
Abstract
Ovarian cancer (OC) is the highest frequent malignant gynecologic tumor with very complicated pathogenesis. The purpose of the present academic work was to identify significant genes with poor outcome and their underlying mechanisms. Gene expression profiles of GSE36668, GSE14407 and GSE18520 were available from GEO database. There are 69 OC tissues and 26 normal tissues in the three profile datasets. Differentially expressed genes (DEGs) between OC tissues and normal ovarian (OV) tissues were picked out by GEO2R tool and Venn diagram software. Next, we made use of the Database for Annotation, Visualization and Integrated Discovery (DAVID) to analyze Kyoto Encyclopedia of Gene and Genome (KEGG) pathway and gene ontology (GO). Then protein-protein interaction (PPI) of these DEGs was visualized by Cytoscape with Search Tool for the Retrieval of Interacting Genes (STRING). There were total of 216 consistently expressed genes in the three datasets, including 110 up-regulated genes enriched in cell division, sister chromatid cohesion, mitotic nuclear division, regulation of cell cycle, protein localization to kinetochore, cell proliferation and Cell cycle, progesterone-mediated oocyte maturation and p53 signaling pathway, while 106 down-regulated genes enriched in palate development, blood coagulation, positive regulation of transcription from RNA polymerase II promoter, axonogenesis, receptor internalization, negative regulation of transcription from RNA polymerase II promoter and no significant signaling pathways. Of PPI network analyzed by Molecular Complex Detection (MCODE) plug-in, all 33 up-regulated genes were selected. Furthermore, for the analysis of overall survival among those genes, Kaplan–Meier analysis was implemented and 20 of 33 genes had a significantly worse prognosis. For validation in Gene Expression Profiling Interactive Analysis (GEPIA), 15 of 20 genes were discovered highly expressed in OC tissues compared to normal OV tissues. Furthermore, four genes (BUB1B, BUB1, TTK and CCNB1) were found to significantly enrich in the cell cycle pathway via re-analysis of DAVID. In conclusion, we have identified four significant up-regulated DEGs with poor prognosis in OC on the basis of integrated bioinformatical methods, which could be potential therapeutic targets for OC patients.
Similar content being viewed by others


Background
Ovarian cancer (OC) is the fifth cause of cancerous death among women all over the world [1]. Although some prognostic biomarkers have been exploited, the overall survival of OC remains weak due to its difficulty in early detection, distant metastasis and rapid dissemination [2, 3]. Therefore, more reliable prognostic biomarkers should be explored as a target for improving the treatment effect and better understanding the underlying mechanism.
Gene chip which was used for more than ten years can quickly detect differentially expressed genes and was proved to be a reliable technique [4] that could make many slice data be produced and stored in public databases. Therefore, a large number of valuable clues could be explored for new research on the base of these data. Furthermore, many bioinformatical studies on OC have been produced in recent years [5], which proved that the integrated bioinformatical methods could help us to further study and better exploring the underlying mechanisms.
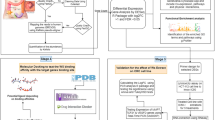
In this study, first, we chosed GSE36668, GSE18520 and GSE14407 from Gene Expression Omnibus (GEO). Second, we applied for GEO2R online tool and Venn diagram software to obtain the commonly differentially expressed genes (DEGs) in the three datasets above. Third, the Database for Annotation, Visualization and Integrated Discovery (DAVID) was used to analyze these DEGs including molecular function (MF), cellular component (CC), biological process (BP) and Kyoto Encyclopedia of Gene and Genome (KEGG) pathways. Fourth, we established protein-protein interaction (PPI) network and then applied Cytotype MCODE (Molecular Complex Detection) for additional analysis of the DEGs which would identify some core genes. Moreover, these core DEGs were imported into the Kaplan Meier plotter online database for the significant prognostic information (P < 0.05). In addition, we furtherly validated the DEGs expression between OV cancer tissues and normal OV tissues via Gene Expression Profiling Interactive Analysis (GEPIA) (P < 0.05). Taken above, only 15 DEGs were qualified. Then, we re-analyzed these 15 DEGs for KEGG pathway enrichment. Finally, four DEGs (BUB1B, BUB1, TTK and CCNB1) were generated and significantly enriched in the cell cycle pathway especially in G2/M phase. In conclusion, the bioinformatic study of our study provides some additional useful biomarkers which could be an effective target for OC patients.
Methods
Microarray data information
NCBI-GEO is regarded as a free public database of microarray/gene profile and we obtained the gene expression profile of GSE36668, GSE18520 and GSE 14407 in ovarian cancer and normal ovarian tissues. Microarray data of GSE36668, GSE18520 and GSE14407 were all on account of GPL570 Platforms ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array) which included 4 OC tissues and 4 normal OV tissues, 53 OC tissues and 10 normal OV tissues and 12 OC tissues and 12 normal OV tissues, respectively.
Data processing of DEGs
DEGs between OC specimen and normal OV specimen were identified via GEO2R online tools [6] with |logFC| > 2 and adjust P value < 0.05. Then, the raw data in TXT format were checked in Venn software online to detect the commonly DEGs among the three datasets. The DEGs with log FC < 0 was considered as down-regulated genes, while the DEGs with log FC > 0 was considered as an up-regulated gene.
Gene ontology and pathway enrichment analysis
Gene ontology analysis (GO) is a commonly used approach for defining genes and its RNA or protein product to identify unique biological properties of high-throughput transcriptome or genome data [7]. KEGG is a collection of databases dealing with genomes, diseases, biological pathways, drugs, and chemical materials [8]. DAVID which is an online bioinformatic tool is designed to identify a large number of genes or proteins function [9]. We could use DAVID to visualize the DEGs enrichment of BP, MF, CC and pathways (P < 0.05).
PPI network and module analysis
PPI information can be evaluated by an online tool, STRING (Search Tool for the Retrieval of Interacting Genes) [10]. Then, the STRING app in Cytoscape [11] was applied to examine the potential correlation between these DEGs (maximum number of interactors = 0 and confidence score ≥ 0.4). In addition, the MCODE app in Cytoscape was used to check modules of the PPI network (degree cutoff = 2, max. Depth = 100, k-core = 2, and node score cutoff = 0.2).
Survival analysis and RNA sequencing expression of core genes
Kaplan Meier-plotter are a commonly used website tool for assessing the effect of a great number of genes on survival based on EGA, TCGA database and GEO (Affymetrix microarrays only) [12]. The log rank P value and hazard ratio (HR) with 95% confidence intervals were computed and showed on the plot. To validate these DEGs, we applied the GEPIA website to analyze the data of RNA sequencing expression on the basis of thousands of samples from the GTEx projects and TCGA [13].
Results
Identifcation of DEGs in ovarian cancers
There were 69 OC tissues and 26 normal OV tissues in our present study. Via GEO2R online tools, we extracted 1516, 1150 and 1670 DEGs from GSE36668, GSE18520 and GSE 14407, respectively. Then, we used Venn diagram software to identify the commonly DEGs in the three datasets. Results showed that a total of 216 commonly DEGs were detected, including 106 down-regulated genes (logFC< 0) and 110 up-regulated genes (logFC> 0) in the OC tissues (Table 1 & Fig. 1).

Authentication of 216 common DEGs in the three datasets (GSE36668, GSE18520 and GSE14407) through Venn diagrams software (available online: http://bioinformatics.psb.ugent.be/webtools/Venn/). Different color meant different datasets. a 110 DEGs were up-regulated in the three datasets (logFC> 0). b 106 DEGs were down-regulated in three datasets (logFC < 0)
DEGs gene ontology and KEGG pathway analysis in ovarian cancers
All 216 DEGs were analyzed by DAVID software and the results of GO analysis indicated that 1) for biological processes (BP), up-regulated DEGs were particularly enriched in regulation of cell cycle, cell division, mitotic nuclear division, protein localization to kinetochore, sister chromatid cohesion and cell proliferation, and down-regulated DEGs in blood coagulation, positive regulation of transcription from RNA polymerase II promoter, palate development, negative regulation of transcription from RNA polymerase II promoter, axonogenesis and receptor internalization; 2) for molecular function (MF), up-regulated DEGs were enriched in protein binding, ATP-dependent microtubule motor activity, protein kinase binding, plus-end-directed, microtubule binding, sequence-specific DNA binding and down-regulated DEGs in RNA polymerase II core promoter proximal region sequence-specific binding, RNA polymerase II transcription factor binding, RNA polymerase II core promoter proximal region sequence-specific binding and growth factor activity, transcriptional repressor activity, transcriptional activator activity; 3) for GO cell component (CC), up-regulated DEGs were significantly enriched in the nucleoplasm, midbody, spindle microtubule, spindle, cytosol and nucleus, and down-regulated DEGs in proteinaceous extracellular matrix anchored component of membrane, extracellular region and extracellular space (Table 2).
KEGG analysis results were shown in Table 3 which demonstrated that up-regulated DEGs were particularly enriched in p53 signaling pathway, cell cycle and progesterone-mediated oocyte maturation while down-regulated DEGs in no significant signaling pathways (P < 0.05).
Protein–protein interaction network (PPI) and modular analysis
A total of 107 DEGs were imported into the DEGs PPI network complex which included 107 nodes and 698 edges, including 60 down-regulated and 47 up-regulated genes (Fig. 2a). There were total 109 of the 216 DEGs which were not contained into the DEGs PPI network (Fig. 2a). Then we applied Cytotype MCODE for further analysis and results showed that 33 central nodes which were all up-regulated genes were identified among the 107 nodes (Fig. 2b).

Common DEGs PPI network constructed by STRING online database and Module analysis. a There were a total of 107 DEGs in the DEGs PPI network complex. The nodes meant proteins; the edges meant the interaction of proteins; green circles meant down-regulated DEGs and red circles meant up-regulated DEGs. b Module analysis via Cytoscape software (degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and max. Depth = 100)
Analysis of core genes by the Kaplan Meier plotter and GEPIA
Kaplan Meier plotter (http://kmplot.com/analysis) was utilized to identify 33 core genes survival data. It was found that 20 genes had a significantly worse survival while 13 had no significant (P < 0.05, Table 4 & Fig. 3). Then, GEPIA was used to dig up the 20 gene expression level between cancerous and normal people. Results reported that 15 of 20 genes reflected high expressed in OC samples contrasted to normal OV samples (P < 0.05, Table 5 & Fig. 4).

The prognostic information of the 33 core genes. Kaplan meier plotter online tools were used to identify the prognositc information of the 33 core genes and 20 of 33 genes had a significantly worse survival rate (P < 0.05)

Significantly expressed 20 genes in OV cancer patients compared to healthy people. To further identify the genes’ expression level between OV cancer and normal people, 20 genes which were related with poor prognosis were analyzed by GEPIA website. 15 of 20 genes had significant expression level in OV cancer specimen compared to normal specimen (*P < 0.05). Red color means tumor tissues and grey color means normal tissues
Re-analysis of 15 selected genes via KEGG pathway enrichment
To understand the possible pathway of these 15 selected DEGs, KEGG pathway enrichment was re-analyzed via DAVID (P < 0.05). Results showed that four genes (BUB1B, BUB1, TTK and CCNB1) markedly enriched in the cell cycle pathway (P = 1.1E-4, Table 6 & Fig. 5).

Re-analysis of 15 selected genes by KEGGpathway enrichment. Fifteen high expressed genes in OV cancer tissues with poor prognosis were re-analyzed by KEGG pathway enrichment. Four genes (BUB1B,BUB1,TTK and CCNB1) were significantly enriched in the cell cycle pathway especially in G2/M phase. Mps1 means TTK. BubR1 means BUB1B. CycB means CCNB1
Discussion
To identify more useful prognostic biomarkers in OV cancer, this study used bioinformatical methods on the basis of three profile datasets (GSE36668, GSE18520 and GSE 14407). Sixty-nine ovarian cancer specimens and twenty-six normal specimens were enrolled in the present research. Via GEO2R and Venn software, we revealed a total of 216 commonly changed DEGs (|logFC| > 2 and adjust P value < 0.05) including 110 up-regulated (Log FC > 0) and 106 down-regulated DEGs (Log FC < 0). Then, Gene Ontology and Pathway Enrichment Analysis using DAVID methods showed that 1) for biological processes (BP), up-regulated DEGs were particularly enriched in regulation of cell cycle, cell division, mitotic nuclear division, protein localization to kinetochore, sister chromatid cohesion and cell proliferation, and down-regulated DEGs in blood coagulation, positive regulation of transcription from RNA polymerase II promoter, palate development, negative regulation of transcription from RNA polymerase II promoter, axonogenesis and receptor internalization; 2) for molecular function (MF), up-regulated DEGs were enriched in ATP-dependent microtubule motor activity, protein binding, plus-end-directed, microtubule binding, sequence-specific DNA binding, protein kinase binding and down-regulated DEGs in transcriptional repressor activity, RNA polymerase II core promoter proximal region sequence-specific binding and growth factor activity, RNA polymerase II core promoter proximal region sequence-specific binding, RNA polymerase II transcription factor binding, transcriptional activator activity; 3) for GO cell component (CC), up-regulated DEGs were significantly enriched in the nucleoplasm, midbody, spindle microtubule, spindle, cytosol and nucleus, and down-regulated DEGs in proteinaceous extracellular matrix anchored component of membrane, extracellular space and extracellular region. For pathway analysis, up-regulated DEGs were particularly enriched in p53 signaling pathway, cell cycle and progesterone-mediated oocyte maturation and while down-regulated DEGs in no noteworthy signaling pathways (P < 0.05). Next, DEGs PPI network complex of 108 nodes and 698 edges was constructed via the STRING online database and Cytoscape software. Then, 33 vital up-regulated genes were screened from the PPI network complex by Cytotype MCODE analysis. Furthermore, through Kaplan Meier plotter analysis, we found that 20 of 33 genes had a significantly worse survival. In validating these 20 genes, 15 genes reflected high expression in OC samples compared with normal samples by GEPIA analysis (P < 0.05). Finally, we re-analyzed 15 genes via DAVID for KEGG pathway enrichment and found that four genes (BUB1B, BUB1, TTK and CCNB1) enriched in cell cycle had a significance (P < 0.05) which could be considered as new effective targets to improve the prognosis of OC patients.
Mitotic checkpoint serine/threonine kinase B (BUB1B), which is seen as a mammalian homolog of yeast Mad3, but they are significantly different because BUB1B has a kinase domain which is not found in Mad3 [14]. In 2004, Kops GJ, et al. reported that apoptotic cell death and massive chromosome loss could occur due to the inhibition of BUB1B kinase activity and reduction of the BUB1B level in human cancer cells [15]. BUB1B has been demonstrated to enhance tumor proliferation and is associated with worse survival rate in several types of cancer, including prostate cancer, breast, gastric and colorectal [16,17,18,19]. Another study proved that knockdown of BUB1B resulted in inhibition of tumor growth in vivo, including the regression of established tumors via postmitotic endoreduplication checkpoint [20] which is the replication of the genome during the cell cycle without the subsequent completion of mitosis and/or cytokinesis [21].
BUB1 which is a serine/threonine kinase and encoded by the BUB1 gene, binds centromeres during mitosis. It has been noted that over-expressed BUB1 is related to several cancers and their worse clinical prognosis. Wang et al. [22] presented that high expression of BUB1 was associated with poor disease-free survival of 203 patients with breast cancer. In addition, Zhao et al. [23] indicated that higher positive percentage of BUB1 protein meant a more advanced stage and a higher differentiation degree of endometrial carcinoma. Furthermore, Pinto et al. [24] demonstrated that over-expression of BUB1 was found to be substantially related to Furhman grade of the tumors and with the number of genomic copy number changes. By isolating daughter cells from mother cells, BUB1 also were vitally responsible for the accurate assignment of chromosomes without establishing the mitotic spindle checkpoint and aligning chromosomes [25, 26].
Monopolar spindle1 (Mps1, also known as TTK), is a bispecific protein kinase that phosphorylates serines/threonines and tyrosines [27]. Mps1 is a core segment of the SAC (spindle assembly checkpoint) and is a key monitoring mechanism to ensure healthy cell proliferation and precise division [28, 29]. In addition to mitotic SAC regulation, Mps1 play roles in other processes, including DNA damage response, centrosome duplication and organ development [30]. Moreover, high expression of Mps1 was easily found in several human malignancies, such as thyroid carcinoma, glioblastoma and breast cancer [31,32,33,34].
CCNB1, G2/Mitotic-specific cyclin B1, is a monitoring protein in mitosis and expressed primarily in G2/M phase which is critical for controlling the cell cycle at the G2/M (mitosis) transition. Recently, increasing evidence demonstrated that CCNB1 was over-expressed in considerable cancers with poor prognosis, including gastric cancer [35], esophageal squamous cell carcinoma [36], non-small cell lung cancer [37] and astrocytomas [38]. Furthermore, it was also pointed out that down-regulation of CCNB1 of mRNA levels and protein could reduce cell proliferation [39]. In 2017, Zhao P, et al. reported that up-regulation of CCNB1 could be an index for pituitary adenomas invasiveness and played a part in the pathology of pituitary adenomas with other monitoring molecules in the cell cycle [40].
Numerous studies have proved that these four genes were related to various types of cancer’s progression, however, very few studies have been reported about these four genes in OV cancer after we searched these four genes in Pubmed website. Therefore, the data in our study could provide useful information and direction for future study in OV cancer.
Conclusions
Taken above, our bioinformatics analysis study identified four DEGs (BUB1B, BUB1, TTK and CCNB1) between OC tissues and normal OV tissues on the base of three different microarray datasets. Results showed that these four genes could play key roles in the progression of OC. However, these predictions should be verified by a series of experiments in the future. Anyway, these data may provide some useful information and direction into the potential bio-markers and biological mechanisms of OC.
Abbreviations
- BP:
-
Biological process
- BUB1B:
-
Mitotic checkpoint serine/threonine kinase B
- CC:
-
Cellular component
- CCNB1:
-
G2/Mitotic-specific cyclin B1
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- DEGs:
-
Differentially expressed genes
- GEO:
-
Gene Expression Omnibus
- GEPIA:
-
Gene Expression Profiling Interactive Analysis
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Gene and Genome
- MCODE:
-
Molecular Complex Detection
- MF:
-
Molecular function
- OC:
-
Ovarian cancer
- OV:
-
Ovarian
- PPI:
-
protein-protein interaction
- STRING:
-
Search Tool for the Retrieval of Interacting Genes
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. Ca-Cancer J Clin. 2017;67(1):7–30.
Weidle UH, Birzele F, Kollmorgen G, Rueger R. Mechanisms and targets involved in dissemination of ovarian Cancer. Cancer Genom Proteom. 2016;13(6):407–23.
Cormio G, Rossi C, Cazzolla A, Resta L, Loverro G, Greco P, et al. Distant metastases in ovarian carcinoma. Int J Gynecol Cancer. 2003;13(2):125–9.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58.
Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47(4):312–9.
Davis S, Meltzer PS. GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–7.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Szasz AM, Lanczky A, Nagy A, Forster S, Hark K, Green JE, et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016;7(31):49322–33.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):w98–w102.
Taylor SS, Ha E, McKeon F. The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J Cell Biol. 1998;142(1):1–11.
Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci U S A. 2004;101(23):8699–704.
Hudler P, Britovsek NK, Grazio SF, Komel R. Association between polymorphisms in segregation genes BUB1B and TTK and gastric cancer risk. Radiol Oncol. 2016;50(3):297–307.
Mansouri N, Movafagh A, Sayad A, Heidary Pour A, Taheri M, Soleimani S, et al. Targeting of BUB1b gene expression in sentinel lymph node biopsies of invasive breast Cancer in Iranian female patients. Asian Pac J Cancer Prev. 2016;17(S3):317–21.
Hahn MM, Vreede L, Bemelmans SA, van der Looij E, van Kessel AG, Schackert HK, et al. Prevalence of germline mutations in the spindle assembly checkpoint gene BUB1B in individuals with early-onset colorectal cancer. Genes Chromosomes Cancer. 2016;55(11):855–63.
Fu X, Chen G, Cai ZD, Wang C, Liu ZZ, Lin ZY, et al. Overexpression of BUB1B contributes to progression of prostate cancer and predicts poor outcome in patients with prostate cancer. Onco Targets Ther. 2016;9:2211–20.
Wan X, Yeung C, Kim SY, Dolan JG, Ngo VN, Burkett S, et al. Identification of FoxM1/Bub1b signaling pathway as a required component for growth and survival of rhabdomyosarcoma. Cancer Res. 2012;72(22):5889–99.
Edgar BA, Orr-Weaver TL. Endoreplication cell cycles: more for less. Cell. 2001;105(3):297–306.
Wang Z, Katsaros D, Shen Y, Fu Y, Canuto EM, Benedetto C, et al. Biological and clinical significance of MAD2L1 and BUB1, genes frequently appearing in expression signatures for breast Cancer prognosis. PLoS One. 2015;10(8):e0136246.
Zhao Q, Bian AP, Zhang Y, Qin L, Shi HR, Su K. Expression of budding uninhibited by benzimidazoles-1 and mitotic arrest deficient-2 in endometrial carcinoma and its significance. Eur J Gynaecol Oncol. 2014;35(1):44–7.
Pinto M, Vieira J, Ribeiro FR, Soares MJ, Henrique R, Oliveira J, et al. Overexpression of the mitotic checkpoint genes BUB1 and BUBR1 is associated with genomic complexity in clear cell kidney carcinomas. Cell Oncol. 2008;30(5):389–95.
Johnson VL, Scott MI, Holt SV, Hussein D, Taylor SS. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J Cell Sci. 2004;117(Pt 8):1577–89.
Yu H, Tang Z. Bub1 multitasking in mitosis. Cell Cycle. 2005;4(2):262–5.
Fisk HA, Mattison CP, Winey M. A field guide to the Mps1 family of protein kinases. Cell Cycle. 2004;3(4):439–42.
Musacchio A. Spindle assembly checkpoint: the third decade. Philos Trans R Soc Lond Ser B Biol Sci. 2011;366(1584):3595–604.
Rutledge SD, Cimini D. Consequences of aneuploidy in sickness and in health. Curr Opin Cell Biol. 2016;40:41–6.
Liu X, Winey M. The MPS1 family of protein kinases. Annu Rev Biochem. 2012;81:561–85.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96.
Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS, Nobel AB, et al. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med. 2006;355(6):560–9.
Suda T, Tsunoda T, Daigo Y, Nakamura Y, Tahara H. Identification of human leukocyte antigen-A24-restricted epitope peptides derived from gene products upregulated in lung and esophageal cancers as novel targets for immunotherapy. Cancer Sci. 2007;98(11):1803–8.
Mizukami Y, Kono K, Daigo Y, Takano A, Tsunoda T, Kawaguchi Y, et al. Detection of novel cancer-testis antigen-specific T-cell responses in TIL, regional lymph nodes, and PBL in patients with esophageal squamous cell carcinoma. Cancer Sci. 2008;99(7):1448–54.
Banerjee SK, Weston AP, Zoubine MN, Campbell DR, Cherian R. Expression of cdc2 and cyclin B1 in helicobacter pylori-associated gastric MALT and MALT lymphoma : relationship to cell death, proliferation, and transformation. Am J Pathol. 2000;156(1):217–25.
Murakami H, Furihata M, Ohtsuki Y, Ogoshi S. Determination of the prognostic significance of cyclin B1 overexpression in patients with esophageal squamous cell carcinoma. Virchows Arch. 1999;434(2):153–8.
Soria JC, Jang SJ, Khuri FR, Hassan K, Liu D, Hong WK, et al. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res. 2000;60(15):4000–4.
Allan K, Jordan RC, Ang LC, Taylor M, Young B. Overexpression of cyclin a and cyclin B1 proteins in astrocytomas. Arch Pathol Lab Med. 2000;124(2):216–20.
Mussnich P, Raverot G, Jaffrain-Rea ML, Fraggetta F, Wierinckx A, Trouillas J, et al. Downregulation of miR-410 targeting the cyclin B1 gene plays a role in pituitary gonadotroph tumors. Cell Cycle. 2015;14(16):2590–7.
Zhao P, Zhang P, Hu W, Wang H, Yu G, Wang Z, et al. Upregulation of cyclin B1 plays potential roles in the invasiveness of pituitary adenomas. J Clin Neurosci. 2017;43:267–73.
Acknowledgements
We would like to thank everyone who take part in this study.
Funding
This work was supported by the National Nature Science Foundation (No. 81571387).
Availability of data and materials
The datasets used and/or analyzed in the present study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
XY Y and JJ Z conceived and designed the idea to this paper; HF and ZY G collected and analyzed the data, and drafted the paper; QL and QH L analyzed the data and revised the final paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Feng, H., Gu, ZY., Li, Q. et al. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J Ovarian Res 12, 35 (2019). https://doi.org/10.1186/s13048-019-0508-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-019-0508-2