
Abstract
Growth hormone deficiency (GHD) is a relatively uncommon and heterogeneous endocrine disorder presenting in childhood with short stature. However, during the neonatal period, the metabolic effects of GHD may to require prompt replacement therapy to avoid possible life-threatening complications. An increasing amount of data suggests the importance of an early diagnosis and treatment of GHD because of its auxological, metabolic, and neurodevelopmental features with respect to the patients diagnosed and treated later in life.
The available results show favourable auxological outcomes for patients with GHD diagnosed and treated with r-hGH early in life compared with those from patients with GHD who do not receive this early diagnosis and treatment. Because delayed referral for GHD diagnosis and treatment is still frequent, these results highlight the need for more attention in the diagnosis and treatment of GHD.
Despite these very encouraging data regarding metabolic and neurodevelopmental features, further studies are needed to better characterize these findings. Overall, the importance of early diagnosis and treatment of GHD needs to be addressed.
Similar content being viewed by others

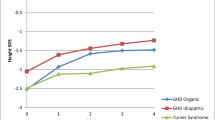
Background
Growth hormone deficiency (GHD) is a relatively uncommon and heterogeneous disorder, in terms of aetiology, pathogenesis, age of diagnosis, and the cause of growth retardation and short stature [1]. The prevalence of GHD in childhood widely varies between 1/3480 and 1/30,000 children [2], even if the milder phenotype has a frequency of nearly 1:2000 [3].
In children, the main manifestation of GHD is represented by growth failure [1], and growth hormone (GH) ameliorates the short- and long-term height prognosis in these patients [4]. However, in children, GH may influence bone mineralization and bring about a large number of metabolic effects, involving lipid and glucose homeostasis and lean and fat mass [5].
The long-term growth response to GH treatment in GHD children may be conditioned by both pre-treatment and treatment-related factors, such as birth weight, baseline height standard deviation score (SDS), age at the onset of treatment, height at the start of puberty, treatment duration, target height (TH), mean frequency of injections, and doses of GH [6,7,8]. However, in a small subset of patients, GHD is recognizable in infancy or early childhood (i.e., < 3 years of age), and some data suggest that an early GHD diagnosis and a concomitant early start of treatment have proven effective in normalizing height [8,9,10,11,12,13,14,15,16,17,18].
It appears to be of great concern to consider early diagnosis and treatment as an advantageous and cost-effective strategy in achieving height improvement and a normal growth pattern during childhood compared with delayed start of treatment [19]. Additionally, the psychological benefits are greater, and the improvement in cost effectiveness is vast [9]. On the whole, published data suggest that the early diagnosis and treatment of GHD may have metabolic and neurodevelopmental consequences in childhood and adulthood [20].
In this review, we retrospectively evaluate data regarding early GHD diagnosis and treatment with r-hGH, examining their auxological, metabolic, and neurodevelopmental features with a focus on patients who have been diagnosed and treated later in life. For this study, relevant papers published in English were identified through systematic searches of the PubMed, EMBASE and Cochrane databases. Keywords in the literature search were entered in all combinations. Searches were augmented by manually reviewing the reference lists of all original articles and all systematic review articles, with each study being evaluated for inclusion.
GH actions in the growth plate
The growth plate is a thin layer of cartilage localized between the epiphyseal and metaphyseal bone at the ends of the long bones. Longitudinal bone growth occurs at the growth plate by endochondral ossification, a process in which cartilage is first formed and then remoulded into bone tissue in a determined temporal and spatial organization. The growth plate consists of three principal layers: the germinal zone, proliferative zone, and hypertrophic zone [21]. At the epiphyseal end of the growth plate, the germinal zone contains the resting progenitors (stem cells), which differentiate into chondrocytes and progress through the proliferative zone. In this matrix-rich setting, the chondrocytes undergo cell divisions and arrange in columns that parallel the longitudinal axis of the bone. Immediately after proliferating, chondrocytes lose their capacity to divide and start to differentiate into prehypertrophic chondrocytes, increasing in size at same time [22]. They then further undergo terminal differentiation into hypertrophic chondrocytes, which have a round appearance and secrete large amounts of matrix proteins [23, 24]. The mineralization process, in combination with low oxygen tension, determines the changes in the environment that allow vascular invasion from the marrow of the metaphysis, thereby enabling the recruitment of osteoclasts and differentiating osteoblasts that remodel the newly formed cartilage into bone tissue [25, 26]. Subsequently, the mineralized chondrocytes undergo programmed apoptosis, leaving a platform for new bone formation. The final effect is that new bone tissue is progressively created at the bottom of the growth plate, resulting in bone elongation. During puberty and its associated rapid growth, the processes of cellular addition and replacement are coupled so that the width of the growth plate remains constant [27], while at the end of the growth period, the growth plate narrows and finally disappears as a result of systemic control and the fusion of the epiphysis with the metaphysis. However, it is now believed that regulation is intrinsic to the growth plate and that growth plate fusion may not precede, but rather follow, the cessation of growth [28, 29].
In children, maintenance of growth is a complex process that is regulated by a multitude of genetic, hormonal, environmental and nutritional factors [30, 31]. The major systemic hormones that regulate longitudinal bone growth during childhood are GH and insulin-like growth factor 1 (IGF-I), thyroid hormone (T3 and T4), glucocorticoids, and during puberty, sex steroids (androgens and oestrogens) [32, 33]. However, two of the most important regulators of postnatal bone growth are GH and IGF-I.
Growth hormone and IGF-I are potent stimulators of longitudinal bone growth [34]. Pituitary adenoma in childhood or adulthood causes excessive GH secretion, leading to gigantism or acromegaly, respectively [35, 36]. Conversely, GH deficiency or insensitivity due to GH-receptor mutations or defects in GH signalling pathways or in the formation of GH-secretory cells all result in severe dwarfism [37,38,39,40,41,42]. Data on patients with untreated isolated GH deficiency suggest that it leads to an average final height SDS of −4.7 (−6.1 to −3.9), [43].
In the early 1950s, Salmon and Daughaday described the somatomedin hypothesis. They proposed that the effects of GH on bone longitudinal growth were indirect and mediated by liver-derived IGF-I [44, 45], which activates chondrocyte proliferation in the growth plate. However, further studies have led researchers to reconsider this theory, since it was also demonstrated that GH can directly stimulate linear bone growth by local action on the growth plate. Isaksson et al. found that local GH injection into the tibial bone significantly stimulates longitudinal growth, whereas the contralateral tibia did not show this increase [46]. Studies in rats demonstrated pronounced tibial bone growth in the GH- or IGF-I-injected growth plate compared with the contralateral vehicle-injected growth plate [47, 48]. Furthermore, local injection of GH regulates the number of chondrocytes expressing IGF-I in rats, suggesting that part of the local action of GH on the growth plate may be mediated by increased local production of IGF-I [49]. Those observations led to the dual effector theory of GH/IGF-1 action at the growth plate, which states that GH acts directly on the germinal zone precursors of the growth plate to stimulate the differentiation of chondrocytes and then amplify local IGF-1 synthesis, which in turn induces the clonal expansion of chondrocyte columns in an autocrine/paracrine manner [50,51,52,53]. In support of the dual-effector theory but also suggesting a direct role of IGF-I on the growth plate, there are studies on mice with an inactivated GHR gene and studies on double knockout mice for GHR and IGF-I. The GHR/IGF-I double mutants were smaller than either the GHR or IGF-I single mutants, suggesting that both GH and IGF-I play a key role in the complex mechanism that leads to longitudinal growth [54].
Metabolic actions of GH
The first reports on significant changes in body composition in child hypopituitary dwarfs were reported over 40 years ago [55, 56]. Afterwards, if we focus on the effect of GHD on body composition, several studies have confirmed its association with an increase in body fat and decreased lean body mass (LBM) in children [57,58,59,60]. Treatment with r-hGH results in an early normalization of body fat percentage within 6 months (58) and a steady increase in LBM during a 2- to 6-year treatment period [57, 60], while after a 6-month discontinuation of therapy, a rapid change in body composition can be already assessed in adult GHD, with an increased amount of fat mass and a decrease in LBM [61]. In particular, GHD adults display disproportionate increases in central abdominal fat. In a study conducted on adolescents with GHD at 6 and 12 months after discontinuation of r-hGH therapy, the authors registered no change in LBM [62]. These data appear to be in contrast with those on adults, but at the end of the study, although linear growth was virtually complete, full body compositional maturation had not occurred yet. It is therefore indicated that GH cessation was associated with a static LBM, while the group receiving GH had a continuing accrual of LMB. Using a CT scan, Rutherford et al. [63] demonstrated that when r-hGH was stopped for 12 months in adolescents treated for isolated GHD, body fat and muscle size increased, while integrity and strength decreased. These patients have increased levels of total and LDL cholesterol [64] and a higher prevalence of impaired glucose tolerance [65]. Additionally, they have been found to have a greater prevalence of atheromatous plaques in the carotid and femoral vessels [66] and a two-fold increase in cardiovascular mortality [67]. It is well known that GH has significant effects on fat, glucose and protein metabolism. A number of studies have shown that GH increases lipolysis and fat oxidation, reduces glucose oxidation and impairs insulin action [68,69,70]. GH lowers protein oxidation and stimulates protein synthesis. It is likely that the protein-sparing effect of GH, together with its effects on stimulating protein synthesis, contribute to the increase in LMB during treatment [71, 72]. However, in acromegaly, energy metabolism is characterized by a trend towards higher carbohydrate oxidation in the basal state and a greater rate of carbohydrate oxidation following oral glucose [73]. The findings of increased carbohydrate oxidation and decreased lipid oxidation are the opposite of those observed after short-term GH administration. IGF-I, but not glucose and/or insulin, was significantly related to basal and post-glucose carbohydrate oxidation. Studies with recombinant IGF-I in men have shown that this growth factor stimulates glucose disposal and increases carbohydrate oxidation [74]. Lewitt et al. have provided strong evidence in vivo that IGF-I and its related binding proteins play a significant role in glucose homeostasis [75].
GH effects on neurodevelopment
In humans, growth hormone binding sites, indicative of GH receptor (GH-R) expression, are located on neurons, astrocytes, oligodendrocytes and microglia, and IGF-I was found to stimulate neuron growth, dendritic arborisation and synaptogenesis [76, 77]. In particular, GH-Rs were demonstrated in their highest concentration in the choroid plexus, thalamus, hypothalamus, pituitary, putamen and hippocampus [77], while IGF-I receptors are highly expressed in the hippocampus, amygdala, caudate nucleus, prefrontal cortex and parahippocampal cortex [76, 78]. There is also evidence that both GH and IGF-I are transported across the blood-brain barrier via transcytosis [79]. In addition, GH mRNA can be found in the central nervous system (CNS), suggesting that GH also acts in an autocrine/paracrine manner [80]. Interestingly, a reduction in GH binding sites within the brain has been observed with increasing age [77, 81]. Moreover, amongst elderly subjects, those with higher concentrations of IGF-I have been demonstrated to perform better on cognitive function tests and have lower rates of cognitive decline, suggesting that the GH-IGF-I axis affects cognitive performance throughout life [82].
The somatotropic axis plays a central role in the development and growth of the CNS. IGF-I intraventricular infusion in rats improves cognitive performance, and the inhibition of IGF-I binding to its receptors has been shown to lead to cognitive impairment, particularly in learning and reference memory [83]. Foetal cord IGF-I and IGF-binding protein-3 (IGFBP-3) concentrations have been related to head circumference at birth [84], and children with growth hormone receptor mutations have varying cognitive phenotypes [85, 86]. Similarly, serum IGF-I concentrations correlate positively with verbal intelligence in childhood [87].
In addition, there are several studies assessing a neuroprotective role of GH and IGF-I, either in combination or alone. For instance, after spinal cord injury, a decreased production of GH and IGF-I can be found [88], and in studies in rats with hypoxic brain damage, Sheepens et al. [89], found augmented immunoreactivity for GH in areas with strong cell loss. After intraventricular application of GH, the amount of neuronal loss was reduced in the frontoparietal cortex, hippocampus and dorsolateral thalamus but was left unchanged in the striatum, reflecting the regional distribution of GH receptors mentioned above, suggesting that these particular neuroprotective effects are mediated directly by GH alone.
Given these previous findings, it is unsurprising that several neuropsychological studies have documented impairments in cognitive functioning (memory and attention) in GHD. Despite a normal intelligence quotient (IQ), short children often show educational impairment, particularly in the domain of reading, spelling and arithmetic, and children with GHD show learning and attention deficits [90] and disturbances in visual-motor integration [91]. An impaired social status in patients with childhood-onset GHD has been reported compared with short and normal controls [92, 93], and women with untreated GHD exhibited lower scores on neuropsychological tests than healthy controls [94]. In a meta-analysis on adults with childhood- or adult-onset GHD, Falletti et al. demonstrated a link between GH and cognitive performance, which improved in GHD patients when growth hormone was replaced [95].
The effects of growth hormone substitution were also studied by Deijen et al., who evaluated the effects of three different doses of GH (1, 2, and 3 IU/m2/day) compared with a placebo treatment over six months, and then in an open-label phase over 18 months. In the first six months, they found improvements in memory function compared with placebo only in the groups with high GH doses, leading to supranormal IGF-I levels. In the next 18 months (open-label phase), they found improvements in memory functions at all doses [96]. This study confirmed the positive findings of GH substitution on cognitive abilities in previously conducted studies with small patient numbers [97, 98]. More recently, Webb EA et al. [99], evaluated the effect of GHD on brain structure and cognition in 15 children (mean age 8.8 years) compared to a group of children with idiopathic short stature. Compared with controls, children with isolated GHD had lower IQ, particularly in the area of verbal comprehension, which is correlated with IGF-I levels, and motor function, thus highlighting the link between GH and cognitive performance. Therefore, poor performance can be ameliorated with GH treatment.
Early diagnosis and treatment of GHD
Although most children with clinical evidence of congenital GHD are diagnosed in the first months or years of life, some are recognized relatively late in childhood, at a time when short stature becomes clearly evident. In contrast, there is a subset of patients whose GHD is recognized before 3 years of age. In these patients, short-term and long-term studies have shown a marked early catch-up growth and a significant amelioration of height with an early start of r-hGH treatment [19]. However, data on the metabolic and neurodevelopmental differences in these two cohorts (early and late diagnosis of GHD and treatment) are very poor [20].
Auxological outcomes of early GH treatment
In the past, reports have suggested that early diagnosis and treatment of GHD patients lead to a greater improvement in height gained, even if the final height remained near 2 SD below the mean population [100,101,102,103,104,105,106]. However, in 1995, Boersma et al. evaluated 26 children (7 with isolated GHD and 19 with GHD in combination with other pituitary deficiencies) who had started GH treatment before the age of 3 years and reported that early treatment of GHD may to lead to adequate catch-up growth, stressing that the early diagnosis and treatment before the statural impairment became evident was essential [14]. In the same paper, Boersma et al. reported how height SDS correlated positively with injection frequency and height SD score at start of treatment, noting that past treatment regimens, for example, 2 or 4 injections per week, could be responsible for failure to complete height gain [14]. In contrast, in children treated with 6 or 7 injections per week, the initial height at the start of treatment was reported to be crucial, since those with an initial height SDS between −2 and −4 showed a remarkable catch-up growth, whereas children with an initial height SDS < −4 did not reach full catch-up growth, although the study follow-up was only 4 years [14].
At the same time, Arrigo et al. studied 23 patients with early-onset GHD, 8 of whom had multiple hypophyseal deficiencies; these patients were treated before 5 years of age with GH and followed up for more than 3 years [15]. The data clearly showed that the height SDS at the end of the study was significantly better than in prior GH treatments, and the predicted height did not differ from the target height [15]. However, in the 6 (26%) patients showing a height < −2 SDS at the end of the study, only 2 showed a height > −4 SDS prior to GH treatment. Conversely, many patients showed a considerable height gain during the study (for example, in patient 1 and 4, the Δheight gain was 6.8 and 6.7 SDS, respectively) [15], not completely confirming the hypothesis of Boersma et al. [14]. It is interesting to note that the authors adjusted the GH doses periodically (every 3 months) during the first years of life [15]. These data were confirmed by another study, involving 23 patients with GHD treatment starting before the age of 5 and extending for at least 9 years until adulthood. These patients attained an adult mean height that did not differ from the midparental height (−0.9 vs. -0.7 SDS), because only three patients failed to achieve a final height within the target range [16]. It is meaningful to note that their height outcome correlated negatively with chronological age at the initiation of GH treatment and positively with height at puberty onset [16], stressing the importance of early diagnosis for both reduced height loss at the beginning of GH treatment and height prognosis at the onset of puberty, which should always be considered in negative terms in relation to stature recovery. However, other data come from 49 French patients with GHD [10], who started GH treatment before 1 year of age and were treated for a mean of 8.0 ± 3.6 years, reaching a mean height of −0.4 SDS with a Δheight gain of 3.11 ± 2.06 SDS (exceeding 4 SDS in 19 patients). The authors reported that the catch-up growth was maximal during the first 3 years and during puberty. Even if the patients showed an acceptable growth spurt, no further catch-up growth was reported for patients who reached or exceeded their target size [10].
Furthermore, in a large prospective long-term study evaluating 19 children with isolated GHD and 30 children with multiple pituitary hormone deficiency treated before the age of 3 years with daily subcutaneous injections for 3–5 years, the authors demonstrated that 84% of the treated patients reached a height above 2 SDS below the mean after 5 years of treatment [17]. These patients were divided into two groups according to their height SDS for chronological height at the start of GH treatment: group A consisted of patients with an initial height within the −2 SDS, whereas group B consisted of patients with initial growth retardation (> −2 SDS) [17]. Both group A (the mean height significantly improved by −2.1 ± 0.6 at the start of GH treatment to 0.5 ± 0.8 after 5 years) and B (from - 3.6 ± 1.0 at the start of the GH treatment to 0.9 ± 1.2 after 5 years) showed a significant change in annual mean height SDS for chronological age during treatment until the fourth year of treatment (the fifth year was not evaluated due to the small number of patients) [17]. However, the evaluation of variables that could predict the total height gain after 5 years of treatment showed that only the height SDS at the start of therapy was significant, indicating that most infants with growth retardation had better catch-up growth [17]. These data strongly supported the need to start early GH treatment in GHD, before growth retardation becomes evident [17].
These results were also confirmed by other studies involving patients treated before the age of 2 [8, 18]. In particular, the authors discussed pre-treatment factors possibly affecting the catch-up growth during GH treatment in GHD children, such as mid-parental height and severity of GHD [107], pre-treatment height and bone age (BA) delay at treatment onset [108]. Interestingly, in a homogeneous population, Wasniewska et al. reported that height gain during this 7-year lasting study appears to be related to the difference between target height (TH) and pre-treatment height and BW [8].
Interestingly, Ranke et al. evaluated 265 children with idiopathic GHD treated before the age of 3, compared with 509 children treated after 7 years of age, and showed that after the first year of GH treatment, the height gain was significantly higher per GH dose unit in very young than in older children. The author suggested that the early detection and treatment of GHD is a cost-effective strategy and results in greater improvements in height and growth velocity compared to a delayed treatment start in childhood [9].
Finally, in another recent study involving 47 GHD patients, in whom the administration of r-hGH was started at or before the age of 2 and consequently followed up to near-adult heights (NAH), the authors reported that treatment was very effective, since at the beginning of the study, and the height was −2.3 SDS, whereas after 5 and 10 years of age and at NAH, the stature reached −0.6, −0.3, and −0.4 SDS, respectively, which was comparable to the TH [12]. Importantly, the authors showed that a normal pattern of linear growth was achieved during childhood (before the age of 10) and that no additional gain in NAH SDS was realized during puberty, consistent with the statement that sensitivity to GH is greater during childhood and that therapeutic efforts to maximize height should be concentrated in the prepubertal years [12].
Metabolic outcomes
Despite the absence of overt abnormal findings at baseline, it is suggested that GHD children, even at a young age, may show subtle metabolic changes that may adversely affect their future metabolic and atherogenic profile.
In these respects, the available data are mainly derived from findings in patients with Prader-Willi syndrome (PWS), a genetic condition frequently presenting hypothalamic dysfunctions responsible for GH and thyroid-stimulating hormone (TSH) deficiencies, central adrenal insufficiency and hypogonadism [109]. It has been demonstrated that the PWS patients receiving early r-hGH treatment (begun prior to the age of 2) show improvements in body composition, motor function, height, and lipid profiles compared to those who were untreated [110]. However, in PWS, early r-hGH treatment seems to be unrelated to a significant increase in fasting insulin and HOMA-IR [111].
These results seem to be confirmed in subjects with GHD and optic nerve hypoplasia who had enrolled in studies around 30 months of age and had undergone early treatment with r-hGH. These subjects showed a reduction in body fat percentage and an improvement in lipid levels compared to untreated controls [112]. However, longitudinal studies involving more cases, especially patients with isolated GHD, are needed.
Interestingly, data derived by studies conducted in GHD dwarf rats have suggested that untreated early-onset GHD may to lead to significant impairment of left ventricular (LV) diastolic function and reductions in cardiac size by adulthood. In this model, early GH substitution increased and sustained the cardiac concentration of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2) and preserved cardiac morphology, limiting the onset of overt diastolic dysfunction [113].
However, it remains to be established whether the previously discussed issues may place patients at higher risk for cardiovascular disease later in life or reverse the long-term effects of r-hGH treatment.
Psychological and neurodevelopmental outcomes
Recent studies have conclusively shown that GH and IGF-1 receptors are located throughout the brain, particularly in regions related to learning and memory, such as the hippocampus [99]. In addition, a large amount of data suggest that GHD and reduced IGF-1 levels may correlate with decreased cognitive ability [99], as shown in animals with GHD presenting with smaller brain volumes with reduced neuronal and glial proliferation [99].
In studies involving animal models with congenital GHD, such as Snell (Pou1f1 deficient) [114], and little (Ghrhr-deficient) mice [115], the authors described the presence of hypomyelination and poor neuronal growth and synaptogenesis. These features appeared to be restored with postnatal GH administration [114; 115]. However, such findings were not confirmed by others [116] or suggested to be related to compensatory upregulated production of local IGF [117].
In humans, GH-treated infants and toddlers seem to be more alert and energetic, as reported by their families, and an increased rate of language and cognitive development was reported in GH-treated patients, even if such results may only be due to increased muscle tone [118]. However, some data are consistent with the finding that children with isolated GHD have improved intellectual development if they are treated before the age of 5 years [119] and present with improved attention, anxiety, social competence, and thought impairment after 3 years of treatment, even if this aspect was more pronounced for isolated GHD patients than for those with idiopathic short stature [120].
Interestingly, in a recent study evaluating the cognitive functioning in 15 prepubertal PWS patients aged 3.5 to 14 years, the authors reported that GH treatment can prevent the deterioration of some cognitive skills in the short term and significantly improve abstract reasoning and visuospatial skills during long-term r-hGH treatment. Furthermore, children with a greater deficit can benefit more from GH treatment [121]. This finding is in line with those of animal studies in which dwarf rats treated early with GH, therefore rendering them GH-replete in puberty but deficient as adults, displayed a spatial learning performance comparable to that of heterozygous animals, whereas a longer supplementation did not confer additional benefits, suggesting a “window period” to be able to obtain such benefits [122].
Regarding the impact of childhood GHD on cognition, published data outline how GHD children with low peak GH to stimulation or low GH levels in nocturnal GH secretion may present reduced performance in a visual motor psychological test [123]. Of note, in another study involving 79 GHD children evaluated after 1 and 3 years of r-hGH treatment, IQ scores at baseline were within the normal range, even if GHD children older than 7 years of age at first assessment had a mean performance IQ slightly below average [124].
Therefore, given the demonstrated effects of GHD on brain structure, cognition and possibly behaviour, an early diagnosis of GHD is a critical component of the work-up of these endocrine disorders.
Conclusions
In conclusion, the available auxological results show a favourable auxological outcome, comparable to the TH, for patients with early-diagnosed GHD treated with r-hGH. GH retains good efficacy in very young patients, and early-diagnosed patients with GHD have a responsiveness to r-hGH that is much greater than that of older patients. Furthermore, early detection and treatment of GHD is a cost-effective strategy. Since delayed referrals for GH diagnosis and treatment still occur frequently, careful evaluation is needed when GHD is suspected, given the importance of early diagnosis and treatment.
Moreover, these preliminary but very interesting data about early GHD diagnosis and treatment and their possible effects on metabolism require confirmation in wider studies. Therefore, data highlighting the action of GH and IGF-1 deficiency on the brain structure, psychological and neurodevelopmental features of GHD patients, as well as the possible effects of GH treatment, seem to assume a “time” dependence of GHD diagnosis, thus stressing the importance of earlier diagnosis and treatment.
References
Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab. 2000;85:3990–3.
Thomas M, Massa G, Craen M, et al. Prevalence and demographic features of childhood growth hormone deficiency in Belgium during the period 1986-2001. Eur J Endocrinol. 2004;151:67–72.
National Institute for Health and Care Excellence. TA188. Human growth hormone (somatropin) for the treatment of growth failure in children: guidance. May 2013. [Internet]. Available from: https://www.nice.org.uk/guidance/ta188. [cited 2016, April 26].
Darendeliler F, Lindberg A, Wilton P. Response to growth hormone treatment in isolated growth hormone deficiency versus multiple pituitary hormone deficiency. Horm Res Paediatr. 2011;76(Suppl 1):42–6. doi:10.1159/000329161.
Ciresi A, Amato MC, Criscimanna A, et al. Metabolic parameters and adipokine profile during GH replacement therapy in children with GH deficiency. Eur J Endocrinol. 2007;156:353–60.
Prince DA, Ranke MB. Final height following growth hormone treatment. In: MB Ranke & R Gunnarsson, editor. Progress in Growth Hormone Therapy – 5 Years of KIGS. Mannheim: J & J Verlag; 1994. p. 129–44.
Achermann JC, Hamdani K, Hindmarsh PC, Brook CG. Birth weight influences the initial response to growth hormone treatment in growth hormone-insufficient children. Pediatrics. 1998;102:342–5.
Wasniewska M, Arrigo T, Cisternino M, et al. Birth weight influences long-term catch-up growth and height prognosis of GH-deficient children treated before the age of 2 years. Eur J Endocrinol. 2000;142:460–5.
Ranke MB, Lindberg A, Albertsson-Wikland K, Wilton P, Price DA, Reiter EO. Increased response, but lower responsiveness, to growth hormone (GH) in very young children (aged 0-3 years) with idiopathic GH Deficiency: analysis of data from KIGS. J Clin Endocrinol Metab. 2005;90:1966–71.
Huet F, Carel JC, Nivelon JL, Chaussain JL. Long-term results of GH therapy in GH-deficient children treated before 1 year of age. Eur J Endocrinol. 1999;140:29–34.
Huet F, Bensignor C, Polak M, Carel JC. Results of early growth hormone treatment in children with hypopituitarism. Bull Acad Natl Med. 2012;196:117–23.
Root AW, Dana K, Lippe B. Treatment of growth hormone-deficient infants with recombinant human growth hormone to near-adult height: patterns of growth. Horm Res Paediatr. 2011;75:276–83. doi:10.1159/000322881.
Carel JC, Huet F, Chaussain JL. Treatment of growth hormone deficiency in very young children. Horm Res. 2003;60(Suppl 1):10–7.
Boersma B, Rikken B, Wit JM. Catch-up growth in early treated patients with growth hormone deficiency. Dutch Growth Hormone Working Group. Arch Dis Child. 1995;72:427–31.
Arrigo T, De Luca F, Bernasconi S, et al. Catch-up growth and height prognosis in early treated children with congenital hypopituitarism. Horm Res. 1995;44(Suppl 3):26–31.
De Luca F, Maghnie M, Arrigo T, Lombardo F, Messina MF, Bernasconi S. Final height outcome of growth hormone-deficient patients treated since less than five years of age. Acta Paediatr. 1996;85:1167–71.
Rappaport R, Mugnier E, Limoni C, et al. A 5-year prospective study of growth hormone (GH)-deficient children treated with GH before the age of 3 years. French Serono Study Group. J Clin Endocrinol Metab. 1997;82:452–6.
Arrigo T, Bozzola M, Cavallo L, et al. Growth hormone deficient children treated from before two years old fail to catch-up completely within five years of therapy. J Pediatr Endocrinol Metab. 1998;11:45–50.
Antoniazzi F, Cavarzere P, Gaudino R. Growth hormone and early treatment. Minerva Endocrinol. 2015;40:129–43.
Alatzoglou KS, Webb EA, Le Tissier P, Dattani MT. Isolated growth hormone deficiency (GHD) in childhood and adolescence: recent advances. Endocr Rev. 2014;35:376–432. doi:10.1210/er.2013-1067.
Hunziker EB, Schnek RK. Physiological mechanisms adopted by chondrocytes in regulating longitudinal bone growth. J Physiol. 1989;44:55–71.
Kember NF. Cell population kinetics of bone growth: the fi rst ten years of autoradiographic studies with tritiated thymidine. Clin Orthop. 1971;76:213–30.
Breur GJ, Turgai J, Vanenkevort BA, Farnum CE, Wilsman NJ. Stereological and serial section analysis of chondrocytic enlargement in the proximal tibial growth-plate of the rat. Anat Rec. 1994;239:255–68.
Buckwalter JA, Mower D, Ungar R, Schaeffer J, Ginsberg B. Morphometric analysis of chondrocyte hypertrophy. J Bone Joint Surg Am. 1986;68A:243–55.
Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–8.
Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1 is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–76.
Hunziker EB, Schenk RK, Cruzorive LM. Quantitation of chondrocyte performance in growth-plate cartilage during longitudinal bone-growth. J Bone Joint Surg Am. 1987;69A:162–73.
Parfitt AM. Misconceptions (1): epiphyseal fusion causes cessation of growth. Bone. 2002;30:337–9.
Nilsson O, Baron J. Fundamental limits on longitudinal bone growth: growth plate senescence and epiphyseal fusion. Trends Endocrinol Metab. 2004;15:370–4.
Hering TM. Regulation of chondrocyte gene expression. Front Biosci. 1999;4:D743–61.
Stevens DA, Williams GR. Hormone regulation of chondrocyte differentiation and endochondral bone formation. Mol Cell Endocrinol. 1999;151:195–204.
Robson H, Siebler T, Shalet SM, Williams GR. Interactions betweenGH, IGFI, glucocorticoids, and thyroid hormones during skeletal growth. Pediatr Res. 2002;52:137–47.
Van Der Eerden BCJ, Karperien M, Wit JM. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24:782–801.
Nilsson O, Marino R, De Luca F, Phillip M, Baron J. Endocrine regulation of the growth plate. Horm Res. 2005;64:157–65.
Daughaday WH. Pituitary gigantism. Endocrinol Metab Clin N Am. 1992;21:633–47.
Ezzat S. Acromegaly. Endocrinol Metab Clin N Am. 1997;26:703–23.
Pfaffle RW, Parks JS, Brown MR, Heimann G. Pit-1 and pituitary function. J Pediatr Endocrinol. 1993;6:229–33.
Rosenfeld R, Allen DB, MacGillivray MH, et al. Growth hormone use in pediatric growth hormone deficiency and other pediatric growth disorders. Am J Manag Care. 2000;6:S805–16.
Savage MO, Burren CP, Blair JC, Woods KA, Metherell L, Clark AJ, Camacho-Hubner C. Growth hormone insensitivity: pathophysiology, diagnosis, clinical variation and future perspectives. Horm Res. 2001;55(Suppl 2):32–5.
Woods KA, Savage MO. Laron syndrome: typical and atypical forms. Bailliere Clin Endocrinol Metab. 1996;10:371–87.
Kofoed EM, Hwa V, Little B, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349:1139–47.
Rosenfeld RG, Rosenbloom AL, GuevaraAguirre J. Growth hormone (GH) insensitivity due to primary GH receptor deficiency. Endocr Rev. 1994;15:369–90.
Wit JM, Kamp GA, Rikken B. Spontaneous growth and response to growth hormone treatment in children with growth hormone deficiency and idiopathic short stature. Pediatr Res. 1996;39:295–302.
Daughaday WH, Hall K, Raben MS, Salmon WD Jr, van den Brande JL, van Wyk JJ. Somatomedin: proposed designation for sulphation factor. Nature. 1972;235:107.
Salmon WD Jr, Daughaday WH. A hormonally controlled serum factor which stimulates sulphate incorporation by cartilage in vitro. J Lab Clin Med. 1957;49:825–36.
Isaksson OG, Jansson JO, Gause IA. Growth hormone stimulates longitudinal bone growth directly. Science. 1982;216:1237–9.
Russell SM, Spencer EM. Local injections of human or rat growth hormone or of purified human somatomedin-C stimulate unilateral tibial epiphyseal growth in hypophysectomized rats. Endocrinology. 1985;116:2563–7.
Schlechter NL, Russell SM, Greenberg S, Spencer EM, Nicoll CS. A direct growth effect of growth hormone in rat hindlimb shown by arterial infusion. Am J Phys. 1986;250:E231–5.
Nilsson A, Isgaard J, Lindahl A, Dahlstrom A, Skottner A, Isaksson OG. Regulation by growth hormone of number of chondrocytes containing IGF-I in rat growth plate. Science. 1986;233:571–4.
Seminara S, Stagi S, Nanni L. Thyroid function during catch-up growth: a focus on the growth plate. In: Preedy V, editor. The Handbook of Growth and Growth Monitoring in Health and Disease. New York: Springer Science & Business Media; 2012. p. 905-16.
Zezulak KM, Green H. The generation of insulin-like growth factor-1-sensitive cells by growth- hormone action. Science. 1986;233:551–3.
Isgaard J, Moller C, Isaksson OG, Nilsson A, Mathews LS, Norstedt G. Regulation of insulin-like growth factor messenger ribonucleic acid in rat growth plate by growth hormone. Endocrinology. 1988;122:1515–20.
Schlechter NL, Russell SM, Spencer EM, Nicoll CS. Evidence suggesting that the direct growth-promoting effect of growth hormone on cartilage in vivo is mediated by local production of somatomedin. Proc Natl Acad Sci U S A. 1986;83:7932–4.
Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol. 2001;229:141–62.
Novak L, Hayles A, Cloutier M. Effect of hGH on body composition in hypopituitary dwarfs. Four compartment analysis and composite body densitometry. Mayo Clinic Proc. 1972;47:241–6.
Parra A, Argote RM, García G, Cervantes C, Alatorre S, Pérez-Pasten E. Body composition in hypopituitary dwarfs before and during human growth hormone therapy. Metabolism. 1979;28:851–7.
Högler W, Briody J, Moore B, Lu PW, Cowell CT. Effect of growth hormone therapy and puberty on bone and body composition in children with idiopathic short stature and growth hormone deficiency. Bone. 2005;37:642–50.
Boot AM, Engels MA, Boerma GJ, Krenning EP, De Muinck Keizer-Schrama SM. Changes in bone mineral density, body composition, and lipid metabolism during growth hormone (GH) treatment in children with GH deficiency. J Clin Endocrinol Metab. 1997;82:2423–8.
Ross J, Czernichow P, Biller BM, Colao A, Reiter E, Kiess W. Growth hormone: health considerations beyond height gain. Pediatrics. 2010;125:e906–18.
Vander Sluis IM, Boot AM, Hop WC, DeRijke YB, Krenning EP, de Muinck Keizer-Schrama SM. Long-termeffects of growth hormone therapy on bone mineral density, body composition, and serum lipid levels in growth hormone deficient children: a 6-year follow-up study. Horm Res. 2002;58:207–14.
Butenandt O, Jocham A, Schwarz HP, Sperlich M, Tschöp M. Childhood onset of GH deficiency: Reassessment of G H status and effects of substitution. Growth Hormon IGF Res. 1998;8(Suppl. A):9–13.
Carroll PV, Drake WM, Maher KT, et al. Comparison of continuation or cessation of growth hormone (GH) therapy on body composition and metabolic status in adolescent with severe GH deficiency at completion of linear growth. J Clin Endocrinol Metab. 2004;89:3890–5.
Rutherford OM, Jones DA, Round JM, Buchanan CR, Preece MA. Changes in skeletal muscle and body composition after discontinuation of growth hormone treatment in growth hormone deficient young adults. Clin Endocrinol. 1991;34:469–75.
De Boer H, Blok GJ, Voerman HJ, Phillips M, Schouten JA. Serum lipid levels in growth hormone deficient men. Metabolism. 1994;43:199–203.
Beyshah SA, Henderseon A, Nithayanathan R, Sharp P, Richmond W. Metabolic abnormalities in growth hormone deficient adults. Carbohydrate tolerance and lipid metabolism. Endocrinol Metab. 1994;1:173–80.
Markussis V, Beyshah SA, Fisher C, Sharp P, Nicholaides AN, Johnston DG. Detection of premature atherosclerosis by high resolution ultrasonography in symptom-free hypopituiary adults. Lancet. 1992;340:1188–92.
Rosen T, Bengtsson B-A. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet. 1990;336:285–8.
Sakharova AA, Horowitz JF, Surya S, Goldenberg N, Harber MP, Symons K, Barkan A. Role of growth hormone in regulating lipolysis, proteolysis, and hepatic glucose production during fasting. J Clin Endocrinol Metab. 2008;93:2755–9.
Vijayakumar A, Novosyadlyy R, Wu Y, Yakar S, LeRoith D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Hormon IGF Res. 2010;20:1–7.
Bidlingmaier M, Strasburger CJ. Growth hormone. Handb Exp Pharmacol. 2010;195:187–200.
Moller N, Jorgensen JOL, Alberti KGMM, Flyvbjerg A, Schmitz O. Short-term effects of growth hormone on fuel oxidation and regional substrate metabolism in normal man. J Clin Endocrinol Metab. 1990;70:1179–86.
Horber F, Marsh H, Haymond M. Differential effects of prednisone and growth hormone on fuel metabolism and insulin antagonism in humans. Diabetes. 1991;40:141–9.
O'Sullivan AJ, Kelly JJ, Hoffman DM, Baxter RC, Ho KKY. Energy metabolism and substrate utilization in acromegaly. J Clin Endocrinol Metab. 1995;80:486–91.
Boulware SD, Tamborlane WV, Matthews LS, Sherwin RS. Diverse effects of insulin-like growth factor I on glucose, lipid and amino acid metabolism. Am J Phys. 1995;262:E130–3.
Lewitt MS, Denyer GS, Cooney GJ, Baxter RC. Insulin-like growth factor binding protein 1 modulates glucose levels. Endocrinology. 1991;129:2254–6.
Bondy CA, Lee WH. Patterns of insulin-like growth factor and IGF receptor gene expression in the brain. Functional implications. Ann N Y Acad Sci. 1993;692:33–43.
Lai Z, Roos P, Zhai O, et al. Age-related reduction of human growth hormone-binding sites in the human brain. Brain Res. 1993;621:260–6.
Araujo DM, Lapchak PA, Collier B, Chabot JG, Quirion R. Insulin-like growth factor-1 (somatomedin-C) receptors in the rat brain: distribution and interaction with the hippocampal cholinergic system. Brain Res. 1989;484:130–8.
Coculescu M. Blood-brain barrier for human growth hormone and insulin-like growth factor-I. J Pediatr Endocrinol Metab. 1999;12:113–24.
Lobie PE, Zhu T, Graichen R, Goh EL. Growth hormone, insulin-like growth factor I and the CNS: localization, function and mechanism of action. Growth Hormon IGF Res. 2000;10(Suppl B):S51–6.
Zhai Q, Lai Z, Roos P, Nyberg F. Characterization of growth hormone binding sites in rat brain. Acta Paediatr Suppl. 1994;406:92–5.
Aleman A, Verhaar HJ, de Haan EH, De Vries WR, Samson MM, Drent ML, et al. Insulin-like growth factor-I and cognitive function in healthy older men. J Clin Endocrinol Metab. 1999;84:471–5.
Markowska AL, Mooney M, Sonntag WE. Insulin-like growth factor-1 ameliorates age-related behavioral deficits. Neuroscience. 1998;87:559–69.
Geary MP, Pringle PJ, Rodeck CH, Kingdom JC, Hindmarsh PC. Sexual dimorphism in the growth hormone and insulin-like growth factor axis at birth. J Clin Endocrinol Metab. 2003;88:3708–14.
Laron Z, Klinger B. Laron syndrome: clinical features, molecular pathology and treatment. Horm Res. 1994;42:198–202.
Kranzler JH, Rosenbloom AL, Martinez V, Guevara-Aguirre J. Normal intelligence with severe insulin-like growth factor I deficiency due to growth hormone receptor deficiency: a controlled study in a genetically homogeneous population. J Clin Endocrinol Metab. 1998;83:1953–8.
Gunnell D, Miller LL, Rogers I, Holly JM. Association of insulin-like growth factor I and insulin-like growth factor-binding protein-3 with intelligence quotient among 8- to 9-year-old children in the Avon Longitudinal Study of Parents and Children. Pediatrics. 2005;116:e681–6.
Tsitouras PD, Zhong YG, Spungen AM, Bauman WA. Serum testosterone and growth hormone/insulin-like growth factor-I in adults with spinal cord injury. Horm Metab Res. 1995;27:287–92.
Scheepens A, Sirimanne ES, Breier BH, Clark RG, Gluckman PD, Williams CE. Growth hormone as a neuronal rescue factor during recovery from CNS injury. Neuroscience. 2001;104:677–87.
Sartorio A, Conti A, Molinari E, Riva G, Morabito F, Faglia G. Growth, growth hormone and cognitive functions. Horm Res. 1996;45:23–9.
Abbott D, Rotnem D, Genel M, Cohen DJ. Cognitive and emotional functioning in hypopituitary short-statured children. Schizophr Bull. 1982;8:310–9.
Rikken B, van Busschbach J, le Cessie S, Manten W, Spermon T, Grobbee R, et al. Impaired social status of growth hormone deficient adults as compared to controls with short or normal stature. Dutch Growth Hormone Working Group. Clin Endocrinol. 1996;44:489–90.
Dean HJ, McTaggart TL, Fish DG, Friesen HG. The educational, vocational, and marital status of growth hormone deficient adults treated with growth hormone during childhood. Am J Dis Child. 1985;139:1105–10.
Bülow B, Hagmar L, Orbaek P, Österberg K, Erfurth EM. High incidence of mental disorders, reduced mental well-being and cognitive dysfunction in hypopituitary women with GH deficiency treated for pituitary disease. Clin Endocrinol. 2002;56:183–93.
Falleti MG, Maruff P, Burman P, Harris A. The effects of growth hormone (GH) deficiency and GH replacement on cognitive performance in adults: a meta-analysis of the current literature. Psychoneuroendocrinology. 2006;31:681–91.
Deijen JB, de Boer H, van der Veen EA. Cognitive changes during growth hormone replacement in adult men. Psychoneuroendocrinology. 1998;23:45–55.
Almqvist O, Thoren M, Saaf M, Eriksson O. Effects of growth hormone substitution on mental performance in adults with growth hormone deficiency: a pilot study. Psychoneuroendocrinology. 1986;11:347–52.
Sartorio A, Molinari E, Riva G, Conti A, Morabito F, Faglia G. Growth hormone treatment in adults with childhood onset growth hormone deficiency: effects on psychological capabilities. Horm Res. 1995;44:6–11.
Webb EA, O'Reilly MA, Clayden JD, et al. Effect of growth hormone deficiency on brain structure, motor function and cognition. Brain. 2012;135(Pt 1):216–27.
Josefsberg Z, Bauman B, Pertzelan A, Laron Z. Greater efficiency of human growth hormone therapy in children below five years of age with growth hormone deficiency. A 5-year follow-up study. Horm Res. 1987;27:126–33.
Bundak R, Hindmarsh PC, Smith PJ, Brook CG. Long-term auxologic effects of human growth hormone. J Pediatr. 1988;112:875–9.
Burns EC, Tanner JM, Preece MA, Cameron N. Final height and pubertal development in 55 children with idiopathic growth hormone deficiency, treated for between 2 and 15 years with human growth hormone. Eur J Pediatr. 1981;137:155–64.
Dean HJ, Friesen HG. Long-term growth of children with growth hormone deficiency and hypoglycemia. Therapeutic Trial of Growth Hormone Committee. Medical Research Council of Canada. J Pediatr. 1989;115:598–600.
Mugnier E, Ployard F, Roy MP, Fermanian J, Rappaport R. Results of treatment with human growth hormone in children with pituitary insufficiency under 7 years of age. Study of 26 cases. Arch Fr Pediatr. 1985;42:671–6.
Zadik Z, Chalew S, Zung A, Landau H, Leiberman E, Koren R, Voet H, Hochberg Z, Kowarski AA. Effect of long-term growth hormone therapy on bone age and pubertal maturation in boys with and without classic growth hormone deficiency. J Pediatr. 1994;125:189–95.
Vanderschueren-Lodeweyckx M, Van den Broeck J, Wolter R, Malvaux P. Early initiation of growth hormone treatment: influence on final height. Acta Paediatr Scand Suppl. 1987;337:4–11.
Cutfield W, Lindberg A, Albertsson Wikland K, Chatelain P, Ranke MB, Wilton P. Final height in idiopathic growth hormone deficiency: the KIGS experience. KIGS International Board Acta Paediatr Suppl. 1999;88:72–5.
Blethen SL, Baptista J, Kuntze J, Foley T, LaFranchi S, Johanson A. Adult height in growth hormone (GH)-deficient children treated with biosynthetic GH. The Genentech Growth Study Group. J Clin Endocrinol Metab. 1997;82:418–20.
Aycan Z, Baş VN. Prader-Willi syndrome and growth hormone deficiency. J Clin Res Pediatr Endocrinol. 2014;6:62–7. doi:10.4274/Jcrpe.1228.
Carrel AL, Myers SE, Whitman BY, Eickhoff J, Allen DB. Long-term growth hormone therapy changes the natural history of body composition and motor function in children with prader-willi syndrome. J Clin Endocrinol Metab. 2010;95:1131–6. doi:10.1210/jc.2009-1389.
Carrell A, Allen DB. Growth hormone Therapy in children with Prader-Willi syndrome. In: Radovick S, MacGillivray MH, editors. Pediatric endocrinology: a practical clinical guide, Second Edition. New York: Springer Science & Business Media; 2013. p. 99-107.
Stewart C, Garcia-Filion P, Fink C, Ryabets-Lienhard A, Geffner ME, Borchert M. Efficacy of growth hormone replacement on anthropometric outcomes, obesity, and lipids in children with optic nerve hypoplasia and growth hormone deficiency. Int J Pediatr Endocrinol. 2016;2016:5. doi:10.1186/s13633-016-0023-9.
Groban L, Lin M, Kassik KA, Ingram RL, Sonntag WE. Early-onset growth hormone deficiency results in diastolic dysfunction in adult-life and is prevented by growth hormone supplementation. Growth Hormon IGF Res. 2011;21:81–8. doi:10.1016/j.ghir.2011.01.003.
Sugisaki T, Noguchi T, Tsukada Y. Cerebral myelinogenesis in the Snell dwarf mouse: stimulatory effects of GH and T4 restricted to the first 20 days of postnatal life. Neurochem Res. 1985;10:767–78.
Noguchi T, Sugisaki T, Nishikawa N, Tsukada Y. Restoration of microcephalic cerebrum with hypomyelination in the growth hormone-deficient mouse (lit): stimulatory effects of GH restricted to the first 20 days of postnatal life. Neurochem Res. 1988;13:249–52.
Lehman DM, Hale DE, Cody JT, Harrison JM, Leach RJ. Molecular, morphometric and functional analyses demonstrate that the growth hormone deficient little mouse is not hypomyelinated. Brain Res Dev Brain Res. 1999;116:191–9.
Sun LY, Evans MS, Hsieh J, Panici J, Bartke A. Increased neurogenesis in dentate gyrus of long-lived Ames dwarf mice. Endocrinology. 2005;146:1138–44.
Myers SE, Whitman BY, Carrel AL, Moerchen V, Bekx MT, Allen DB. Two years of growth hormone therapy in young children with Prader-Willi syndrome: physical and neurodevelopmental benefits. Am J Med Genet A. 2007;143A:443–8.
Laron Z, Galatzer A. Effect of hGH on head circumference and IQ in isolated growth hormone deficiency. Early Hum Dev. 1981;5:211–4.
Stabler B, Siegel PT, Clopper RR, Stoppani CE, Compton PG, Underwood LE. Behavior change after growth hormone treatment of children with short stature. J Pediatr. 1998;133:366–73.
Siemensma EP, Tummers-de Lind van Wijngaarden RF, Festen DA et al. Beneficial effects of growth hormone treatment on cognition in children with Prader-Willi syndrome: a randomized controlled trial and longitudinal study. J Clin Endocrinol Metab. 2012;97:2307-14. doi:10.1210/jc.2012-1182.
Nieves-Martinez E, Sonntag WE, Wilson A, et al. Early-onset GH deficiency results in spatial memory impairment in mid-life and is prevented by GH supplementation. J Endocrinol. 2010;204:31–6. doi:10.1677/JOE-09-0323.
Andronikof-Sanglade A, Fjellestad-Paulsen A, Ricard-Malivoir S, Evain-Brion D. Specific abnormalities in a visual motor psychological test in short children with abnormal growth hormone secretion. Acta Paediatr. 1997;86:154–9.
Puga González B, Ferrández Longás A, Oyarzábal M, Nosas R, Grupo Colaborativo Español. The effects of growth hormone deficiency and growth hormone replacement therapy on intellectual ability, personality and adjustment in children. Pediatr Endocrinol Rev. 2010;7:328–38.
Acknowledgements
The authors acknowledge A.Fa.D.O.C. onlus - Association of families with children affected by growth deficit and other endocrine disorders.
Funding
Not applicable
Availability of data and materials
Not applicable
Financial competing interests
The authors do not have any financial and non-financial competing interests in relation to this manuscript. Editorial assistance was provided by Airon Communications, Milan, Italy, with financial support from Novo Nordisk, in compliance with international guidelines for good publication practice.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Stagi, S., Scalini, P., Farello, G. et al. Possible effects of an early diagnosis and treatment in patients with growth hormone deficiency: the state of art. Ital J Pediatr 43, 81 (2017). https://doi.org/10.1186/s13052-017-0402-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-017-0402-8