
Abstract
Background
X-linked hypophosphatemic rickets (XLH) is the first cause of inherited hypophosphatemia and is caused by mutation in the PHEX gene, resulting in excessive expression of the phosphaturic factor FGF23. Symptoms are mainly related to rickets in children and osteomalacia in adults and cause several complications that can be highly invalidating. Due to its rarity, XLH is poorly known and diagnosis is frequently delayed. Conventional treatment is based on oral phosphate salts supplementation and activated vitamin D analogs, which however, cannot cure the disease in most cases.
Objective
Due to the low prevalence of XLH, an experts’ opinion survey was conducted across Italian centers to collect data on XLH and on its management.
Methods
A questionnaire was developed by a group of experts to collect data on XLH epidemiology, diagnosis and treatment in Italy.
Results
Data from 10 Italian centers (nine of which pediatric) on 175 patients, followed between 1998 and 2017, were included in the survey. Most patients were followed since childhood and 63 children became adults during the investigated period. The diagnosis was made before the age of 1 and between 1 and 5 years in 11 and 50% of cases, respectively. Clinically apparent bone deformities were present in 95% of patients. These were ranked moderate/severe in 75% of subjects and caused growth stunting in 67% of patients. Other frequent complications included bone pain (40%), dental abscesses (33%), and dental malpositions (53%). Treatment protocols varied substantially among centers. Nephrocalcinosis was observed in 34% of patients. Tertiary hyperparathyroidism developed in 6% of patients.
Conclusions
XLH remains a severe condition with significant morbidities.
Similar content being viewed by others
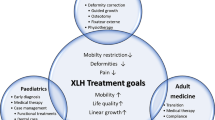


Background
X-linked hypophosphatemic rickets (XLH) (OMIM 307800) is a rare disease caused by mutations in the PHEX gene. The gene encodes for an endopeptidase that is primarily expressed on the surface of osteoblast, osteocytes, odontoblasts and cementoblasts [1, 2]. The disease follows an X-linked mode of transmission with dominant expression, and represents the first cause of hypophosphatemic rickets [1]. Female carriers usually present symptoms similar to male patients [3]. Unless patients are identified by family screening, they are usually diagnosed in the first years of life after noticing bowing of the lower legs, delayed walking, abnormal walking gait, and/or growth stunting. The disease may be initially misdiagnosed for vitamin D deficiency, but biochemical investigations and in some cases, lack of response to vitamin D supplements usually allow the diagnosis. Typically, patients show at diagnosis low serum phosphate levels secondary to increased phosphaturia, elevated alkaline phosphatase, normal parathyroid hormone (PTH) levels and low-normal urine calcium. X-rays demonstrate typical features of rickets without significant bone resorption, as opposed to rickets secondary to vitamin D deficiency [1, 4]. Growth plates of long bones have a cupped appearance, with increased thickness and irregularities. Positive family history often facilitates the diagnosis, but de novo mutations occur frequently and some adults lack overt features of XLH [5].
Other frequent symptoms in children include rachitic rosary, dolichocephaly with parietal flattening and frontal bossing, and bone pain. More rarely, patients are diagnosed with craniosynostosis, type 1 Chiari malformations, intracranial hypertension or hearing impairment [5, 6]. The majority of children with overt XLH grow asymmetrically. They have slightly decreased sitting height and markedly decreased leg length. Patients often have dental symptoms secondary to dental malpositions and/or to hypomineralization of dentine and hypoplasia of the enamel, which cause frequent abscesses [1, 5, 6].
Adult patients usually present with symptoms secondary to defective bone mineralization (osteomalacia) and often complain of bone and joint pain [7]. They frequently develop pseudofractures, as well as invalidating osteoarthropathies that involve predominantly the ankles, knees and sacroiliac joints [7]. They can also develop enthesopathy, which is caused by calcification of tendons and ligaments. These are particularly prominent in the ankles, knees, pelvic joints and spine [8, 9].
The above symptoms highlight the severity of the disease, which may not be recognized due to its rarity. For this same reason clinical and biochemical data on large cohorts of patients are scanty.
On these premises, we have conducted a survey in ten Italian reference centers for the diagnosis and treatment of XLH, to evaluate the epidemiology of the disease, diagnostic challenges, treatment attitudes, organization of care and disease burden.
Methods
A questionnaire was developed by a group of experts to collect retrospective data on XLH in Italy. The experts met initially in September 2016 and subsequently twice to elaborate the questionnaire, analyze the initial results and solve open issues to finalize data collection. The survey was limited to data that could be collected reliably from medical charts. Single patient data were not transferred; only aggregate data from each center were collected, to protect privacy. Patients were included in the study if the following data were retrievable from clinical charts: diagnosis, epidemiology and treatment pattern. Patients were excluded if such data were not available.
The diagnosis of XLH was based on genetic confirmation of a PHEX mutation, or, if not available, was based on symptoms, family history, radiological evidence of rickets, and laboratory tests.
Ten units were identified in North and Central Italy after screening pediatric, endocrinology and nephrology units for their expertise in XLH, as well as regional registries for rare diseases.Footnote 1 The centers included in the study were selected based on: (i) number of patients treated every year and inclusion in the National Network for rare diseases. The questionnaire investigated the following disease areas: epidemiology, diagnosis, disease severity, treatment, organization of care.
Quality of life data were not available in the charts, therefore, the impact of the disease burden was estimated by the clinicians, using a 5-point scale (1: low impact, 5: high impact), inspired from the EUROQOL questionnaire, exploring various dimensions of daily life (functional capacity, mobility, usual activities, pain/discomfort).
Descriptive statistical analyses were used to illustrate data. Results are expressed as mean ± standard deviation (SD), median and range or percentage, as appropriate.
Results
Data from 175 patients that were followed between 1998 and 2017 in the 10 participating centers were included in the survey (Table 1).
Most centers were pediatric units, therefore data on adult patients were limited. Twenty-two patients were diagnosed before 1998 and followed afterwards. Of the 153 patients (75% males and 25% females) diagnosed after 1998, 135 were pediatric (0–17 years of age). For most criteria, data were available for all patients; however, some data could not be traced back from all medical charts or were restricted to pediatric patients.
Diagnosis
The distribution of the age at diagnosis is reported in Table 2. Such info was available for 150 patients only; of those, over 60% were diagnosed between 1 and 5 years No trends towards earlier diagnosis in recent years was observed by comparing patients diagnosed after and before 2012 (data not shown).
A genetic test was performed in 65 patients and a pathogenic PHEX mutation was confirmed in 56 cases (86%). For the reminder of the cohort, the diagnosis was based on clinical grounds. Physicians from the 10 centers were hence asked to rank the most important features leading to the diagnosis of XLH. These included rickets, hypophosphatemia and, when available, a positive family history. Interestingly, 17% of patients had been diagnosed with vitamin D deficiency prior to the diagnosis of XLH. Overall, positive family history was present in 33% (54/164) of patients in which the information was available.
Symptoms
Most (94,8%) patients had lower leg deformities, which were ranked severe in 28%, moderate in 47%, and mild or absent in the remainder 25% of patients. “Severe” deformities referred to overt genu valgus or genu varum that impaired normal walking, “moderate” to less important deformities with mild or no walking impairment, and “mild” to minor limb deformities with regular joint alignment (Fig. 1).

Severity of symtoms
Bone pain was reported in two third (65,6%) of patients but was ranked as mild to moderate in most (79,3%) cases. More than half (67%) of patients had growth retardation (height lower than − 2 SD). Scoliosis or osteopenia was seldom reported. Pathological fractures were observed in nearly one third of patients. Craniosynostosis was reported in 5% of the cohort. Dental malpositions were present in half of patients (53%) and approximately one third (33%) had experienced at least one dental abscess. Hearing impairment and neurological problems were infrequent (observed in 5 and 9% of cases, respectively - Table 3).
Medical and orthopedic treatment
All centers prescribed conventional treatment to all pediatric patients, including oral phosphate supplements and active vitamin D analogues, namely calcitriol and alfacalcidol. The latter was generally preferred in young children because liquid solutions are commercially available in Italy. In some centers non-active vitamin D supplements were also prescribed. Doses were modified according to age, but the dose range was considerably wide. For example, the dose of calcitriol for school age children ranged from 0.25 to 2.0 mcg/day. In some centers, doses of vitamin D was calculated according to weight, while other centers used fixed doses and adapted treatment according to the biochemical response. Phosphate salt prescription was equally very variable. For school age children, the dose varied from 15 to 60 mg/Kg/day. Phosphate was prescribed in different pharmacologic preparations, including using IV solution for oral administration, galenic solutions (Joulie or modified Joulie solutions), and commercially available tablets or liquid solutions, some of which were imported from other European countries. The frequency of uptake also varied, but most centers prescribed four doses per day in preadolescent patients and usually reduced the frequency to 2–3 doses per day in adolescents. Two centers did not recommend systematic treatment in adults, while the remaining centers advocated to continue low dose treatment with phosphate supplements and/or vitamin D, irrespectively to the presence of symptoms.
Likewise, metabolic targets for optimal disease control were, to some extent, different between centers. Unanimously, normalization of alkaline phosphatase was the main biochemical marker of a satisfactory treatment, with optimal target values within the normal range and acceptable values below 1.5-fold the upper limit of normal range for age. All experts tried to keep serum calcium and PTH levels within the normal range. In six centers, serum phosphate levels did not represent a target for treatment. Nonetheless, four centers aimed at serum phosphate levels above a given threshold, ranging 2–3 mg/dl.
Complications of conventional treatment include nephrocalcinosis and hyperparathyroidism. Retrospective evaluation of renal ultrasound did not allow ranking the severity of nephrocalcinosis. Likewise, hyperparathyroidism was difficult to assess and for consistency, only patients requiring surgery or treatment with cinacalcet for tertiary hyperparathyroidism were recorded. Overall, nephrocalcinosis was reported in 34% of patients, while tertiary hyperparathyroidism was developed in 6%.
From the orthopedic standpoint, half of patients had undergone at least one surgical procedure. These included osteotomies and epiphysiodesis, with the latter procedure being more frequently used in recent years. More detailed analyses were not possible due to the multicentric nature of the survey and changes in the clinical practice and indications for surgery over the years, including the type of procedure.
Professional caregivers
Most pediatric patients were followed by pediatric endocrinologists (7/10), pediatricians with a special interest in XLH (2/10) and pediatric nephrologists (1/10). In all cases a multidisciplinary team, including orthopedic surgeons, radiologists, dentists, and possibly geneticists were activated. Neurologists, neurosurgeons, physical therapists were also involved in the team, though less frequently.
During the time frame of the survey, 63 patients had reached adult age. Of these, 31 continued to be followed in pediatric centers (11 aged 18–25 years, and 22 aged > 25 years). The remaining adult patients were addressed to adult endocrinologists, nephrologists or rheumatologists.
Disease burden
Quality of life (QoL) data were not available due to the retrospective nature of the study. Nonetheless, experts participating in the study were asked to estimate the impact of the disease on patients, using a 5-point scale (1: low impact, 5: high impact), inspired from the EUROQOL questionnaire, which explored several dimensions of daily life (functional capacity, mobility, usual activities, pain/discomfort). The estimated impact of the disease on daily life ranked on average 4.5 for patients with severe disease, 3.7 for patients with moderate disease and 1.8 for those with mild symptoms. “Severe” disease corresponded to patients with severe leg deformities as defined above and/or major complications, “moderate” to moderate bone changes and limited complications, such as dental malpositions, and “mild” to the remaining patients. As expected, rickets secondary to vitamin D deficiency ranked best (1.8). Osteopetrosis ranked worst (4.61) and XLH had and intermediate score of 3.5 (Table 4). These estimates are clearly limited by the retrospective nature of the survey and reflect physician’s opinion and not patient perception.
Discussion
XLH is a rare inherited disease, characterized by rickets and abnormal renal phosphate losses. Unrestricted expression of the phosphaturic hormone fibroblast growth factor 23 (FGF23) secondary to PHEX mutations cause most symptoms. Animal studies demonstrate that inactivation of PHEX increases FGF23 levels, which downregulates sodium-dependent phosphate transporters and 1 alpha hydroxylation of 25(OH)-vitamin D in renal proximal cells. The mechanisms by which PHEX mutations cause FGF23 production are still poorly understood. However, experimental and clinical data show that neutralizing antibodies against FGF23 prevent most XLH symptoms, thus demonstrating that this hormone is the principal culprit in this disease [10, 11].
Symptoms are mainly related to rickets in children and osteomalacia in adults, which cause different complications. XLH during childhood manifests mainly with bone deformities (bowed legs, genu varum or valgus, rachitic rosary, dolichocephaly), short stature, bone pain and dental abnormalities. The diagnosis is often delayed, which can significantly impact on long-term outcome [5, 6].
Conventional therapy includes supplementation with active vitamin D analogues and with oral phosphate salts. In most patients, symptoms improve, but the disease is not cured by conventional therapy [2]. This is most likely related to direct effects of high FGF23 levels on peripheral tissues, which are not inhibited by conventional treatment and may even be enhanced, since FGF23 levels increase in treated children [2]. Moreover, treatment is limited by side effects, in particular hyperparathyroidism and nephrocalcinosis that frequently develop when using high doses of therapy.
The present study was conducted to evaluate the current status in the diagnosis and treatment of XLH in Italy. Overall, results are in line with available data in the literature, although with some noticeable differences.
The prevalence of XLH is estimated between 1.2–3.0/60,000 [12, 13]. The expected number of prevalent pediatric patients with XLH in Italy is 187–470, considering an overall population of 9.374.322 persons aged 0–17 years. Our survey included 150 children diagnosed over a period of 20 years in 10 Italian centers with high experience in treating XLH. This number is lower than expected, indicating that the disease is either underdiagnosed or often treated in less specialized centers.
The diagnosis of XLH is based on clinical, radiological and biochemical grounds and can be confirmed by demonstrating a PHEX mutation. A positive X-linked family history usually confirms the diagnoses. When genetic analysis is not available and family history is absent, the disease is presumed, but cannot be demonstrated. In other cohorts, positive family history is observed in approximately 60% of cases [14, 15], as opposed to 33% in our survey. Most patients in which genetic testing was performed however, had a positive mutation, including patients with negative family history. It is unlikely that de novo mutations are more frequent in Italy. Most likely, parents of many patients were poorly symptomatic and/or reliable family history could not be traced back from medical records.
Our patients had overt symptoms with similar frequencies to those reported in the literature, with the exception of Chiari malformation and craniosynostosis. These are probably underdiagnosed because radiologic investigations, including brain MRI, are not routinely performed in most centers.
Conventional treatment usually improves bone pain and other XLH features but carries the risk of causing hyperparathyroidism and/or nephrocalcinosis. In the clinical practice, PTH levels, urinary calcium excretion and renal ultrasound are evaluated at regular interval to adapt treatment. Overall, nephrocalcinosis has been reported in the literature in 30–70% of patients [13, 15,16,17]. In the vast majority of cases, renal function is preserved, but very long-term data are not available yet. Most publications report a positive relationship between oral phosphate doses and nephrocalcinosis, whereas the association with vitamin D therapy is less strong [13, 17,18,19]. No increase in the supersaturation of either calcium oxalate or phosphate in urines was observed in one study including 20 patients with XLH [20]. Conceivably, the incidence observed in the Italian cohort is lower than previously reported (34%) because physicians have learned to balance treatment to avoid this complication. Hydrochlorothiazide has been shown to decrease calciuria in XLH [21] and potassium citrate may help preventing calcium precipitation. The prescription of these treatments was not included in the survey.
The incidence of hyperparathyroidism was more difficult to estimate because PTH levels are known to fluctuate overtime. Due to the retrospective nature of the survey only tertiary hyperthyroidism was evaluated and was diagnosed in 6% of patients.
Likewise, indication for orthopedic surgery in XLH are not clearly established and surgical attitudes have changed over the years [22,23,24,25]. If possible, osteotomies are performed preferentially after completion of growth [22, 26]. The only consistent finding in our survey was the increased use of epiphisiodesis in most recent years in all centers, while the overall number of surgeries did not change substantially overtime.
As expected, XLH has a negative impact on patient’s life. Most patients treated with conventional therapy are not cured and continue to experience complications related to their disease. The majority of children in this survey had normal daily activities despite their physical impairment. The impact in adults could not be estimated, due to the limited number of patients. However, data in the literature show that QoL in these patients can be severely affected [7, 27].
Conclusion
This survey investigated the epidemiology, diagnosis and treatment of XLH in Italy. Despite its limitations, it highlights the severity of the disease and lack of homogeneity in treatment attitudes among specialists. Most importantly, results show a number of unmet needs and the limitations of conventional treatment that cannot cure the disease. In addition, these may cause adverse events, particularly when high dosages are used.
New therapies with anti-FGF23 antibodies are likely to modify the prognosis of XLH in the future [28,29,30].
Notes
Participating centers included: Bambino Gesù Children’s Hospital – Rome (F.E. and M.C.), Borgo Roma Hospital-University of Verona – Verona (F.A.), Istituto Auxologico Italiano IRCCS – Milan (M.L.B. and I.C.), IRCCS Cà Granda Foundation-Maggiore Policlinico Hospital – Milan (C.E.V.), Giannina Gaslini Institute – Genoa (N.D.I. and M.M.), S. Orsola Malpighi University Hospital – Bologna (A.C., A.B. and F.B.), Regina Margherita Children’s Hospital, University of Turin – Turin (L.D.S. and D.T.), University Hospital of Pisa – Pisa (G.B.), IRCCS San Raffaele Hospital-Vita-Salute San Raffaele University – Milan (G.W. and S.M.), Careggi University Hospital-University of Florence – Florence (M.L.B.).
Abbreviations
- FGF23:
-
Fibroblast growth factor23
- PTH:
-
ParaThyroid hormone
- QoL:
-
Quality of life
- SD:
-
Standard deviation
- XLH:
-
X-Linked Hypophosphatemic
References
Haffner D, Waldegger S. Disorders of phosphorus metabolism. In: Geary DF, Schaefer F, editors. Pediatric kidney disease. Berlin Heidelberg: Springer-Verlag; 2016. p. 953–72.
Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, et al. Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res. 2008;23(6):939–48.
Whyte MP, Schranck FW, Armamento-Villareal R. X-linked hypophosphatemia: a search for gender, race, anticipation, or parent of origin effects on disease expression in children. J Clin Endocrinol Metab. 1996;81(11):4075–80.
Carpenter TO, Shaw NJ, Portale AA, Ward LM, Abrams SA, Pettifor JM. Rickets. Nat Rev Dis Primers. 2017;3:17101.
Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician's guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26(7):1381–8.
Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 2014;3(1):R13–30.
Che H, Roux C, Etcheto A, Rothenbuhler A, Kamenicky P, Linglart A, et al. Impaired quality of life in adults with X-linked hypophosphatemia and skeletal symptoms. Eur J Endocrinol. 2016;174(3):325–33.
Connor J, Olear EA, Insogna KL, Katz L, Baker S, Kaur R, et al. Conventional therapy in adults with X-linked hypophosphatemia: effects on Enthesopathy and dental disease. J Clin Endocrinol Metab. 2015;100(10):3625–32.
Liang G, Katz LD, Insogna KL, Carpenter TO, Macica CM. Survey of the enthesopathy of X-linked hypophosphatemia and its characterization in Hyp mice. Calcif Tissue Int. 2009;85(3):235–46.
Liu S, Zhou J, Tang W, Menard R, Feng JQ, Quarles LD. Pathogenic role of Fgf23 in Dmp1-null mice. Am J Physiol Endocrinol Metab. 2008;295(2):E254–61.
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561–8.
Endo I, Fukumoto S, Ozono K, Namba N, Inoue D, Okazaki R, et al. Nationwide survey of fibroblast growth factor 23 (FGF23)-related hypophosphatemic diseases in Japan: prevalence, biochemical data and treatment. Endocr J. 2015;62(9):811–6.
Rafaelsen S, Johansson S, Raeder H, Bjerknes R. Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, phenotypes, and treatment complications. Eur J Endocrinol. 2016;174(2):125–36.
Gaucher C, Walrant-Debray O, Nguyen TM, Esterle L, Garabedian M, Jehan F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet. 2009;125(4):401–11.
Makitie O, Doria A, Kooh SW, Cole WG, Daneman A, Sochett E. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2003;88(8):3591–7.
Berndt M, Ehrich JH, Lazovic D, Zimmermann J, Hillmann G, Kayser C, et al. Clinical course of hypophosphatemic rickets in 23 adults. Clin Nephrol. 1996;45(1):33–41.
Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silink M. Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med. 1991;325(26):1843–8.
Keskin M, Savas-Erdeve S, Sagsak E, Cetinkaya S, Aycan Z. Risk factors affecting the development of nephrocalcinosis, the most common complication of hypophosphatemic rickets. J Pediatr Endocrinol Metab. 2015;28(11–12):1333–7.
Vaisbich MH, Koch VH. Hypophosphatemic rickets: results of a long-term follow-up. Pediatr Nephrol. 2006;21(2):230–4.
Patzer L, van't Hoff W, Shah V, Hallson P, Kasidas GP, Samuell C, et al. Urinary supersaturation of calcium oxalate and phosphate in patients with X-linked hypophosphatemic rickets and in healthy schoolchildren. J Pediatr. 1999;135(5):611–7.
Alon U, Chan JC. Effects of hydrochlorothiazide and amiloride in renal hypophosphatemic rickets. Pediatrics. 1985;75(4):754–63.
Gizard A, Rothenbuhler A, Pejin Z, Finidori G, Glorion C, de Billy B, et al. Outcomes of orthopedic surgery in a cohort of 49 patients with X-linked hypophosphatemic rickets (XLHR). Endocr Connect. 2017;6(8):566–73.
Kocaoglu MBF, Sen C, Eralp L, Balci HI. Combined technique for the correction of lower-limb deformities resulting from metabolic bone disease. . 2011; Jan;93(1). J Bone Joint Surg Br. 2011;93(1):52–6.
Matsubara HTH, Kabata T, Sakurakichi K, Watanabe K, Tomita K. Deformity correction for vitamin D-resistant hypophosphatemic rickets of adults. Arch Orthop Trauma Surg. 2008;128(10):1137–43.
Sharkey MS, Grunseich K, Carpenter TO. Contemporary medical and surgical management of X-linked Hypophosphatemic rickets. J Am Acad Orthop Surg. 2015;23(7):433–42.
Horn A, Wright J, Bockenhauer D, Van't Hoff W, Eastwood DM. The orthopaedic management of lower limb deformity in hypophosphataemic rickets. J Child Orthop. 2017;11(4):298–305.
Beck-Nielsen SS, Brusgaard K, Rasmussen LM, Brixen K, Brock-Jacobsen B, Poulsen MR, et al. Phenotype presentation of hypophosphatemic rickets in adults. Calcif Tissue Int. 2010;87(2):108–19.
Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. 2014;124(4):1587–97.
Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. 2018;378(21):1987–98.
Insogna KL, Briot K, Imel EA, Kamenicky P, Ruppe MD, Portale AA, et al. A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of Burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: week 24 primary analysis. J Bone Miner Res. 2018.
Acknowledgements
Not applicable.
Funding
The following authors received consultation fees from Kyowa Kirin SrL: Emma F., Cappa M., Antoniazzi. F, Bianchi M.L., Chiodini I., Eller Vainicher C., Di Iorgi N., Maghnie M., Cassio A., Balsamo A., Baroncelli G.I., Mora S., Brandi M.L., Weber G.
Meetings to elaborate the questionnaire and discuss the manuscript were organized by MAP Provider SrL that received consultation fee from Kyowa Kirin srl.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
The following authors contributed in providing retrospective data and interpretation of results: EF, CM, AF, BML, CI, EVC, DIN, MM, CA, BA, BF, deSL, TD, BGI, MS, BML, WG. Authors: LEP and DAA. contributed in the conduct of the analysis, elaboration of results and manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Emma, F., Cappa, M., Antoniazzi, F. et al. X-linked hypophosphatemic rickets: an Italian experts’ opinion survey. Ital J Pediatr 45, 67 (2019). https://doi.org/10.1186/s13052-019-0654-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-019-0654-6