surgery or tracheal intubation. Although inconsistent, the incidence of HAP may reach up to 25% in patients undergoing cardiac surgery, thus leading to increased morbidity and hospital length of stay [1]. The delay observed between surgery and the clinical onset of pneumonia, leading to late initiation of antibiotics, might represent one possible explanation for this increased morbidity. Therefore, early identification of patients who are the most likely to develop HAP could allow initiation of preemptive treatments prior to the clinical onset of pneumonia.
ARDS is another frequent condition among ICU patients. It results from an inflammatory state that affects fluid leakage and leukocyte recruitment into the air spaces, thus leading to major hypoxemia. Although many results from clinical trials have led to improved treatment of ARDS, this condition is still responsible for high levels of mortality. There has for several years been a shift of paradigm about the management of ARDS, considering that preemptive therapeutic approaches, targeting lung inflammation before its clinical onset, could lead to major improvement of its prognosis [2]. Thus, a better understanding of molecular regulatory pathways leading to ARDS could be of high interest, especially in acute systemic inflammatory states such as sepsis, which has been identified as a risk factor for ARDS [3, 4].
Sepsis is a life-threatening condition commonly observed in critically ill patients, associated with high mortality rates reaching levels above 25% in septic shock mainly resulting from the development of multiorgan failure. However, sepsis is a heterogeneous condition, encompassing at the same time patients with various prognoses, with either favorable evolutions or poor outcomes [5, 6]. The detection of subjects who exhibit the highest risk of poor outcomes among septic patients might help to improve their prognosis through early adaptation of their management.
Endocan, or endothelial cell specific molecule-1, is a 50-kDa proteoglycan mainly expressed by lung endothelial cells, whose secretion in the bloodstream is upregulated by proinflammatory cytokines (IL-1β, TNF-α) and bacterial LPS [7]. The biological role of endocan results from its ability to link with the integrin LFA-1, leading to the inhibition of its interaction with its endothelial ligand ICAM-1 [8,9,10]. This phenomenon leads to an inhibition of leukocyte diapedesis, explaining a potential anti-inflammatory effect of endocan during acute lung injury. Several convergent reports confer to indicate blood endocan as a predictive marker for HAP and ARDS development.
In this review, we present a summary of the structure, expression, and functions of endocan. We also report its potential applications in the prediction and prognosis of ARDS and HAP, as well as in the prognosis of sepsis.
Main text
Structure of the esm1 gene
The human endocan cDNA was first cloned in our laboratory in 1996 from a HUVEC cDNA library (Genbank accession number X89426). Cloning of its gene was achieved in 1999 (Genbank accession numbers AJ401091 and AJ401092). The mouse endocan cDNA and gene were also cloned first in our laboratory in 1999 and 2000 (Genbank accession numbers AJ249354 and AJ416379).
Initially called esm1 for endothelial cell specific molecule-1, the molecule was renamed endocan in 2001 based on its specific endothelial expression and its proteoglycan nature. Thus, the gene name remains esm1, and its product name is endocan.
The human esm1 gene is located on chromosome 5 at position q11.2. It covers 12 kilobases and is organized into three exons (Fig. 1). The esm1 genes from other species span equivalent sizes and possess the same number of exons [7].

Comparative structure and expression of human and mouse endocan
The mRNA from all species encodes for a propeptide including a signal peptide of 19 amino acids and a mature polypeptide of typically 165 amino acids in the vast majority of mammalians.
Homologies
No clear structural evidence allows one to include endocan in the existing family of proteoglycans: no lectin site is present, and there is no central domain containing leucine-rich repeats, a characteristic of the small leucine-rich proteoglycans [11]. However, endocan is rich in cysteines with 10.9% of the protein encompassing cysteine residues [6]. Interestingly, all 18 cysteines are concentrated in the 110-amino-acid N-terminal region of the protein (Fig. 1). Endocan shows a 15–28% identity with members of the insulin-like growth factor (IGF) binding-protein superfamily, characterized by their cysteine-rich content and their affinity for IGF [7, 12]. However, no evidence for the binding of IGF by endocan has yet been demonstrated [8]. The best protein similarities are found with the human β2 integrins (cysteine-rich tandem repeats: 27.6% identity and 54.6% similarity) [7].
Polymorphism
Based on the search for variants of the human esm1 gene on the ExAC site, the risk of finding a homozygous variant is estimated among the European population to be less than 2 individuals per million, as summarized in Table 1. Note that this risk is equivalent in the Asian population. On the other hand, the risk is twice as high in the African population and five times higher in the Latino population.
Structure of endocan
The human endocan is a soluble proteoglycan consisting of a mature protein of 165 amino acids and a chondroitin/dermatan sulfate glycan chain of 15–40 kDa covalently linked to the protein on serine 137 [7, 13, 14]. Circulating human endocan is fully glycosylated.
The protein core contains several domains respectively encoded by exons 1, 2, and 3, as shown in Fig. 1 [13, 15].
The mouse endocan encodes for a mature polypeptide of 165 amino acids with 74% homology with its human counterpart and all conserved structural domains (Fig. 1) [16]. But recombinant and endogenous circulating mouse endocan are both much less glycosylated than human endocan.
Expression, distribution, and catabolism of endocan
Human endocan is produced specifically by the vascular endothelium and preferentially by the pulmonary and kidney endothelial cells [7]. Tumoral cells can sometimes express endocan (grade III–IV glioblastoma or pituitary adenomas) [17, 18]. Endocan does not belong to the endothelial glycocalix as other proteoglycans like syndecans and circulates freely at about 1 ng/ml in the bloodstream [19, 20].
Mouse endocan was found expressed in endothelial tip cells in several retinal angiogenesis models [21], and in tumor endothelial cells from xenogenic tumors [22]. However, mouse endocan also appears to be produced by some nonendothelial cells, not yet defined, from the lung, kidney, and spleen [22]. Mouse endocan also circulates freely at a baseline concentration of 1 ng/ml [16].
The catabolism of endocan is not well established. One catabolic pathway involves a proteolytic degradation by neutrophil cathepsin G, especially generating one major catabolite of 14 kDa detected in serum from healthy subjects and septic patients [23]. Endocan might also be eliminated through hepatic metabolism, as the increase of blood endocan inversely correlates with the degree of liver deficiency [24].
Endocan gene regulation
Endocan is continually synthetized and secreted under physiological conditions. However, its synthesis and secretion are both upregulated upon endothelial cell stimulation by TNF-α, VEGF, and glycated serum albumin [25, 26]. These increases are observed in vitro 1 h after stimulation for endocan mRNA and 4 h after stimulation for endocan secretion. Then, endocan mRNA and secretion remain high for 24 h, before progressively decreasing to baseline levels [26]. In contrast, basal and TNF-α-induced endocan synthesis and secretion are inhibited by activators of JAK2/STAT3-snail pathways, such as IFN-γ [26], or by activators of PI3 kinase/Akt/FOXO1-ZEB pathways such as PDGF, angiotensin II, endothelin 1, insulin, or hyperglycemia [27, 28].
Inflammation
Upregulation of endocan by proinflammatory cytokines led to suspecting a role in regulating the inflammatory reaction. Indeed, it was initially found that endocan binds to LFA-1 with high affinity (kDA = 18 nM) and inhibits its interaction with the adhesion molecule ICAM-1 [8]. This was later confirmed by intravital microscopy and biomimetic microfluidic assay showing that endocan alleviates leukocyte rolling and transmigration, but not firm adhesion [10].
In animal models, calpain inhibitor pretreatment of endotoxemic rats reduces the severity of endoxemia at 6 h, and is associated with an increase of blood endocan levels and a reduction in leukocyte rolling [29]. In a murine endotoxin-induced acute lung injury model, pretreatment with endocan decreases TNF-α, IFN-γ, IL-1β, and IL-6 levels and alleviates pulmonary epithelial cell apoptosis [30]. Blockade of endocan in mice bearing tumor xenografts induces a stromal inflammation and slows tumor growth [9]. An esm1 gene deficiency impairs leukocyte extravasation at the transmigration step. In a peritonitis assay, esm1−/− mice exhibited a significant decrease in leukocyte extravasation when compared with control littermates [31]. By intravital microscopy, the decreased leukocyte extravasation observed in esm1−/− mice was solely attributable to reduced transmigration but not to adhesion or rolling [10].
This apparent discrepancy between functions of human and mouse endocan may be explained by the differences between their respective cellular sources and glycanation levels [32]. Indeed, by contrast with its human counterpart, mouse endocan is spontaneously produced by nonendothelial cells in the lung, kidney, and spleen. In healthy mice, the main circulating form of endocan is mainly nonglycosylated and nonendothelial, which is quite different from the main human circulating form, fully coming from endothelial cells and fully glycosylated. Each of these forms is suspected to possess opposite biological properties depending on the glycanic status: anti-inflammatory for the glycosylated form, proinflammatory for the nonglycosylated form.
Mouse endocan sounds more complex than its human counterpart. The glycanic status and the function of the true endothelial cell-derived mouse endocan is presently unknown.
Clinical implications
The lung is sensitive to various infectious, toxic, pharmacologic, or immunologic agents. In response, the lung develops an inflammatory reaction which can compromise gas exchange and the patient’s outcome. During the course of inflammation, the pulmonary endothelial cells are solicited to participate in its regulation. In turn, blood endocan increases mainly through de-novo synthesis and secretion [26]. This increase of blood endocan occurs early and independently of any alveolocapillary lesion.
Endocan and community-acquired pneumonia
Very little is known about the expression of endocan during the course of pneumonia. A first study showed increased plasma endocan levels in 82 community-acquired pneumonia patients, which moderately correlate with clinical severity scores such as the Pneumonia Severity Index (r = 0.554, p < 0.001), CURB-65 (r = 0.510, p < 0.001), and Acute Physiology and Chronic Health Evaluation II (r = 0.447, p < 0.001), but not with CRP, white blood cells, or neutrophils [33]. The kinetics of endocan during the first 24 h after the diagnosis of pneumonia exhibited no change for the first 3 h, but decreased 24 h later [34]. No correlations were found between endocan and IL-6, IL-10, NGAL, Resistin, MPO, ICAM-1, and VCAM-1 either at 0 h or 24 h, except for VCAM-1 at 24 h (ρ = 0.52, p = 0.009) [34].
Prediction and prognosis of hospital-acquired pneumonia
Postoperative pneumonia (POP) is a hospital-acquired pneumonia that develops at least 48 h after surgery. The incidence of POP varies depending on the patient’s comorbidities and the type of surgery, but also on the diagnostic criteria used. It ranges from 2 to 25% of operated patients in cardiac surgery [1]. In addition, POP extends the average hospital length of stay from 4 to 9 days [1]. The diagnosis of POP remains difficult, based essentially on a bundle of clinical, radiological, and biological arguments. In a study enrolling patients undergoing cardiac surgery, C-reactive protein (CRP) values measured in patients evolving to VAP did not differ before day 3 following surgery from levels observed in subjects in whom VAP did not occur [35]. In another study, procalcitonin (PCT) did not allow accurate discrimination of POP until at least 3 days after cardiac surgery [36].
A total of 330 patients undergoing cardiac surgery with and without the use of cardiopulmonary bypass (CPB) have been investigated for blood endocan. All showed increased endocan levels peaking at 6–24 h post surgery, which then slowly declined yet were not returned to baseline concentrations at day 5 [37,38,39,40]. The endocan level in cardiac surgery with CPB peaked earlier at 6 h and was higher than that in patients undergoing off-pump surgery [40, 41]. No correlation was found between endocan levels at 6 h post surgery and the duration of CPB [40]. Higher perioperative concentrations of endocan were observed in patients with the longest duration of norepinephrine support (p = 0.007) [38]. Patients developing POP exhibited higher blood endocan levels before surgery and at 6 h post surgery than those who did not develop POP (Table 2) [42, 43]. Thus, endocan allowed earlier identification of patients evolving to POP than PCT or CRP [35, 36].
Blood endocan levels at 12 h, 24 h, or 48 h post surgery were less discriminant. In routine practice, the average time to a formal diagnosis of pneumonia after cardiac surgery is 4.6 ± 3.7 days [43]. Blood endocan > 15 ng/ml 6 h after cardiac surgery had a sensitivity of 80% and a specificity of 100% to predict POP [42].
Ventilator-associated pneumonia (VAP) is another hospital-acquired pneumonia that develops 48 h or longer after mechanical ventilation. The incidence of VAP increases with the duration of mechanical ventilation. VAP causes an increase of the mortality rate estimated at around 13% [44]. Endocan has been reported as a potential predictor of poor outcomes in VAP. Indeed, in a study enrolling 42 patients with VAP, blood endocan measured at day 1 and day 7 after the diagnosis of VAP was observed at higher levels in nonsurvivors than in survivors (day 1, 12.50 ± 2.74 ng/ml vs 10.35 ± 1.86 ng/ml, p < 0.05; day 7, 24.33 ± 7.14 ng/ml vs 5.93 ± 1.56 ng/ml, p < 0.001) [45].
Diagnosis and prognosis of sepsis
Sepsis remains a devastating problem in critically ill patients despite modern treatments, with an estimated mortality rate greater than 25% in patients suffering from septic shock [5, 6]. Early recognition and treatment might prevent the development of multiorgan failure, septic shock, and sepsis-related death [6]. Currently, only a combined evaluation of clinical signs as well as hemodynamic and laboratory parameters appears to improve objective and correct diagnostic decision-making for severe sepsis and septic shock [46].
Endothelial cells play a key role in the pathogenesis of sepsis by producing cytokines and chemotactic agents, and by expressing surface adhesion molecules, which induce migration of circulating leukocytes into tissues. Indeed, both endocan and angiopoietin-2 are endothelial-specific biomarkers which increase during sepsis, worsening into multiple organ dysfunction syndrome (MODS), and decrease when sepsis improves [47]. Endocan showed correlation with VCAM-1 and E-selectin but not with ICAM-1 [48]. Severe sepsis with endocan remaining > 6.28 ng/ml at days 1, 4, and 7 is associated with poor prognosis. Indeed, for each 1 ng/ml elevation of endocan levels, the fatality rate increased by 11.1% [48].
PCT represents the major biomarker used routinely for diagnosis/prognosis/monitoring of sepsis. The diagnosis/prognosis values of endocan have been compared to those of PCT in a total of 555 patients (Table 3). Accordingly, endocan appeared as a consistent good diagnostic criterion as well as PCT. However, the correlation between endocan and PCT remained poor. Indeed, no significant correlation was found between endocan and PCT in patients with bacteriemia [49, 50]. In severe sepsis, Scherpereel et al. [19] found no significant correlation between endocan and PCT, while Pauly et al. [51] reported a significant yet poor correlation between endocan and PCT (r = 0.17, p = 0.04).
Furthermore, endocan appeared more accurate than PCT in the prognosis of sepsis. In two independent studies, ROC analysis showed that the area under the curve (AUC) of endocan to predict mortality at day 28 was observed at 0.91 by Zhao and Dong (vs 0.76 for PCT) [52] and at 0.63 by Pauly et al. (vs 0.58 for PCT) [51]. Moreover, the AUC for mortality at 6 months was found to be 0.65 for endocan and 0.59 for PCT [51]. Finally, the endocan and PCT AUC to predict mortality at ICU discharge was respectively observed as 0.71 and 0.66 [53].
Prediction and prognosis of ARDS
ARDS is a complex condition resulting from many dysregulated biological pathways. Several subtypes can be identified that respond differently to treatments according to the predominant etiology and mechanism of lung inflammation [54]. For example, blood IL-8 and TNF-α have been suggested for identifying a subtype of ARDS with exacerbated vascular permeability which can benefit from conservative fluid management [55].
ARDS should be considered as a heterogeneous syndrome where the potential interest of biomarkers to initiate targeted treatments is emerging. As a marker of lung endothelial response, endocan is expected to exert a protective role against acute lung inflammation. A total of 446 patients with or without ARDS at ICU admission have been tested currently for endocan. In polytrauma, severe sepsis, or septic shock patients without ARDS at admission, high levels of endocan were found predominantly in patients who did not develop ARDS [47, 56,57,58]. Consistently, blood endocan levels below 5.5 ng/ml in these critically ill patients without ARDS at admission were associated with a high risk of ARDS occurrence within 3 days (Table 4). In septic patients, endocan exhibited higher values than the Lung Injury Prediction Score (LIPS) to predict ARDS, with the AUC respectively calculated at 0.93 (95% CI 0.87–1.00; p < 0.001) for endocan and 0.55 (95% CI 0.39–0.71; p = 0.59) for the LIPS [58]. Conversely, high endocan levels in severe sepsis were associated with the need for mechanical ventilation independently of ARDS. However, this result might reflect a more important overall severity of sepsis rather than the development of lung inflammation [59].
In patients with ARDS at admission, endocan was significantly higher in nonsurvivors than in survivors (median (IQR) 5.01 (2.98–8.44) ng/ml vs 3.01 (2.36–4.36) ng/ml, p = 0.017). Endocan could predict mortality of ARDS independently with a hazard ratio of 1.374 (95% CI 1.150–1.641) and an AUC of 0.715 (p = 0.017) by ROC analysis [60]. In another series of 90 patients with ARDS at admission, endocan concentrations on the day after ARDS diagnosis were significantly higher in patients with poor evolution than in those with good evolution, with the median (IQR) respectively observed as 12.0 (6.8–18.6) vs 7.2 (5.4–12.5) (p < 0.01) [61]. Consistently, high endocan levels (> 13 ng/ml) in mechanically ventilated ARDS correlated with failure to withdraw from mechanical ventilation [62].
The aforestated observations suggest that endocan’s kinetics may be summarized as follows in the natural history of ARDS: endocan correlates with the overall severity of acute systemic inflammation; insufficient endocan levels observed in such cases prior to the development of lung injury seem to predict a higher risk of respiratory failure, consecutively to an insufficient anti-inflammatory effect; and increased endocan levels in constituted lung inflammation, as observed in ARDS, seem to correlate with poor outcomes. Altogether, these data suggest a delayed increase rather than an absence of an increase of blood endocan in patients who finally develop ARDS. Furthermore, this increase seems all the more marked when the ARDS is severe (Fig. 2).

Hypothetical model of endocan kinetics in acute systemic and lung inflammatory states. ARDS acute respiratory distress syndrome, ICU intensive care unit, SIRS systemic inflammatory response syndrome
Other critically ill patients
In critically ill hematology patients, endocan levels higher than 4.46 ng/ml at admission were associated with a higher cumulative probability of renal replacement therapy requirement (p = 0.006), a higher requirement of mechanical ventilation (p = 0.01), and a higher requirement of vasopressors throughout the ICU stay (p < 0.0001) [63]. Total endocan levels at admission were also independently associated with ICU mortality (odds ratio 1.39, 95% CI 1.06–1.83, p = 0.018).
Conclusions
A better description of the phenotypical characteristics of ICU patients should help to differentiate patients with high risk of poor evolution from those who are likely to evolve favorably. Several authors underline the importance to separate these different phenotypes of patients, as they may respond differently to the same treatment: because they exhibit a phenotype corresponding to a particular physiopathological mechanism [55]; or because patients with the worst outcomes are the most likely to take the maximal benefit from a given treatment [64].
This approach may help to understand why most novel therapies that have been tested in the treatment of sepsis, HAP, or ARDS over the past decade have failed to show any benefit for ICU patients.
In sepsis, the association between the development of ARDS and paradoxically low endocan levels prior to its onset might be useful to identify patients exhibiting excessive leukocyte recruitment. This could correspond to a particular phenotype that could take maximal benefit from therapies modulating unbalanced inflammation, such as corticosteroids. Conversely, excessive endocan levels in patients with constituted ARDS could also be the witness of unbalanced pulmonary inflammation, thus indicating particular treatments such as corticosteroids or restrictive vascular filling.
In HAP, increased levels of endocan, combined with other parameters, could allow one to target patients potentially eligible for early antimicrobial therapy, started before the onset of pneumonia.
Of course, it is currently unknown whether abnormal endocan levels will eventually help to guide the early initiation of personalized treatments in ICU patients. However, current data give a rationale to explore these hypotheses.
Abbreviations
- ARDS:
-
Acute respiratory distress syndrome
- AUC:
-
Area under the curve
- CPB:
-
Cardiopulmonary bypass
- CRP:
-
C-reactive protein
- HAP:
-
Hospital-acquired pneumonia
- HUVEC:
-
Human umbilical vein endothelial cell
- ICU:
-
Intensive care unit
- IGF:
-
Insulin-like growth factor
- LIPS:
-
Lung Injury Prediction Score
- MODS:
-
Multiple organ dysfunction syndrome
- PCT:
-
Procalcitonin
- POP:
-
Postoperative pneumonia
- VAP:
-
Ventilator-associated pneumonia
References
Hortal J, Giannella M, Pérez MJ, Barrio JM, Desco M, Bouza E, et al. Incidence and risk factors for ventilator-associated pneumonia after major heart surgery. Intensive Care Med. 2009;35:1518–25.
Levitt JE, Matthay MA. Clinical review: Early treatment of acute lung injury—paradigm shift toward prevention and treatment prior to respiratory failure. Crit Care. 2012;16:223.
Gajic O, Dabbagh O, Park PK, Adesanya A, Chang SY, Hou P, et al. Early identification of patients at risk of acute lung injury: evaluation of lung injury prediction score in a multicenter cohort study. Am J Respir Crit Care Med. 2011;183:462–70.
Trillo-Alvarez C, Cartin-Ceba R, Kor DJ, Kojicic M, Kashyap R, Thakur S, et al. Acute lung injury prediction score: derivation and validation in a population-based sample. Eur Respir J. 2011;37:604–9.
Vincent J-L, Marshall JC, Namendys-Silva SA, François B, Martin-Loeches I, Lipman J, et al. Assessment of the worldwide burden of critical illness: the intensive care over nations (ICON) audit. Lancet Respir Med. 2014;2:380–6.
Cohen J, Vincent J-L, Adhikari NKJ, Machado FR, Angus DC, Calandra T, et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15:581–614.
Lassalle P, Molet S, Janin A, Heyden JV, Tavernier J, Fiers W, et al. ESM-1 is a novel human endothelial cell-specific molecule expressed in lung and regulated by cytokines. J Biol Chem. 1996;271:20458–64.
Béchard D, Scherpereel A, Hammad H, Gentina T, Tsicopoulos A, Aumercier M, et al. Human endothelial-cell specific molecule-1 binds directly to the integrin CD11a/CD18 (LFA-1) and blocks binding to intercellular adhesion molecule-1. J Immunol. 2001;167:3099–106.
Yassine H, De Freitas CN, Depontieu F, Scherpereel A, Awad A, Tsicopoulos A, et al. The non glycanated endocan polypeptide slows tumor growth by inducing stromal inflammatory reaction. Oncotarget. 2015;6:2725–35.
Zheng X, Soroush F, Long J, Hall ET, Adishesha PK, Bhattacharya S, et al. Murine glomerular transcriptome links endothelial cell-specific molecule-1 deficiency with susceptibility to diabetic nephropathy. PLoS One. 2017;12:e0185250.
Iozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–52.
Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev. 1999;20:761–87.
Béchard D, Gentina T, Delehedde M, Scherpereel A, Lyon M, Aumercier M, et al. Endocan is a novel chondroitin sulfate/dermatan sulfate proteoglycan that promotes hepatocyte growth factor/scatter factor mitogenic activity. J Biol Chem. 2001;276:48341–9.
Sarrazin S, Lyon M, Deakin JA, Guerrini M, Lassalle P, Delehedde M, et al. Characterization and binding activity of the chondroitin/dermatan sulfate chain from Endocan, a soluble endothelial proteoglycan. Glycobiology. 2010;20:1380–8.
Scherpereel A, Gentina T, Grigoriu B, Sénéchal S, Janin A, Tsicopoulos A, et al. Overexpression of endocan induces tumor formation. Cancer Res. 2003;63:6084–9.
Depontieu F, de Freitas CN, Gourcerol D, Giordano J, Grigoriu B, Delehedde M, et al. Development of monoclonal antibodies and ELISA specific for the mouse vascular endocan. J Immunol Methods. 2012;378:88–94.
Matano F, Yoshida D, Ishii Y, Tahara S, Teramoto A, Morita A. Endocan, a new invasion and angiogenesis marker of pituitary adenomas. J Neuro-Oncol. 2014;117:485–91.
Maurage C-A, Adam E, Minéo J-F, Sarrazin S, Debunne M, Siminski R-M, et al. Endocan expression and localization in human glioblastomas. J Neuropathol Exp Neurol. 2009;68:633–41.
Scherpereel A, Depontieu F, Grigoriu B, Cavestri B, Tsicopoulos A, Gentina T, et al. Endocan, a new endothelial marker in human sepsis. Crit Care Med. 2006;34:532–7.
Gaudet A, Chenevier-Gobeaux C, Parmentier E, Delobel J-E, Dubucquoi S, Mathieu D, et al. Endocan is a stable circulating molecule in ICU patients. Clin Biochem. 2017;50(15):870–7.
del Toro R, Prahst C, Mathivet T, Siegfried G, Kaminker JS, Larrivee B, et al. Identification and functional analysis of endothelial tip cell-enriched genes. Blood. 2010;116:4025–33.
Abid MR, Yi X, Yano K, Shih S-C, Aird WC. Vascular endocan is preferentially expressed in tumor endothelium. Microvasc Res. 2006;72:136–45.
De Freitas CN, Legendre B, Parmentier E, Scherpereel A, Tsicopoulos A, Mathieu D, et al. Identification of a 14 kDa endocan fragment generated by cathepsin G, a novel circulating biomarker in patients with sepsis. J Pharm Biomed Anal. 2013;78–79:45–51.
Nault J-C, Guyot E, Laguillier C, Chevret S, Ganne-Carrie N, N’Kontchou G, et al. Serum proteoglycans as prognostic biomarkers of hepatocellular carcinoma in patients with alcoholic cirrhosis. Cancer Epidemiol Biomark Prev. 2013;22:1343–52.
Nirala BK, Perumal V, Gohil NK. Glycated serum albumin stimulates expression of endothelial cell specific molecule-1 in human umbilical vein endothelial cells: Implication in diabetes mediated endothelial dysfunction. Diab Vasc Dis Res. 2015;12:290–7.
Bechard D, Meignin V, Scherpereel A, Oudin S, Kervoaze G, Bertheau P, et al. Characterization of the secreted form of endothelial-cell-specific molecule 1 by specific monoclonal antibodies. J Vasc Res. 2000;37:417–25.
Daly C, Pasnikowski E, Burova E, Wong V, Aldrich TH, Griffiths J, et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc Natl Acad Sci U S A. 2006;103:15491–6.
Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev. 2004;18:1060–71.
Tissier S, Lancel S, Marechal X, Mordon S, Depontieu F, Scherpereel A, et al. Calpain inhibitors improve myocardial dysfunction and inflammation induced by endotoxin in rats. Shock Augusta Ga. 2004;21:352–7.
Zhang X, Zhuang R, Wu H, Chen J, Wang F, Li G, et al. A novel role of endocan in alleviating LPS-induced acute lung injury. Life Sci. 2018;202:89–97.
Rocha SF, Schiller M, Jing D, Li H, Butz S, Vestweber D, et al. Esm1 modulates endothelial tip cell behavior and vascular permeability by enhancing VEGF bioavailability. Circ Res. 2014;115:581–90.
De Freitas CN, Lassalle P. Highlight on mouse endocan. Circ Res. 2015;116:e69–70.
Kao S-J, Chuang C-Y, Tang C-H, Lin C-H, Bien M-Y, Yu M-C, et al. Plasma endothelial cell-specific molecule-1 (ESM-1) in management of community-acquired pneumonia. Clin Chem Lab Med. 2014;52:445–51.
Smart L, Bosio E, Macdonald SPJ, Dull R, Fatovich DM, Neil C, et al. Glycocalyx biomarker syndecan-1 is a stronger predictor of respiratory failure in patients with sepsis due to pneumonia, compared to endocan. J Crit Care. 2018;47:93–8.
Jiao J, Wang M, Zhang J, Shen K, Liao X, Zhou X. Procalcitonin as a diagnostic marker of ventilator-associated pneumonia in cardiac surgery patients. Exp Ther Med. 2015;9:1051–7.
Jebali MA, Hausfater P, Abbes Z, Aouni Z, Riou B, Ferjani M. Assessment of the accuracy of procalcitonin to diagnose postoperative infection after cardiac surgery. Anesthesiology. 2007;107:232–8.
Madhivathanan PR, Fletcher N, Gaze D, Thomson R, Chandrasekaran V, Al-Subaie N, et al. Perioperative kinetics of endocan in patients undergoing cardiac surgery with and without cardiopulmonary bypass. Cytokine. 2016;83:8–12.
Bouglé A, Allain P-A, Favard S, Ait Hamou N, Carillion A, Leprince P, et al. Postoperative serum levels of Endocan are associated with the duration of norepinephrine support after coronary artery bypass surgery. Anaesth Crit Care Pain Med. 2018.
Stoppelkamp S, Veseli K, Stang K, Schlensak C, Wendel HP, Walker T. Identification of predictive early biomarkers for sterile-SIRS after cardiovascular surgery. PLoS One. 2015;10:e0135527.
Poinsot P-A, Barrucand B, Ecarnot F, Lassalle P, Besch G, Chocron S, et al. Kinetics of endocan in patients undergoing cardiac surgery with and without cardiopulmonary bypass. Cytokine. 2018;110:328–32.
Brettner F, Chappell D, Schwartz L, Lukasz A, Kümpers P, Becker BF, et al. Vascular endothelial dysfunction during cardiac surgery: on-pump versus off-pump coronary surgery. Eur Surg Res. 2017;58:354–68.
Perrotti A, Chenevier-Gobeaux C, Ecarnot F, Barrucand B, Lassalle P, Dorigo E, et al. Relevance of endothelial cell-specific molecule 1 (endocan) plasma levels for predicting pulmonary infection after cardiac surgery in chronic kidney disease patients: the Endolung pilot study. Cardiorenal Med. 2017;8:1–8.
Perrotti A, Chenevier-Gobeaux C, Ecarnot F, Bardonnet K, Barrucand B, Flicoteaux G, et al. Is endocan a diagnostic marker for pneumonia after cardiac surgery? The ENDOLUNG study. Ann Thorac Surg. 2017;105(2):535–41.
Melsen WG, Rovers MM, Groenwold RHH, Bergmans DCJJ, Camus C, Bauer TT, et al. Attributable mortality of ventilator-associated pneumonia: a meta-analysis of individual patient data from randomised prevention studies. Lancet Infect Dis. 2013;13:665–71.
El Halim Ashraf A, Sayed M. Serum endocan role in diagnosis and prognosis of ventilator associated pneumonia. Egypt Soc Chest Dis Tuberc. 2015;64:865–9.
Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013;41:580–637.
Ioakeimidou A, Pagalou E, Kontogiorgi M, Antoniadou E, Kaziani K, Psaroulis K, et al. Increase of circulating endocan over sepsis follow-up is associated with progression into organ dysfunction. Eur J Clin Microbiol Infect Dis. 2017;36(10):1749–56.
Hsiao S-Y, Kung C-T, Tsai N-W, Su C-M, Huang C-C, Lai Y-R, et al. Concentration and value of endocan on outcome in adult patients after severe sepsis. Clin Chim Acta. 2018;483:275–80.
Seo K, Kitazawa T, Yoshino Y, Koga I, Ota Y. Characteristics of serum endocan levels in infection. PLoS One. 2015;10:e0123358.
Zuwala-Jagiello J, Simon K, Kukla M, Murawska-Cialowicz E, Gorka-Dynysiewicz J, Grzebyk E, et al. Increased circulating endocan in patients with cirrhosis: relation to bacterial infection and severity of disease. J Physiol Pharmacol. 2017;68:273–82.
Pauly D, Hamed S, Behnes M, Lepiorz D, Lang S, Akin I, et al. Endothelial cell-specific molecule-1/endocan: diagnostic and prognostic value in patients suffering from severe sepsis and septic shock. J Crit Care. 2016;31(1):68–75.
Zhao R, Dong S. Clinical value of serum endocan and procalcitonin in early diagnosis and prognosis evaluation of sepsis. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2017;29:321–6.
Mihajlovic DM, Lendak DF, Brkic SV, Draskovic BG, Mitic GP, Novakov Mikic AS, et al. Endocan is useful biomarker of survival and severity in sepsis. Microvasc Res. 2014;93:92–7.
Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA, et al. Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med. 2014;2:611–20.
Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, et al. ARDS subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med. 2017;195(3):331–8.
Mikkelsen ME, Shah CV, Scherpereel A, Lanken PN, Lassalle P, Bellamy SL, et al. Lower serum endocan levels are associated with the development of acute lung injury after major trauma. J Crit Care. 2012;27:522.e11–7.
Palud A, Parmentier-Decrucq E, Pastre J, De Freitas CN, Lassalle P, Mathieu D. Evaluation of endothelial biomarkers as predictors of organ failures in septic shock patients. Cytokine. 2015;73:213–8.
Gaudet A, Parmentier E, Dubucquoi S, Poissy J, Duburcq T, Lassalle P, et al. Low endocan levels are predictive of acute respiratory distress syndrome in severe sepsis and septic shock. J Crit Care. 2018;47:121–6.
Mangat M, Amalakuhan B, Habib S, Reyes LF, Hinojosa CA, Rodriguez AH, et al. High endocan levels are associated with the need for mechanical ventilation among patients with severe sepsis. Eur Respir J. 2017;50:1700013.
Tang L, Zhao Y, Wang D, Deng W, Li C, Li Q, et al. Endocan levels in peripheral blood predict outcomes of acute respiratory distress syndrome. Mediat Inflamm. 2014;2014:625180.
Orbegozo D, Rahmania L, Irazabal M, Mendoza M, Annoni F, De Backer D, et al. Endocan as an early biomarker of severity in patients with acute respiratory distress syndrome. Ann Intensive Care. 2017;7:93.
Tsangaris I, Tsantes A, Vrigkou E, Kopterides P, Pelekanou A, Zerva K, et al. Angiopoietin-2 levels as predictors of outcome in mechanically ventilated patients with acute respiratory distress syndrome. Dis Markers. 2017;2017:6758721.
Zafrani L, Resche-Rigon M, De Freitas CN, Gaudet A, Mathieu D, Parmentier-Decrucq E, et al. Endothelial cell-specific molecule-1 in critically ill patients with hematologic malignancy. Crit Care Med. 2018;46:e250–7.
Iwashyna TJ, Burke JF, Sussman JB, Prescott HC, Hayward RA, Angus DC. Implications of heterogeneity of treatment effect for reporting and analysis of randomized trials in critical care. Am J Respir Crit Care Med. 2015;192:1045–51.
Acknowledgements
Not applicable.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors contributed to writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
NDFC and LP are employees of Lunginnov. PL is cofounder of Lunginnov. The remaining authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
De Freitas Caires, N., Gaudet, A., Portier, L. et al. Endocan, sepsis, pneumonia, and acute respiratory distress syndrome. Crit Care 22, 280 (2018). https://doi.org/10.1186/s13054-018-2222-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-018-2222-7