
Abstract
Background
R-spondin2 (Rspo2) is a secreted agonist of the canonical Wnt/β-catenin signaling pathway. Rspo2 plays a key role in development of limbs, lungs and hair follicles, and more recently during ovarian follicle development. Rspo2 heterozygous deficient female mice become infertile around 4 months of age mimicking primary ovarian insufficiency (POI). The study aimed to investigate the regulation of RSPO2 and its potential involvement in pathophysiology of POI.
Methods
We cloned the RSPO2 promoter and performed transcriptional assays to determine if RSPO2 can be regulated by NOBOX, an ovarian transcription factor. Then, we evaluated 100 infertile women after obtaining a detailed history of the disease and follicle-stimulating hormone measurements, besides karyotype determination and fragile-X premutation syndrome investigation. All exons, intron-exon boundaries and untranslated regions of the RSPO2 gene were identified by sequencing, and the results were statistically analyzed.
Results
We found that RSPO2 can be regulated by NOBOX via the presence of NOBOX Binding Element in its promoter. Among 9 identified variants in POI women, 4 of them were equally homozygous, 4 have never been described (c.-359C > G, c.-190G > A, c.-170 + 13C > T and c.-169-8 T > A), only one c.557 T > C was predicted to alter a single amino acid in the RSPO2 protein (p.Leu186Pro).
Conclusions
RSPO2 is a novel target gene of the NOBOX key transcription factor, confirming its important role during the follicular growth in ovary. However, RSPO2 mutations are rare or uncommon in women with POI.
Similar content being viewed by others


Background
Primary Ovarian Insufficiency (POI) is an important human disease model that provided substantial understanding into the factors involved in differentiation and ovarian development. This rare disease is characterized by amenorrhea with elevated gonadotropin levels, and affects 1% of women before the age of 40 years [1]. POI encompasses a heterogeneous spectrum of conditions, through two major mechanisms, follicle dysfunction and follicle depletion [2]. Although, the majority of cases remain idiopathic, POI can be triggered by autoimmune disease, viral, iatrogenic or genetic causes [1]. The disorder may be associated in syndromic diseases, such as Turner’s syndrome, X-Fragile syndrome or BPES syndrome [3]. Genetic component such as X-chromosome abnormalities, deletions, FMR1 premutations, BMP15 variants, were identified as the first genetic causes of the pathophysiology [4]. To date, about 30 autosomal genes were identified as involved in the pathogenesis of POI, and more recently a digenic form has been observed in large cohort of POI women [5]. For instance, recent publications reported mutations in genes involved in meiosis like STAG3 [6], HFM1 [7] or SYCE1 [8] and in transcription factors including FIGLA [9] or SOHLH2 [10]. However, except the high prevalence of NOBOX [11] and BMP15 mutations [4] no gene has been shown implicated in more than 6% and 3%, respectively, of POI cases.
Most POI are idiopathic, thus it seems to be important to research additional factors. Potential candidates could be members of R-spondin (RSPO) family that are ligands activating Wnt pathway [12]. Among them, R-spondin1 acts on ovarian differentiation and RSPO1 mutations have been found in patients with sex differentiation disorders [13]. It has recently been suggested that Rspo2, another member of this family, could be important in regulating ovarian follicle development [14]. Rspo2 is expressed in mouse oocytes from primary follicles to growing follicles. An ex vivo and in vivo treatment of mouse ovary with Rspo2 promoted development of follicles. Moreover, Rspo2 heterozygous deficient female mice become infertile around 4 months of age, then mimicking POI [15,16,17].
So, the aim of this study was to analyze RSPO2 regulation by NOBOX, a master-transcription factor of ovarian folliculogenesis and to determine whether RSPO2 genetic variation is associated with idiopathic POI women.
Methods
Cloning of RSPO2 promoter and luciferase assays
RSPO2 promoter containing two Nobox Binding Elements (NBE, region −3560 to −2606) was obtained by PCR and subcloned into SacI and XhoI sites of pGL4.26 Luc/miniP vector (Promega). COS7 cells seeded on 96-well plates were transfected with plasmids encoding the human wild-type (WT) or R303X-NOBOX transcription factor, firefly luciferase under control of the RSPO2 promoter, and Renilla luciferase (internal control) as previously reported [11] This R303X-NOBOX mutant is devoid of a large part of the homeodomain and used as a negative control [11]. Firefly and Renilla luciferase activities were measured consecutively with dual-luciferase assays (Promega) and a TriStar reader (Berthold), and are expressed as relative light units. Each assay was performed independently three times and included six replicates.
Statistical analysis
Statistical analysis was performed using PRISM 5 software (GraphPad). Data were analyzed using one-way ANOVA with Kruskal-Wallis post-tests. Data are shown as mean ± SEM. (**), p value < 0.01 and (****), p < 0.0001.
Patients
One hundred idiopathic POI women were recruited following Institutional Review Board approval (reference PHRC No. A0R03 052, approved by Bicêtre Ethical committee (CPP Ile-de-France VII #PP 16–024). All the participants gave their written informed consent for hormonal and genetic analyses, in keeping with the provisions of the French Bioethics Law and the Declaration of Helsinki and after approval by the Bicêtre and Reims Hospital ethic committees. Inclusion criteria were cessation of menstrual cycles for more than 4 months before 40 years of age, with at least two serum FSH concentrations of >30 IU/L. No Turner syndrome, X chromosome caryotypic anomalies, neither FRM1 premutation were present in the patients. Autoimmune and metabolic causes (such as galactosemia) of POI were excluded in this series. As a control population, 100 healthy women, 20–39 years of age were included in the present study belonging to the VARIETE cohort which was an open, prospective, national, multicenter, nonrandomized study of healthy volunteers, designed to establish normative data for hormones (ClinicalTrials.gov Identifier: NCT01831648). All women had normal age of puberty onset, body mass index, and regular spontaneous menstrual cycles (27–31 days). None had clinical signs that could suggest POI. All healthy subjects gave their written informed consent to participate in the study, which was approved by the Paris-Sud Ethics committee before the beginning of the study.
Hormonal measurements
FSH and LH plasma levels were measured by radioimmunoassay (Immunotech Beckman Coulter, France) as previously reported [18]. Estradiol was determined after previous plasma extraction (DiaSorin, Italy; Schering CisBio International, France; and Immunotech Beckman Coulter, respectively) [19]. Serum inhibin B levels were measured with a commercial ELISA (Serotec, Kidlington, Oxford, United Kingdom) with a detection limit of 6 ng/L and inter- and intraassay coefficients of variation of 13 and 6%, respectively (at 15 ng/L). Serum AMH was measured by an enzymatically amplified two-site immunoassay (AMH Gen II ELISA; Beckman Coulter, Marseilles, France). All steps of procedure were conducted according to the manufacturer’s recommendations. The lower detection limit of the assay was 0.7 pmol/L. The mean interassay coefficient of variation (CV) was 8.6%, and the mean intraassay CV was 4.0% at 22 pmol/L.
DNA sequencing and mutational analysis of genomic DNA
Genomic DNA was isolated from human blood leukocytes using standard methods, and the exons of the RSPO2 gene were sequenced in both directions in 100 women with POI. Reference sequences were obtained from Ensembl (ENSG00000147655 for genomic DNA, ENST00000276659 for messenger RNA) and the National Center for Biotechnology Information for messenger RNA NM_178565.4.
Mutational analysis of genomic DNA was studied by sequencing all six exons and exon–intron boundaries after amplification of RSPO2 gene. The entire coding sequence and intron-exon boundaries were amplified from genomic DNA using 6 sets of primers (Table 1). After a 5-min hot start, DNA was amplified for 35 cycles (95 °C for 45 s, 56–65 °C for 1 min, and 72 °C for 45 s) in 1× buffer with 2.5 mM MgCl2, 0.2 mM dNTPs, 0.06 μM concentrations of each primer, 100 ng of genomic DNA, and 1.25 U of Dream Taq (Fermentas) in a total volume of 50 μL. The amplified samples were subjected to agarose gel electrophoresis to ensure single bands. PCR products were purified, then sequenced on an automated sequencer (3100 Applied Biosystems) using the same primers as for PCR by Sanger Method.
Nucleotide numbering reflects cDNA numbering (http://www.HGVS.org/varnomen) with +1 corresponding to the A of the ATG translation initiation codon (codon 1) in the reference sequence.
Ethnically-matched controls from the 1000 Genomes Project Database [20] and the Exome Aggregation Consortium [21] were used for individual variant and gene mutation frequencies in this study.
Molecular modeling
PolyPhen (Polymorphism Phenotyping, http://genetics.bwh.harvard.edu/pph2/; http://www.HGVS.org/varnomen) and Sorting Intolerant From Tolerant (SIFT; http://sift.bii.a-star.edu.sg/) software packages as well as evolutionary conservation were used to predict the pathogenic nature of the coding sequence alteration.
Alternative Splice Site Predictor (ASSP) (http://wangcomputing.com/assp/), Netgen2 (http://www.cbs.dtu.dk/services/NetGene2/), NNSplice.v.0.9 (http://www.fruitfly.org/seq_tools/splice.html), Human Splicing Finder v.2.4.1 (http://www.umd.be/HSF/) software were used to predict splicing site of intronic variations.
Results
RSPO2 is a novel target gene of NOBOX
The ovarian Rspo2 expression was reported very low in Nobox-null mice suggesting that it is controlled by this transcription factor [22]. In silico analysis of Rspo2 promoter revealed the presence of two NOBOX Binding Element (NBE) (Fig. 1a). A 954 bp fragment of the human RSPO2 promoter, containing two NBE, was cloned in a luciferase reporter plasmid (RSPO2-promoter).

Identification of RSPO2 as a NOBOX target and in vitro assay of NOBOX transcriptional activity using RSPO2 promoter as reporter gene. a Schematic map of 954 bp promoter sequence of human RSPO2 including 2 NOBOX binding elements (NBE) located at −3370 and −2898 upstream of the transcription site. b NOBOX activity is shown as the luciferase activity above baseline, which is defined as the activity observed in cells transfected with empty vector (Ctl). The transcriptional activity of wild-type NOBOX (NOBOX WT) was studied using RSPO2 promoter transfected in COS7 cells. As a negative control, the NOBOX deleted homeodomain mutant (NOBOX-303X) was used. Results are presented as mean ± SEM of 3 independent experiments each performed in sixplicate. **P < .01 ****P < .0001. RLU, relative light units
To measure the effects of NOBOX on RSPO2 transactivation promoter, COS7 cells were transfected with RSPO2-promoter and wild-type (WT)-NOBOX or R303X-NOBOX, a mutant devoid of a large part of the homeodomain find in POI patient [11]. We observed a significant increased activation of RSPO2-promoter with WT-NOBOX suggesting that NOBOX can bind RSPO2 promoter (Fig. 1b). As a negative control R303X-NOBOX was unable to activate the promoter (Fig. 1b).
Clinical characteristics of the patients
One hundred POI patients completed the study. Median age was 30 (13–39) at the time of the diagnosis. Clinical and hormonal characteristics of these patients are summarized in Table 2. Most of the patients (n = 80) presented with normal puberty and secondary amenorrhea whereas 20 presented with primary amenorrhea with variable pubertal development (from absence (5), partial (11) to complete development (4)). Among patients with secondary amenorrhea, symptoms appear from 15 to 39 year-old with a median age at 29.
Identification of SNPs in the RSPO2 locus
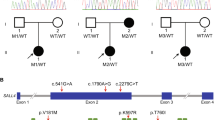
Analysis of the RSPO2 gene in 100 POI patients revealed the presence of 9 sequence variations identified in the non-coding and coding portions (Fig. 2 and Table 3). All 9 variations were heterozygous and 4 of them were equally homozygous. Among them, 4 of these SNPs have never been described (c.-359C > G, c.-190G > A, c.-170 + 13C > T and c.-169-8 T > A). Of the 9 identified SNPs, only one c.557 T > C was predicted to alter a single amino acid in the RSPO2 protein (p.Leu186Pro). This variant was found heterozygous in 51% and homozygous in 20% of patients and was also described as very common in controls by Exome variant server and the 1000 Genomes. It is predicted as non-deleterious by Polyphen and SIFT softwares and hence clearly could be considered as not disease-causing.

Schematic representation of RSPO2 gene and RSPO2 protein. a R-spondin2 gene contains 6 exons (reference sequence was based on NC_000008.11 and ENSG00000147655). Locations of 9 variations are indicated by an arrow. b R-spondin2 mRNA includes exons 2 to 6 encoding the protein (reference sequence was based on NM_178565 and ENST00000276659). c RSPO2 encompasses a signal peptide (SP), two cysteine-rich furin like repeats (FL) and a thrombospondin type 1 repeat domain (TR) (reference sequence was based on NP_848660)
Otherwise, 4 variations (c.-170 + 13C > T, c.-169-8 T > A, c.94 + 26G > A and c.616 + 43_616 + 44insTG) were found in intron-exon junction. The in silico analysis using Alternative Splice Site Predictor (ASSP), Netgen2, NNSplice.v.0.9, Human Splicing Finder v.2.4.1 did not display the generation of a new splicing site. The remaining 4 variations located in 5’UTR non coding sequence (c.-500A > G, c.-359C > G, c.-190G > A and c.-138 A > G) were rare and did not cause the production of a new start codon (ATG).
Discussion
More than 80% of POI causes are unknown until now, it is of interest that multiple approaches are required to discover new responsible candidate genes. Mouse forward genetics is a powerful tool for the identification of genes and pathways important in biological process and has been very valuable for the characterization of key players in ovarian folliculogenesis [23]. Recently transgenic mouse studies supported the role of Rspo2 in follicle development. Human RSPO2 gene is located on chromosome 8 and encodes a member of the R-Spondin family of secreted proteins involved in β-catenin activation through the canonical Wnt pathway leading to T-cell factor-dependent gene activation [24,25,26]. Footless mutant mice with a transgene insertion in the Rspo2 gene showed many malformations [15], although Footless heterozygous animals were indistinguishable from wild-type animals through weaning, female animals were only fertile until 4 months of age. Similarly, heterozygous Rspo2 mutant female mice were 25% sterile at 4 months and 85% sterile at 5 months [25]. This infertile phenotype associated with the oocyte Rspo2 expression and follicular growth function led us to consider this gene as a possible candidate gene in POI. It is likely that the initial waves of follicle growth were maintained in Rspo2 heterozygous mutants, but inappropriate levels of Rspo2 in oocytes eventually led to the failure of follicle development during late reproductive life. It was recently shown that the administration of Rspo2 agonist promotes human early follicle development after xenografted ovarian cortical pieces in immunodeficient mice [14]. Loss of function mutations in RSPO2 has not previously been associated with any phenotype in women.
Of interest, we described a number of mutations of the oocyte-specific homeobox gene NOBOX associated with POI patients [11, 27]. Interestingly, the ovarian Rspo2 expression was reported very low in Nobox-null mice [22]. Otherwise, by an in silico analysis of RSPO2 promoter the presence of NOBOX Binding Element (NBE) is revealed. Here, the transcriptional assays showed that NOBOX was able to activate the RSPO2 expression. This suggests that RSPO2 is a novel target gene of NOBOX, a master regulator of the folliculogenesis. It was, thus, of interest to look at RSPO2 variants and to evaluate the role of RSPO2 in POI patients.
Among 100 independent idiopathic POI patients, we identified 9 Single Nucleotide Polymorphisms (SNPs) in RSPO2, four of them had never been described and one caused an amino-acid change but none of them seems to be deleterious when analyzed in silico.
The variations of RSPO2 gene found in this cohort seem to be polymorphisms with current technics, and RSPO2 mutations are therefore rare. However, RSPO2 appears to have an important role in ovary and it should be interesting to search RSPO2 mutations in a larger cohort of POI patients. Moreover, RSPO2 is also a local intraovarian factor expressed at specific stages of follicular development. Taking into account, it would be rare to find a pathogenic variant or polymorphism in the peripheral blood of patients with POI. The disruption mediated through RSPO2 is most likely a local disruption which is temporally associated with folliculogenesis. Since it is challenging to study RSPO2 variations in ovarian tissue, we cannot exclude some somatic variants. Identically, the search of mutations in genes largely involved in different folliculogenesis steps such as NANOS3, FOXO3a and PRLR [28,29,30] has also been performed without any success. Moreover, many factors and regulators involved in folliculogenesis are intraovarian and work in a paracrine manner. The expression in oocytes and granulosa cells is more important than in somatic cells. Therefore, the paper demonstrating that R-spondin2 acts in folliculogenesis [14] and the present study support and reinforce the importance of RSPO2 in this process.
Future high-throughput sequencing of such genes in women with idiopathic ovarian insufficiency should provide a better idea of the contribution that oocyte genes make to nonsyndromic POI.
Conclusion
RSPO2 is described as a key actor of the folliculogenesis regulation. In the present study, we demonstrated that RSPO2 is regulated by NOBOX a master transcription factor, suggesting that RSPO2 can be mutated in women affected by primary ovarian insufficiency. However, our analysis has not allowed to detect RSPO2 pathogenic variants. We conclude that mutations in the coding regions of RSPO2 are not common causes in POI.
Abbreviations
- AMH:
-
Anti-Müllerian hormone
- DNA:
-
deoxyribonucleic acid
- FSH:
-
Follicle Stimulating Hormone
- LH:
-
Luteinizing Hormone
- NBE:
-
Nobox Binding Element
- POI:
-
primary ovarian insufficiency
- Rspo:
-
R-spondin
- SNP:
-
Single nucleotide polymorphism
- WT:
-
wild-type
References
De Vos M, Devroey P, Fauser BC. Primary ovarian insufficiency. Lancet. 2010;376:911–21.
Nelson LM. Clinical practice. Primary ovarian insufficiency. N Engl J Med. 2009;360:606–14.
Skillern A, Rajkovic A. Recent developments in identifying genetic determinants of premature ovarian failure. Sex dev. 2008;2:228–43.
Persani L, Rossetti R, Cacciatore C. Genes involved in human premature ovarian failure. J Mol Endocrinol. 2010;45:257–79.
Bouilly J, Beau I, Barraud S, Bernard V, Azibi K, Fagart J, et al. Identification of multiple gene mutations accounts for a new genetic architecture of primary ovarian insufficiency. J Clin Endocrinol Metab. 2016;101:4541–50.
Caburet S, Arboleda VA, Llano E, Overbeek PA, Barbero JL, Oka K, et al. Mutant cohesin in premature ovarian failure. N Engl J Med. 2014;370:943–9.
Wang J, Zhang W, Jiang H, Wu BL. Primary ovarian insufficiency C. Mutations in HFM1 in recessive primary ovarian insufficiency. N Engl J Med. 2014;370:972–4.
de Vries L, Behar DM, Smirin-Yosef P, Lagovsky I, Tzur S, Basel-Vanagaite L. Exome sequencing reveals SYCE1 mutation associated with autosomal recessive primary ovarian insufficiency. J Clin Endocrinol Metab. 2014;99:E2129–32.
Zhao H, Chen ZJ, Qin Y, Shi Y, Wang S, Choi Y, et al. Transcription factor FIGLA is mutated in patients with premature ovarian failure. Am J Hum Genet. 2008;82:1342–8.
Qin Y, Jiao X, Dalgleish R, Vujovic S, Li J, Simpson JL, et al. Novel variants in the SOHLH2 gene are implicated in human premature ovarian failure. Fertil Steril. 2014;101:1104–9. e6
Bouilly J, Bachelot A, Broutin I, Touraine P, Binart N. Novel NOBOX loss-of-function mutations account for 6.2% of cases in a large primary ovarian insufficiency cohort. Hum Mutat. 2011;32:1108–13.
de Lau WB, Snel B, Clevers HC. The R-spondin protein family. Genome Biol. 2012;13:242.
Tomaselli S, Megiorni F, De Bernardo C, Felici A, Marrocco G, Maggiulli G, et al. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat. 2008;29:220–6.
Cheng Y, Kawamura K, Takae S, Deguchi M, Yang Q, Kuo C, et al. Oocyte-derived R-spondin2 promotes ovarian follicle development. FASEB J. 2013;27:2175–84.
Bell SM, Schreiner CM, Hess KA, Anderson KP, Scott WJ. Asymmetric limb malformations in a new transgene insertional mutant, footless. Mech Dev. 2003;120:597–605.
Ahmed NN, Grimes HL, Bellacosa A, Chan T, Tsichlis PN. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci U S A. 1997;94:3627–32.
Nam JS, Park E, Turcotte TJ, Palencia S, Zhan X, Lee J, et al. Mouse R-spondin2 is required for apical ectodermal ridge maintenance in the hindlimb. Dev Biol. 2007;311:124–35.
Trabado S, Maione L, Bry-Gauillard H, Affres H, Salenave S, Sarfati J, et al. Insulin-like peptide 3 (INSL3) in men with congenital hypogonadotropic hypogonadism/Kallmann syndrome and effects of different modalities of hormonal treatment: a single-center study of 281 patients. J Clin Endocrinol Metab. 2014;99:E268–75.
Bouligand J, Ghervan C, Tello JA, Brailly-Tabard S, Salenave S, Chanson P, et al. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742–8.
Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nat. 2016;536:285–91.
Choi Y, Qin Y, Berger MF, Ballow DJ, Bulyk ML, Rajkovic A. Microarray analyses of newborn mouse ovaries lacking Nobox. Biol Reprod. 2007;77:312–9.
Edson MA, Nagaraja AK, Matzuk MM. The mammalian ovary from genesis to revelation. Endocr Rev. 2009;30:624–712.
Kazanskaya O, Glinka A, del Barco BI, Stannek P, Niehrs C, Wu W. R-Spondin2 is a secreted activator of Wnt/beta-catenin signaling and is required for Xenopus myogenesis. Dev Cell. 2004;7:525–34.
Nam JS, Turcotte TJ, Smith PF, Choi S, Yoon JK. Mouse cristin/R-spondin family proteins are novel ligands for the frizzled 8 and LRP6 receptors and activate beta-catenin-dependent gene expression. J Biol Chem. 2006;281:13247–57.
Kamata T, Katsube K, Michikawa M, Yamada M, Takada S, Mizusawa H. R-spondin, a novel gene with thrombospondin type 1 domain, was expressed in the dorsal neural tube and affected in Wnts mutants. Biochim Biophys Acta. 1676;2004:51–62.
Bouilly J, Roucher-Boulez F, Gompel A, Bry-Gauillard H, Azibi K, Beldjord C, et al. New NOBOX mutations identified in a large cohort of women with primary ovarian insufficiency decrease KIT-L expression. J Clin Endocrinol Metab. 2015;100:994–1001.
Qin Y, Zhao H, Kovanci E, Simpson JL, Chen ZJ, Rajkovic A. Mutation analysis of NANOS3 in 80 Chinese and 88 Caucasian women with premature ovarian failure. Fertil Steril. 2007;88:1465–7.
Vinci G, Christin-Maitre S, Pasquier M, Bouchard P, Fellous M, Veitia RA. FOXO3a variants in patients with premature ovarian failure. Clin Endocrinol. 2008;68:495–7.
Bachelot A, Bouilly J, Liu Y, Rebourcet D, Leux C, Kuttenn F, et al. Sequence variation analysis of the prolactin receptor C-terminal region in women with premature ovarian failure. Fertil Steril. 2010;94:2772–5.
Acknowledgements
We thank the patients and their family members who generously participated in the genetic studies.
Funding
This work was supported by INSERM. The study of controls subjects was supported by a grant from Programme Hospitalier de Recherche Clinique, French Ministry of Health, N° P081216 / IDRCB 2009-A00892–55 (Variété), AP-HP.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Author information
Authors and Affiliations
Contributions
JB, IB and SB designed and performed the experiments, JB, IB and SB interpreted the results, and JB, SB and NB drafted the manuscript. SB, VB, JY, BD, provided samples and clinical data and made critical revisions to the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
POI cohort has been recruited following Institutional Review Board approval, reference PHRC No. A0R03 052, approved by Bicêtre Ethical committee (CPP Ile-de-France VII #PP 16–024).
Consent for publication
Not applicable.
Competing interests
The authors report no financial or commercial conflicts of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bouilly, J., Beau, I., Barraud, S. et al. R-spondin2, a novel target of NOBOX: identification of variants in a cohort of women with primary ovarian insufficiency. J Ovarian Res 10, 51 (2017). https://doi.org/10.1186/s13048-017-0345-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-017-0345-0