
Abstract
Metabolites are intermediate products of cellular metabolism catalysed by various enzymes. Metabolic remodelling, as a biochemical fingerprint of cancer cells, causes abnormal metabolite accumulation. These metabolites mainly generate energy or serve as signal transduction mediators via noncovalent interactions. After the development of highly sensitive mass spectrometry technology, various metabolites were shown to covalently modify proteins via forms of lysine acylation, including lysine acetylation, crotonylation, lactylation, succinylation, propionylation, butyrylation, malonylation, glutarylation, 2-hydroxyisobutyrylation and β-hydroxybutyrylation. These modifications can regulate gene expression and intracellular signalling pathways, highlighting the extensive roles of metabolites. Lysine acetylation is not discussed in detail in this review since it has been broadly investigated. We focus on the nine aforementioned novel lysine acylations beyond acetylation, which can be classified into two categories: histone acylations and nonhistone acylations. We summarize the characteristics and common functions of these acylation types and, most importantly, provide a glimpse into their fine-tuned control of tumorigenesis and potential value in tumour diagnosis, monitoring and therapy.
Similar content being viewed by others

Background
Metabolites are intermediate products of cellular metabolism catalysed by various enzymes inherent to cells, and their interactions within a biological system are collectively known as the metabolome [1]. Metabolic remodelling, as a well-recognized hallmark of malignancy, generates sufficient energy to fuel the exponential growth and proliferation of tumour cells [2,3,4]. Tumour-inducing metabolic disturbances result in aberrant accumulation of metabolites, also termed oncometabolites [5]. In addition to being involved in metabolic pathways and energy supply, metabolites trigger oncogenic signalling via noncovalent interactions with macromolecules, mainly involved in competitive inhibition or allostery [1]. In one noteworthy example, the signalling molecule succinate binds to and activates succinate receptor 1 (SUCNR1) to promote tumour cell proliferation and metastasis [6,7,8]. Importantly, metabolites have been shown to possess nonmetabolic functions because of direct protein modifications, which are distinguished from other modifications, such as ubiquitination and sumoylation. These modifications greatly enrich the level of protein modification and expand the functions of metabolites.
Chemical modifications of proteins are also referred to as posttranslational modifications (PTMs). DNA instructs the synthesis of proteins from only 20 primary amino acids; however, PTMs increase the functional diversity of proteins and the proteome [9, 10]. These modifications, which include lysine acylation, can regulate protein activity, turnover, localization, and dynamic interactions with cellular molecules, such as other proteins, nucleic acids, lipids and cofactors [11, 12]. For instance, the succinylation of glutaminase (GLS) at lysine residue 164 (K164) marks GLS for ubiquitination at residue K158 and facilitates its subsequent degradation [13]. Additionally, the enzymatic activity of glutaryl-CoA dehydrogenase (GCDH) is suppressed by glutarylation, causing the lysine oxidation level to decrease [14]. These modifications in turn can influence almost all aspects of normal cell biology and pathogenesis. Notably, PTMs are, beyond all doubt, of supreme importance in tumorigenesis.
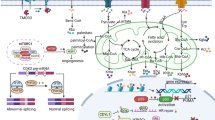
In the past few decades, lysine acetylation (Kac), an abundant, reversible, and highly regulated PTM, has irrefutably become a ‘hot spot’ in the field of epigenetic modification and is precisely modulated by proteins involved in its deposition (writers), binding (readers) and removal (erasers) [15,16,17,18]. Owing to the rapid development and application of mass spectrometry technology [9, 19], a large panel of novel lysine acylation types have successively been identified, including lysine crotonylation (Kcr) [20], lactylation (Kla) [21], succinylation (Ksucc) [22], propionylation (Kpr) [23], butyrylation (Kbu) [23], malonylation (Kma) [24], glutarylation (Kglu) [25], 2-hydroxyisobutyrylation (Khib) [26] and β-hydroxybutyrylation (Kbhb) [27]. They stem from major metabolites including glucose, amino acids and fatty acids (Fig. 1). These modifications can be classified into two main categories: histone acylation and nonhistone acylation. Histone acylation mainly affects the epigenome via transcriptional regulation of gene expression, and nonhistone acylation mainly modulates protein functions such as protein stability, activity and localization, as well as protein-protein interactions. In this review, we comprehensively introduce the nine novel metabolite-induced cognate lysine acylations beyond acetylation and elucidate their dramatic roles in metabolomic-epigenetic and metabolomic-proteomic signalling in tumours (Table 1).

The metabolic association of lysine acylation. Three major metabolic resources(glucose, fatty acid and amino acids) generate abundant intermediate product within cells, such as lactate, Succ-CoA, Ac-CoA and BHB, which provide acyl-groups to covalently modify proteins. Corresponding metabolites of Kcr, Kbu, Kpr, Kglu, Khib mainly emanate from fatty acid oxidation and amino acid metabolism whereas those of Kla, Kma, Kbhb and Ksucc are derived from glucose metabolism
Crotonylation
Characteristics and functions of Kcr
Scientists discovered that Kcr, a novel histone mark, functions distinctively from Kac [20]. Notably, Kcr can be modulated enzymatically by P300/CBP with sodium crotonate serving as the substrate [70] and metabolically on the basis of the concentration of the intermediate metabolite crotonyl-CoA, which increases during mitochondrial or peroxisomal fatty acid oxidation, as well as lysine and tryptophan metabolism [28]. Later, PCAF, MOF, KAT6A, HBO1 and Tip60 were identified as catalysts of Kcr [33, 71,72,73,74], and the opposite reaction is mediated by the histone deacetylases HDAC3 [75] and SIRT1–3 [76], with SIRT3 being the major decrotonylase in living cells [77]. Notably, a recent study asserted that class I HDACs are the principal histone decrotonylases [78]. Interestingly, Kcr of HDAC1 itself undermines its deacetylation activity on histone substrates, hinting at the intricacy of PTM regulation [70]. The AF9 YEATS domain has been reported to be a ‘reader’ of histones modified by Kcr with a binding preference for crotonylated lysine sites over lysine acetylation sites in histone H3 [79, 80]. In addition to AF9, the YEATS domains of Taf14 and YEATS2, the double PHD finger (DPF) domains of MOZ and DPF2, and the second bromodomain of TAF1 are capable of binding crotonyl marks [81,82,83,84].
P300-mediated histone Kcr has been reported to stimulate gene transcription to a greater degree than histone Kac [28]. However, histone decrotonylation leads to global transcriptional repression and impairs promoter recruitment of its reader proteins in mammalian cells [78]. Histone Kcr marks enhancers and transcriptional start sites (TSSs) of active sex chromosome-linked genes in postmeiotic male germ cells by conferring resistance to transcriptional repressors, which enables successful escape of transcriptional meiotic sex chromosome inactivation (MSCI) [85, 86] and regulates the differentiation of male germ cells [20, 29]. Chromodomain Y-like protein CDYL acts as a crotonyl-CoA hydratase to reduce histone Kcr levels and regulate spermatogenesis [30]. According to previous findings that histone Kcr participates in the DNA damage response [31] and CDYL promotes homology-directed repair (HDR) [87, 88], scientists further revealed a key role of CDYL-regulated RPA1 crotonylation in the HDR process [32]. In addition, Kcr of EB1, a core and scaffold microtubule plus-end tracking protein, ensures accurate spindle positioning in mitosis [33]. Kcr-AF9 YEATS interactions are promoted by crotonate pretreatment, positively regulating gene expression in the inflammatory response [80]. In acute kidney injury (AKI), histone Kcr counters inflammation and mitochondrial stress to protect renal function via transcriptional regulation, demonstrating its potential therapeutic value in renal diseases [34]. Low levels of histone Kcr in the medial prefrontal cortex promote stress-induced depressive behaviours in rodents by regulating gene transcription, thus laying a foundation for innovative epigenetic treatment in depression [35]. In addition, histone Kcr can be targeted to eradicate HIV since the previously latent HIV is reactivated upon hypercrotonylation induced by crotonyl-CoA production [36]. Additionally, histone Kcr has been demonstrated to facilitate telomere maintenance [37] and the differentiation [38] of stem cells.
Role of Kcr in cancer
Kcr is considerably reduced in prostate tumours and has a positive correlation with tumour grade. Furthermore, in-depth investigations have documented that histone hypocrotonylation induced by bromodomain-containing protein 4 (BRD4) inhibitors can abrogate cell proliferation and migration, thus showing the potential value of histone hypocrotonylation for therapeutic intervention in prostate cancer [89]. Histone Kcr is also abundant in gut cells, especially H3K18cr, which is associated with increased expression of genes involved in cancer and regulation of cell cycle progression [90]. Moreover, HDAC2, which is elevated in colon cancer and related to tumorigenesis, exhibits decrotonylase activity and thus downregulates histone Kcr levels, meaning that Kcr may participate in cancer suppression [90,91,92]. Interestingly, the YEATS domain, which has a higher affinity for Kcr sites than other acylation sites [79, 81, 82], plays an important role in leukaemia [93, 94], and inhibition of this domain can attenuate oncogene transcription in leukaemia [94]. Hence, we surmise that the strong epigenetic impact of the YEATS domain on leukaemia may be partially due to its interaction with crotonylated histone. In addition, K420cr of enolase 1 (ENO1) increases its activity, which fosters the proliferation, migration, and invasion of colorectal cancer cells [95]. Wan et al. found that global Kcr levels are reduced in liver, stomach, and kidney carcinomas but increased in thyroid, oesophageal, pancreatic and lung carcinomas. In particular, increased Kcr levels hamper hepatoma cell motility and proliferation [96]. The highly upregulated proteins with Kcr marks in small cell lung cancer are crucial regulators of tumour metastasis or the tumour microenvironment or are involved in wound healing, functions that are all hallmarks of cancer [97,98,99]. Additionally, since defective double-strand break (DSB) repair leads to genomic instabilities that may augment tumorigenesis [100], Kcr-mediated DNA damage repair [32] and accurate spindle positioning [33] are conducive to maintaining genomic integrity and blocking tumour development.
Lactylation
Characteristics and functions of Kla
Lactate-derived Kla is a newly identified PTM that closely couples metabolism, especially the ‘Warburg effect’, with epigenetic modification. Under lactate increases induced by hypoxia and bacterial challenges, histone Kla is stimulated and directly promotes gene transcription [21]. A recent study showed that lactyl-CoA, not lactate or (R)-S-lactoylglutathione, demonstrates significant and specific binding to P300 to mediate histone Kla [101]. In addition, protein Kla is realized in a nonenzymatically modulated manner and has been comprehensively described. In protein Kla, the acyl group is derived from the secondary glycolytic intermediate lactoylglutathione (LGSH), and elevated LGSH indicates an increase in Kla-modified glycolytic enzymes and significantly represses glycolytic output. Therefore, the role of Kla in glycolysis can be interpreted as a feedback mechanism in which hyperglycaemic conditions promote LGSH generation and Kla, thus suppressing glycolytic flux and cell growth [102].
The levels of acetyl-CoA and lactate are increased by Gli-like transcription factor 1 (Glis1), which directly binds to and opens chromatin at glycolytic genes while closing chromatin at somatic genes. Therefore, H3K27ac and H3K18la on the promoters of pluripotency genes have been shown to activate their expression and induce pluripotency acquisition, forming an epigenome–metabolome–epigenome signalling cascade for cell reprogramming [39]. Because elevated histone Kla induced by lactate production is located on the promoters of profibrotic genes in alveolar macrophages, lactate along with histone Kla may be an unconventional and putative target to treat lung fibrosis [40]. In addition, in the late phase of M1 (proinflammatory) macrophage polarization, increased histone Kla induces homeostatic gene expression and facilitates the acquisition of the M2 (anti-inflammatory)-like phenotype, which may inhibit macrophage activation, dampen the inflammatory response and prevent further tissue damage [21]. This transition also occurs in colitis dictated by a Toll-like receptors (TLRs) signalling adapter via histone Kla [41].
Role of Kla in cancer
More recently, pioneering work has provided the first compelling insight into the role of histone Kla in ocular melanoma. H3K18la promotes the expression of YTHDF2, which recognizes N6-methyladenosine (m6A)-modified PER1 and TP53 mRNAs and enhances their degradation, thus driving oncogenesis [103]. Considering the finding that increased histone Kla in the late phase of M1 macrophage polarization drives an M2-like phenotype, which is characteristic of tumour-associated macrophages (TAMs) [21, 101], an indirect role for Kla in cancer growth and proliferation has been proposed, and histone Kla may become a potential tumour therapy target. Furthermore, because lactate is an oncometabolite produced by the ‘Warburg effect’ and a global epigenetic regulator, lactate-induced Kla may be involved in tumorigenesis [21]. For example, a study established that lactate promotes cell proliferation and modulates cellular metabolism at least in part through histone lactylation-mediated gene expression in non-small cell lung cancer cells [104]. Additionally, the increase in lactate in breast cancer significantly boosts the transcription of tumour-related genes such as key oncogenes and cell cycle genes, which may be partially ascribed to histone Kla on relevant gene promoters. However, this scenario warrants further investigation [105].
Succinylation
Characteristics and functions of Ksucc
Ksucc, the covalent deposition of a succinyl group on the ε-amino group of lysine, was first identified and verified in Escherichia coli and can induce significant chemical changes in proteins to promote important cellular functions [22]. Histone Ksucc marks are distributed widely in HeLa cells, mouse embryonic fibroblasts (MEFs), Drosophila S2 cells, and Saccharomyces cerevisiae cells, showing that histone Ksucc is highly conserved among species. H2AK95, H2BK116, H2AK95 and H2BK120 are unique Ksucc sites that have rarely been reported for other PTMs, most of which occur in the globular domain (GD) and the C-terminus of histones, where they are more accessible for participation in chemical reactions [106]. Nonenzymatic Ksucc is mediated by succinyl-CoA, which serves as the entry point of odd-numbered fatty acids and branched-chain amino acids into the tricarboxylic acid (TCA) cycle [107, 108], while enzymatic Ksucc can also be mediated by succinyltransferases, such as the α-ketoglutarate dehydrogenase complex (KGDHC) [109], GCN5 [110,111,112], histone acetyltransferase 1 (HAT1) [113] and carnitine palmitoyltransferase 1A (CPT1A) [114,115,116], and desuccinylases, such as SIRT5 [117, 118] and SIRT7 [119]. The GAS41 YEATS domain is the ‘reader’ of the Ksucc mark on H3K122 [120].
Hypersuccinylation in chromatin is caused by succinyl-CoA accumulation and results in enhanced transcriptional responses. In other words, histone Ksucc is an inextricable link between the TCA cycle status and epigenetic transcriptional control [42]. Truly, histone Ksucc can impinge upon nucleosome dynamics and foster DNA unwrapping from the histone surface, thus allowing proteins, such as transcription factors, to easily access buried regions of nucleosomal DNA [43]. In view of the prominent role of succinyl-CoA in the TCA cycle, relevant Ksucc-modified proteins play roles in numerous metabolic pathways, including the oxoacid metabolism, purine/pyrimidine metabolism, glycolysis/gluconeogenesis, and pyruvate metabolism pathways, in many species [108, 118, 121,122,123,124,125]. Additionally, Ksucc of core mitochondrial enzymes undermines their stability and activity, thus impairing mitochondrial respiration, mitophagy and metabolic flexibility [44]. In congenital metabolic disorders with succinyl-CoA ligase (SCL) deficiency [126], global protein hypersuccinylation caused by succinyl-CoA accumulation may partially contribute to hereditary mitochondrial encephalomyopathy [127]. Ksucc is also involved in solubilizing amyloid plaques and tangles and deterring the aggregation of neurofilaments [45,46,47,48], consistent with its notable decline in the Alzheimer’s disease (AD) brain [128,129,130,131]. In cardiovascular diseases, hypersuccinylation of ECHA [i.e., caused by SIRT5 knockout (KO)] impairs its activity and ATP production to cause hypertrophic cardiomyopathy and increased mortality under chronic pressure overload [49, 50]. Moreover, augmented Ksucc of succinate dehydrogenase (SDH) boosts its activity and exacerbates ischaemia-reperfusion injury in SIRT5-KO mouse hearts [51,52,53]. Additionally, Ksucc plays an active role in drug resistance [121], fungal pathogenicity [132], hypothyroxinemia [133], colitis [134] and other pathological activities.
Role of Ksucc in cancer
A large number of investigations have revealed the role of Ksucc as a double-edged sword in tumorigenesis. Researchers have found that Ksucc exerts a positive effect on tumorigenesis through histone and nonhistone modifications. In human pancreatic ductal adenocarcinoma (PDAC), GCN5 catalyses the H3K79succ modification on the promoter region of YWHAZ to promote its expression, which reinforces the proliferation, migration and invasion of PDAC cells [111]. This GCN5-mediated H3K79succ modification is also involved in liver cancer [112] and glioblastoma [110] formation. Furthermore, Ksucc profoundly modifies nonhistone proteins and fosters tumorigenesis. In gastric cancer (GC), Ksucc of S100A10 and lactate dehydrogenase A (LDHA) is significantly increased in GC with inhibited degradation, enhancing tumour invasion and migration [115, 116], while desuccinylation of 2-oxoglutarate dehydrogenase (OGDH) by SIRT5 dampens its activity and subsequently suppresses tumorigenesis in GC [135]. K569succ of caldesmon (CALD1), which is closely related to tumour metastasis, is profoundly decreased in GC and may be a viable biomarker for GC [136]. In addition, K311succ of GLS in PDAC [137], K118succ of LDHA in prostate cancer [138], K123succ of Cu/Zn superoxide dismutase (SOD1) in lung cancer [139], K433succ of pyruvate kinase M2 (PKM2) in colon cancer [140], Ksucc of H3K122 and phosphoglyceromutase 1 (PGAM1) catalysed by HAT1 [113] and hypersuccinylation induced by R-2-hydroxyglutarate [141] all have a far-reaching and positive impact on tumorigenesis.
However, emerging evidence also suggests that SIRT5-mediated desuccinylation could potentiate tumorigenesis by inactivating succinate dehydrogenase complex subunit A (SDHA) in renal cell carcinoma [142], stabilizing GLS in breast cancer [13, 143], activating citrate synthase (CS) and serine hydroxymethyltransferase (HMT2) in colon cancer [144, 145] and inhibiting PKM2 in lung cancer [146]. These studies collectively suggest an avenue for targeting SIRT5 in a tumour therapeutic strategy executed via pharmacological inhibition [147]. In addition, hyposuccinylation induced by reduced succinyl-CoA levels has been demonstrated in oesophageal squamous cell cancer (ESCC) and may alter ESCC metabolism and promote cell migration [148]. Similarly, in renal cell carcinoma, Ksucc has also been found to be connected with tumour energy metabolism [149]. Moreover, in colon cancer cells treated with dichloroacetate (DCA), a glycolysis inhibitor, elevated proteins labelled with Ksucc may have important functions in turning the ‘Warburg effect’ [150, 151] into normal oxidative phosphorylation to mediate the antitumour effect of DCA [152].
Collectively, these studies show that the function of Ksucc in tumour development is context-dependent and that the regulatory pathway that is predominant may be critical in determining whether Ksucc functions as a tumour suppressor or tumour promoter.
Propionylation and butyrylation
Characteristics and functions of Kpr and Kbu
Kpr and Kbu have been concurrently identified on histones in mammalian cells, as well as in yeast core histones, indicating the evolutionary conservation of these modifications in eukaryotic cells [23, 153, 154]. Propionyl-CoA, obtained through the catabolism of odd-numbered fatty acids and branched-chain amino acids, is the putative substrate for Kpr, whereas butyryl-CoA derived from even-chain fatty acids provides a butyryl group for Kbu [23, 155]. P300, CREB-binding protein (CBP) and HBO1 can catalyse Kpr and Kbu as ‘writers’ [23, 73, 156]. Kpr can also be mediated by the GNAT superfamily (GCN5 and PCAF) and MYST family (MOF, MOZ and HB01) [157, 158]. SIRT1–3, histone deacetylases, can remove the propionyl and butyryl groups from lysine residues in the presence of NAD+ [153, 156, 159]. As a ‘reader’ of Kpr and Kbu, the bromodomain of BRD4 can bind these acyl groups with binding affinities that are significantly lower than its binding affinity for Kac [160], while the YEATS domain has enhanced affinities for Kpr and Kbu over Kac [80]. The bromodomains of BRD9 and CECR2 and the second bromodomain of TAF1 also recognize the longer butyryl mark and thus promote protein modification [84].
Histone Kpr and Kbu can be reversibly modulated upon metabolic perturbation, being inhibited upon glucose deprivation and rescued by glucose replenishment [54], providing a novel epigenetic regulatory mark of cell metabolism. Moreover, histone Kpr and Kbu stimulate transcription to an extent similar to that of histone Kac [54, 55, 57]. For instance, in spermatogenic cells, the oscillation of histone Kac and Kbu at gene TSSs may be a determinant in maintaining high transcriptional activities during spermatogenesis [58]. Researchers have identified that abnormal accumulation of these modifications at the total protein level caused by defective acyl-CoA metabolism may greatly impact protein functions and shed light on the pathophysiology of congenital metabolic disorders [60]. In addition, Kpr of bovine carbonic anhydrase II (BCA II) has been implicated in promoting amyloid fibrillation and protein aggregation, partially contributing to the formation of systemic amyloidosis and neurodegenerative diseases [56].
Role of Kpr and Kbu in cancer
Extensive studies provide a glimpse into the potential roles of Kpr in cancer diagnosis and therapy. The sharp reduction in Kpr of H3K23 in the U937 leukaemia cell line during monocytic differentiation implies that the initial hyperpropionylation in U937 cells might be a stage-specific marker during haematopoiesis and leukaemogenesis and may serve as a diagnostic indicator for leukaemia therapy [153]. It has also been reported that human bromodomain- and PHD finger-containing protein 1 (BRPF1) variants that are typically expressed in multiple cancers [161, 162] impair BRPF1-dependent activation of KAT6A for propionylation of H3K23, indicating the previously unrevealed therapeutic strategy of targeting the BRPF1 mutation and Kpr in cancers [74]. Histone deacetylase inhibitors (HDACis), as antitumour drugs, can induce not only histone Kac but also Kpr in glioma cells, suggesting that Kpr may be measured to monitor HDACi pharmacological actions and their influence on interactions in malignant cells [163]. Additionally, propionate, a short-chain fatty acid (SCFA) that originates from bacterial fermentation of dietary fibres in the colon [164], has been shown to induce overall Kpr, which upregulates MICA/B surface expression and suppresses the development of colon cancer [165].
Analogously, histone Kbu can be upregulated in tumour cells by HDACis, such as suberoylanilide hydroxamic acid (SAHA) in neuroblastoma [166], largazole-7 in colorectal cancer [167], and sodium butyrate (NaB) in Ewing sarcoma [168], indicating its unique influences on the antitumour activities of HDACis. Lysine butyrylated sites on core histones have been identified in ESCC cell lines, including H3K18, H3K23, H3K79 and H4K77 [169]. In addition, one of the potent delivery ligands for tumour therapy is GnRH-III[4Lys(Bu), 8Lys (Dau = Aoa)] with Kbu at position 4, which has a higher binding affinity, higher enzymatic stability, higher cellular uptake and higher tumour growth inhibitory activity than GnRH-III bioconjugates with Kac [170, 171].
Malonylation
Characteristics and functions of Kma
Kma was first identified as an evolutionarily conserved PTM in prokaryotes and eukaryotes in 2011 [24]. In 2012, scientists found malonyllysine sites in histones of HeLa and S. cerevisiae cells [106]. Several histone Kma marks have been found in C-terminal GDs, signifying the distinction between the roles of Kma and Kac in cellular regulation [61]. Considering that the citrate-derived metabolite malonyl-CoA is the precursor of de novo fatty acid synthesis and a critical inhibitor of fatty acid oxidation [172] and because it supplies substrates for Kma [24], the elevation of Kma upon inhibited fatty acid synthase activity may be ascribed to the accumulation of malonyl-CoA [173]. SIRT5 catalyses lysine demalonylation reactions in human cells [24, 117], while SIRT2, as a yeast sirtuin homologue, might remove this modification in budding yeast or fission yeast [174].
In diabetes-induced neural tube defects (NTDs), histone Kma is elevated both in vitro and in vivo, offering new insights into the pathological effect of histone Kma in human NTDs [62]. In addition, global profiling of proteins modified by Kma in E. coli demonstrated that this modification was intimately associated with energy metabolism, especially with fatty acid synthase and the TCA cycle [173]. Moreover, hypermalonylation in SIRT5-KO mice modified and suppressed glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to redirect glucose away from oxidation towards glycogen synthesis or the pentose phosphate pathway (PPP) in primary hepatocytes [175]. In lipopolysaccharide (LPS)-stimulated macrophages, increasing malonyl-CoA induced increased Kma levels at the 213 site of GAPDH, which spurred proinflammatory cytokine production by modulating GAPDH activity and mRNA-binding capacity, which are inflammatory signals in macrophages [59]. In malonyl-CoA decarboxylase (MCD)-deficient disease, increased Kma due to malonyl-CoA accumulation impairs mitochondrial function and fatty acid oxidation, suggesting that Kma plays a role in the pathophysiology of malonic aciduria [60, 61]. In addition, the Sirt5-Kma pathway may contribute to altered chondrocyte metabolism during osteoarthritis development since SIRT5-deficient chondrocytes exhibit increased Kma levels and decreased glycolysis and mitochondrial respiration rates [63].
Role of Kma in cancer
SIRT5 can not only demalonylate and inactivate SDHA to cause multidrug resistance in wild-type Kras colorectal carcinomas (CRCs) [176] but also mediate TPI demalonylation and impair its formation and activity, causing recurrence of mutant Kras CRC [177]. These findings may lead to new possibilities for achieving long-term CRC remission through suppression of SIRT5-mediated demalonylation in combination with chemotherapy. Moreover, by elevating malonyl-CoA levels, fatty acid synthase inhibition has been implicated in the promotion of mTOR lysine malonylation, which impairs angiogenesis, underscoring the importance of mTOR Kma in tumorigenesis since it may be an effective weapon against pathological neovascularization [178].
Glutarylation
Characteristics and functions of Kglu
Pioneering work led to the first report of Kglu in 2014. Kglu modifies metabolic enzymes and mitochondrial proteins [25]. Kglu can be nonenzymatically driven with glutaryl-CoA, which is an important intermediate in lysine and tryptophan metabolism [25]. The removal of a glutaryl group is known to be primarily performed by SIRT5 and SIRT7 in an NAD+-dependent manner, while P300 and GCN5 are known as ‘writers’ of Kglu [25, 64].
Of particular interest, the core histone H2B has been revealed to have three Kglu sites (H2BK5, H2BK116, and H2BK120), which may be critical for chromatin-mediated processes, such as the regulation of gene expression [25]. Indeed, H4K91glu can destabilize nucleosome structure by interrupting histone-histone interactions to create a more relaxed environment for DNA accessibility and gene transcription [64]. A feedback loop in which glutaryl-CoA is produced within the lysine/tryptophan oxidation pathway impedes GCDH function via glutaryl modification and downregulates lysine catabolism [14]. Mutations in the GCDH gene cause the neurometabolic disorder glutaric aciduria type 1 (GA1), featuring protein hyperglutarylation, which impairs enzymatic activity and protein interactions to disrupt mitochondrial heterogeneity [65]. Protein Kglu has also been revealed to maintain sperm motility and may be involved in the aetiology of asthenozoospermia [66].
Role of Kglu in cancer
SIRT5 overexpression is significantly correlated with poor prognosis in CRC, and scientists have found that glutamate dehydrogenase 1 (GLUD1) can be deglutarylated and activated via SIRT5 to promote cellular glutaminolysis, thus enhancing colorectal carcinogenesis. This finding suggests that SIRT5 and Kglu are promising targets for the selective killing of cancer cells [179]. However, another study revealed that SIRT5 protects cells from oxidative damage via isocitrate dehydrogenase 2 (IDH2) desuccinylation and glucose-6-phosphate dehydrogenase (G6PD) deglutarylation, and SIRT5-KO upregulates cellular susceptibility to oxidative stress [180], which is vital to tumorigenesis [181, 182]. The abovementioned evidence shows the complexity of SIRT5 and Kglu regulation of tumour progression.
2-Hydroxyisobutyrylation
Characteristics and functions of Khib
Khib has been found to be widely distributed on both histone and nonhistone proteins in HeLa cells, MEFs, Drosophila S2 cells, yeast (S. cerevisiae) cells and developing rice seeds [26, 183], verifying its evolutionarily conserved and dynamic role among many species. Among 63 human and mouse histone Khib sites, nearly one-half are neither Kac sites nor Kcr sites [26], uncovering the discriminative effects of Khib on cellular modulation. Khib is likely derived from 2-hydroxyisobutyrate donors, one of which may be 2-hydroxyisobutyryl-CoA (Hib-CoA). The Khib pathway reprogrammes epigenetic networks in response to dynamic changes in the cellular metabolite Hib-CoA [26]. The acetyltransferase Esa1p in budding yeast and its homologue Tip60 in humans as well as P300 can add a 2-hydroxyisobutyryl group to substrate proteins [184, 185], whereas demodification can be achieved by HDAC1–3 [26], Rpd3p (class I HDAC), Hos3p (class II HDAC) [186] and CobB [187].
H4K8hib is enriched at the TSSs of genes and is associated with active gene transcription in meiotic and postmeiotic cells [26]. H4K8hib is a dynamic modification that orchestrates glucose level changes within cells and establishes a link between histone modification and carbon metabolism [186].
Role of Khib in cancer
HDAC3-mediated de-2-hydroxyisobutyrylation of H4K8 on the covalently closed circular DNA (cccDNA) minichromosome of hepatitis B virus (HBV) can be enhanced by interferon-α to inhibit HBV transcription and replication in hepatoma cells [188], which may hinder liver cancer formation. Quantitative proteome studies have identified core histones modified by Khib, Kbu, Kpr, and Ksucc in HSP90 inhibitor-treated bladder cancer cells, and the results indicated an affinity between epigenetic modifications and HSP90 inhibitor-mediated antitumour effects [189]. Since oncomutations of linker histones (LHs; H1/H5) occur mainly near PTM sites and because the most common site in the GD of histone H1.2 is modified by Khib, these mutations are thought to potentially block the primary PTM sites and interfere with their reading, writing and/or erasing processes, thus regulating gene expression in cancer cells [190]. Moreover, the reduction in the K281hib level on ENO1 induced by aspirin leads to inhibited ENO1 activity and is critical for attenuating glycolysis and the proliferation of hepatoma cells [191]. Protein expression within the actin cytoskeleton regulatory pathway and Khib modification levels are significantly altered in oral squamous cell carcinoma, which may indicate their importance for tumour progression [192]. Additionally, Khib-modified proteins are abundant in carbohydrate metabolism pathways, especially metabolic pathways in cancer [193].
β-Hydroxybutyrylation
Characteristics and functions of Kbhb
Kbhb, a widespread histone mark in human and mouse cells, can be dramatically induced under prolonged fasting conditions and diabetic ketoacidosis [27] because an elevated ketone body, β-hydroxybutyrate (BHB), is a substrate in the generation of β-hydroxybutyryl-CoA that can induce P300-mediated histone Kbhb [194, 195]. In contrast to the unspecialized Zn-dependent HDAC1–3 removal of the β-hydroxybutyryl group [196], human SIRT3 displays class-selective histone de-β-hydroxybutyrylase activities favouring H3K4, H3K9, H3K18, H3K23, H3K27, and H4K16, but not H4K5, H4K8, or H4K12, suggesting a potential regulatory mechanism involving hierarchical gene repression under metabolic alterations [197]. However, treatment with NaBut, 4-PBA or SAHA, all of which are classical HDACis, can promote histone Kbhb to an even greater extent than BHB [194].
Kbhb is abundant in the GD of the LHs H1.4 and H1.5, influencing the interaction of LHs with both DNA and the nucleosome, similar to Khib [190]. Because Kbhb, especially H3K9bhb, elevates and upregulates gene expression in starvation-responsive metabolic pathways in the mouse liver during prolonged fasting, a feedback mechanism is obviously formed; thus, metabolite production altered by cellular energy conditions can influence histone Kbhb and relevant gene expression to maintain homeostasis [27]. In addition, ketogenesis proceeds through an unusual metabolic pathway that links the epigenetic modification required for memory development of CD8+ T memory cells by prompting BHB production and deposition of H3K9bhb marks on relevant genes [67]. In obese diabetic mice treated with dapagliflozin, a selective competitive inhibitor of sodium/glucose cotransporter 2 (SGLT2), elevated β-hydroxybutyric acid levels mediated Kbhb of H3K9 to promote the expression of the adiponectin gene in adipocytes, partially accounting for the molecular mechanisms by which SGLT2 inhibitors protect against cardiovascular events in diabetic patients [198]. BHB-induced Kbhb of H3K9 can antagonize glomerulosclerosis induced by diabetes and alleviate depressive behaviours by modulating specific gene expression through promoters [68, 69].
Role of Kbhb in cancer
Metastasis-associated protein 2 (MTA2) was upregulated in hepatocellular carcinoma (HCC) cells and induced the accumulation of βHB, increased the level of H3K9bhb, and exerted a cascading effect on HCC cell stemness and progression [199]. P53 encoded by the Tp53 gene acts as an essential tumour suppressor and promotes cell growth arrest and apoptosis [200, 201]. A recent study revealed that p53 can be modified by Kbhb at K120, K319, and K370 and can be inactivated in BHB-treated tumour cells. Specifically, Kbhb of p53 results in reduced p53 acetylation and expression of the downstream genes p21 and PUMA, thereby facilitating cell growth. It can also be deduced that Kbhb of P53 partially explains the role of ketone bodies in tumour biology [202]. In addition, P53 can also be modified by Ksucc, Kpr, Kbu, and Kcr, indicating that these modifications may affect P53-mediated tumour inhibition [23, 156, 203, 204].
Conclusions and future perspectives
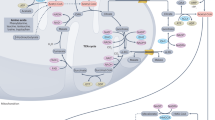
A growing body of breakthrough discoveries on novel acylation types is creating new opportunities for deeper investigations into the nonmetabolic roles of metabolites in tumorigenesis. Histone acylation inextricably links the epigenome with the metabolome via transcriptional modulation, and nonhistone acylation alters protein functions to transmit proteomic-metabolomic signals (Table 2). Accordingly, despite metabolites being downstream products of manifold biological activities, metabolites have profound impacts on upstream biosystems and serve as ‘drivers’ of diverse biological processes [1]. Because acylation can be influenced by the availability of acyl-CoA through both enzymatic and nonenzymatic mechanisms [205], a shift in cellular utilization of energy sources in tumours induces aberrant acylation [206], which has been acknowledged as a general mechanism of cancer cell regulation and is highly conserved and deleterious [207]. Specifically, altered acylation affects tumour metabolism to promote tumorigenesis by influencing gene expression and cell signalling in multiple tumour types (Fig. 2). The abovementioned increase in research on acylation types that we reviewed based on acylation category directly and indirectly paves the way for a greater understanding of acylation-driven feedback loops.

Regulatory roles of lysine acylation in tumorigenesis. Lysine acylation exerts profound effect on diverse tumour formation. In glioblastoma, H3K79succ promotes gene expression and tumour growth. In lung cancer, Ksucc of SOD1 impedes its anti-tumor effect while desuccinylation of PKM2 boost tumour growth. In breast cancer, hypersuccinylation causes GLS degradation and impedes glutamine consumption of tumour cells. In renal cell carcinoma, SDHA is desuccinylated and fosters tumour proliferation. In prostate cancer, Ksucc of LDHA increases its activity in promoting tumour metastasis and histone hypocrotonylation induced by BRD4 inhibitors hampers tumour proliferation and migration. In ocular melanoma, H3K18la drives oncogenesis. In ESCC, restoring Ksucc level restricts cell growth, migration and invasion. In GC, elevated Ksucc of S100A10 and LDHA enhances tumour invasion and migration whereas desuccinylation of OGDH suppresses tumorigenesis. In colorectal cancer, Ksucc of PKM2 and Kcr of ENO1 promotes cell survival and tumour development and desuccinylation of CS and SHMT2 accelerates colon cancer growth. Kpr induced by propionate suppresses the development of colon cancer. Demalonylation of SDHA and TPI prompts the recurrence of CRC and GLUD1 can be deglutaryled to promote colorectal carcinogenesis. In liver cancer, Ksucc of H3K122 and PGM1 are required for liver cancer growth. Repressed Khib of ENO1 causes proliferation defective of liver cancer cells and H3K9bhb participates in the promotion of HCC stemness and progression. Increasing Kcr level leads to undermined liver cancer cell migration and proliferation. In PDAC, Ksucc of H3K79, H3K122, PGM1 and GLS promotes cell proliferation, migration and invasion
The potential crosstalk among lysine acylations also draws our attention since their corresponding metabolic sources are intertwined with each other. For instance, ccrotonyl-CoA is obtained from both glutaryl-CoA and butyryl-CoA, and propionyl-CoA can also be transformed into succinyl-CoA (Fig. 1). Furthermore, because regulatory enzymes, including writers, erasers and readers of acylation, overlap, these modifications are likely to change concurrently in metabolic disturbances or any other pathological state and may function together to mediate cellular signalling. For example, SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defence [180]. It is also worth noting that acylations and other PTMs often modify the same protein such as P53, H3, LDHA and ENO1 (Fig. 2), in an agonistic or antagonistic manner. The β-hydroxybutyrylation of p53 results in reduced p53 acetylation, which affects the transcriptional activity of p53 on its target gene [202]. In addition, H2A-K119 malonylation inhibits H2A S121 phosphorylation, showing novel crosstalk between lysine malonylation and serine/threonine phosphorylation [174]. We assume that there must be more precise and specific interplay between the different acylations and other PTMs, and substantial effort will be required to map out and carefully dissect these interactions.
However, since acetylation occurs ubiquitously in diverse cellular biological processes, its important role in tumour regulation cannot be fully understood. Notably, many regulatory enzymes for the aforementioned novel acylation types were first found to catalyse acetylation or deacetylation. Therefore, the acetylation level can concomitantly change in conjunction with other acylation levels upon the modulation of enzymatic activity, which may confuse the outcomes. For example, Kac is significantly elevated in parallel to other acylation types and cannot be excluded as a cause for any of the antitumour effects of HDACis [163, 166,167,168]. In addition, it is essential to identify whether these novel types of acylation function synergistically and comprise a ‘nonacetyl’ group or behave independently in cellular regulation. Furthermore, because one specific acylation can modify different enzymes in various metabolic pathways or on different sites in the same protein, such as Ksucc of PKM2 [140, 146] and GLS [13, 137], the sophisticated modification paradigm may cause different, even completely opposite, results. Therefore, it is important to specify whether these acylation types form a complicated network that includes diverse proteins in a single disease and determine ways to prevent unwanted effects and strengthen desired influences in cancer therapy. In contrast to the abundant recognition of nonhistone acylation, discoveries of histone acylation remain scant. Further efforts to identify new histone sites with novel acylation types are urgently needed to expand the scope of knowledge on epigenetic alterations affecting tumour heterogeneity [208], since precise and effective personalized therapy using epidrugs has emerged [209]. After resolving these conundrums, lysine acylation will likely play a burgeoning role in tumour diagnosis, monitoring and/or treatment in the clinic.
Despite considerable efforts to understand the relevance of metabolite-derived PTMs in the cellular context, more progress is needed in the future to identify their tremendous impact, which will ultimately expand the roles of metabolites in tumorigenesis.
Availability of data and materials
Not applicable.
Abbreviations
- Ac-CoA:
-
Acetyl-CoA or acetyl coenzyme A
- AD:
-
Alzheimer’s disease
- α-KG:
-
α-ketoglutarate
- α-KGDH:
-
α-ketoglutarate dehydrogenase complex
- AKI:
-
Acute kidney injury
- BCA II:
-
Bovine carbonic anhydrase II
- BHB:
-
β-hydroxybutyrate
- BMDM:
-
Bone marrow-derived macrophage
- BRPF1:
-
Bromodomain- and PHD finger-containing protein 1
- BRD4:
-
Bromodomain-containing protein 4
- CALD1:
-
Caldesmon
- CBP:
-
CREB-binding protein
- cccDNA:
-
Covalently closed circular DNA
- CDYL:
-
Chromodomain Y-like protein
- CPT1A:
-
Carnitine palmitoyltransferase 1A
- CRC:
-
Colorectal carcinomas
- CS:
-
Citrate synthase
- DCA:
-
Dichloroacetate
- DPF:
-
Double PHD finger
- DPF2:
-
Also known as BAF45d
- DSB:
-
Double-strand break
- ENO1:
-
Enolase 1
- ESCC:
-
Oesophageal squamous cell cancer
- FASN:
-
Fatty acid synthase
- GA1:
-
Glutaric aciduria type 1
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GC:
-
Gastric cancer
- GCDH:
-
Glutaryl-CoA dehydrogenase
- GCN5:
-
General control of amino acid synthesis 5-like 2, also known as KAT2A
- GD:
-
Globular domain
- Glis1:
-
Gli-like transcription factor 1
- GLS:
-
Glutaminase
- GLUD1:
-
Glutamate dehydrogenase 1
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- HAT1:
-
Histone acetyltransferase 1
- HBO1:
-
Also known as KAT7 or MYST2
- HBV:
-
Hepatitis B virus
- HCC:
-
Hepatocellular carcinoma
- HDACs:
-
Histone deacetylases
- HDACis:
-
Histone deacetylase inhibitors
- HDR:
-
Homology directed repair
- 2-HG:
-
2-hydroxyglutarate
- Hib-CoA:
-
2-hydroxyisobutyryl-CoA
- H3K122:
-
Histone H3 lysine 122
- H3K18:
-
Histone H3 lysine 18
- H3K23:
-
Histone H3 lysine 23
- H3K27:
-
Histone H3 lysine 27
- H3K4:
-
Histone H3 lysine 4
- H3K79:
-
Histone H3 lysine 79
- H3K9:
-
Histone H3 lysine 9
- H4K8:
-
Histone H4 lysine 8
- H4K12:
-
Histone H4 lysine 12
- H4K77:
-
Histone H4 lysine 77
- IDH2:
-
Isocitrate dehydrogenase 2
- IFN-α:
-
Interferon-α
- K164:
-
Lysine residue 164
- Kac:
-
Lysine acetylation
- KAT6A:
-
Lysine acetyltransferase 6A
- KATs:
-
Lysine acetylatransferases
- Kbu:
-
Butyrylation
- Kcr:
-
Lysine crotonylation
- Kbhb:
-
Lysine β-hydroxybutyrylation
- KGDHC:
-
α-ketoglutarate dehydrogenase complex
- Kglu:
-
Lysine glutarylation
- Khib:
-
Lysine 2-hydroxyisobutyrylation
- Kla:
-
Lysine lactylation
- Kma:
-
Lysine malonylation
- KO:
-
Knockout
- Kpr:
-
Lysine propionylation
- Ksucc:
-
Lysine succinylation
- LDHA:
-
Lactate dehydrogenase A
- LGSH:
-
Lactoylglutathione
- LHs:
-
Linker histones
- LPS:
-
Lipopolysaccharide
- m6A:
-
N6-methyladenosine
- MCD:
-
Malonyl-CoA decarboxylase
- MEF:
-
Mouse embryonic fibroblasts
- MOF:
-
Also known as KAT8
- MOZ:
-
Also known as KAT6A
- MSCI:
-
Meiotic sex chromosome inactivation
- MTA2:
-
Metastasis-associated protein 2
- NaB:
-
Sodium butyrate
- NAD + :
-
Nicotinamide adenine dinucleotide
- NTD:
-
Neural tube defects
- OGDH:
-
2-oxoglutarate dehydrogenase
- PCAF:
-
p300/CBP associated factor
- PDAC:
-
pancreatic ductal adenocarcinoma
- PGAM1:
-
Phosphoglyceromutase 1
- PKM2:
-
Pyruvate kinase isozyme M2
- PPP:
-
Pentose phosphate pathway
- PTMs:
-
Post-translational modifications
- ROS:
-
Reactive oxygen species
- SAHA:
-
Suberoylanilide hydroxamic acid
- SCFA:
-
Short-chain fatty acid
- SCL:
-
Succinyl-CoA ligase
- SDH:
-
Succinate dehydrogenase
- SDHA:
-
Succinate dehydrogenase complex, subunit A
- SGLT2:
-
Sodium/glucose cotransporter 2
- SHMT2:
-
Serine hydroxymethyltransferases
- SIRT:
-
Sirtuin
- SOD1:
-
Cu/Zn superoxide dismutase
- SUCNR1:
-
Succinate receptor 1
- TAMs:
-
Tumor-associated macrophages
- TCA:
-
Tricarboxylic acid
- TIP60:
-
Tat-interacting protein, also known as histone acetyltransferase KAT5
- TLR:
-
Toll-Like receptors
- TSSs:
-
Transcriptional start sites
References
Rinschen MM, Ivanisevic J, Giera M, Siuzdak G. Identification of bioactive metabolites using activity metabolomics. Nat Rev Mol Cell Biol. 2019;20(6):353–67.
Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368:6487.
Li J, Eu JQ, Kong LR, Wang L, Lim YC, Goh BC, et al. Targeting metabolism in Cancer cells and the tumour microenvironment for Cancer therapy. Molecules. 2020;25(20):4831.
Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20(1):28.
Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123(9):3652–8.
Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, et al. Cancer-derived succinate promotes macrophage polarization and Cancer metastasis via succinate receptor. Mol Cell. 2020;77(2):213–27 e5.
Zhao T, Mu X, You Q. Succinate: an initiator in tumorigenesis and progression. Oncotarget. 2017;8(32):53819–28.
Matlac DM, Hadrava Vanova K, Bechmann N, Richter S, Folberth J, Ghayee HK, et al. Succinate mediates tumorigenic effects via succinate receptor 1: potential for new targeted treatment strategies in succinate dehydrogenase deficient Paragangliomas. Front Endocrinol (Lausanne). 2021;12:589451.
Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21(3):255–61.
Deribe YL, Pawson T, Dikic I. Post-translational modifications in signal integration. Nat Struct Mol Biol. 2010;17(6):666–72.
Seo J, Lee KJ. Post-translational modifications and their biological functions: proteomic analysis and systematic approaches. J Biochem Mol Biol. 2004;37(1):35–44.
Karve TM, Cheema AK. Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J Amino Acids. 2011;2011:207691.
Greene KS, Lukey MJ, Wang X, Blank B, Druso JE, Miao-chong JL, et al. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc Natl Acad Sci. 2019;116(52):26625–32.
Bhatt DP, Mills CA, Anderson KA, Henriques BJ, Lucas TG, Francisco S, et al. Deglutarylation of GCDH by SIRT5 controls lysine metabolism in mice. bioRxiv. 2020:2020.06.28.176677. https://doi.org/10.1101/2020.06.28.176677.
Allfrey VG, Faulkner R, Mirsky A. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. P Natl Acad Sci USA. 1964;51(5):786.
Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15(8):536–50.
Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, et al. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics. 2009;8(2):215–25.
Shvedunova M, Akhtar A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol. 2022. https://doi.org/10.1038/s41580-021-00441-y.
Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein post-translational modifications with mass spectrometry. Nat Methods. 2007;4(10):798–806.
Tan M, Luo H, Lee S, Jin F, Yang Jeong S, Montellier E, et al. Identification of 67 histone Marks and histone lysine Crotonylation as a new type of histone modification. Cell. 2011;146(6):1016–28.
Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–80.
Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol. 2010;7(1):58–63.
Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, et al. Lysine Propionylation and Butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6(5):812–9.
Peng C, Lu ZK, Xie ZY, Cheng ZY, Chen Y, Tan MJ, et al. The first identification of lysine Malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 2011;10(12):M111 012658.
Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19(4):605–17.
Dai L, Peng C, Montellier E, Lu Z, Chen Y, Ishii H, et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat Chem Biol. 2014;10(5):365–70.
Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, et al. Metabolic regulation of gene expression by histone lysine beta-Hydroxybutyrylation. Mol Cell. 2016;62(2):194–206.
Sabari Benjamin R, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong Ha E, et al. Intracellular Crotonyl-CoA stimulates transcription through p300-catalyzed histone Crotonylation. Mol Cell. 2015;58(2):203–15.
Montellier E, Rousseaux S, Zhao Y, Khochbin S. Histone crotonylation specifically marks the haploid male germ cell gene expression program. BioEssays. 2012;34(3):187–93.
Liu S, Yu H, Liu Y, Liu X, Zhang Y, Bu C, et al. Chromodomain protein CDYL acts as a Crotonyl-CoA Hydratase to regulate histone Crotonylation and spermatogenesis. Mol Cell. 2017;67(5):853–66 e5.
Abu-Zhayia ER, Machour FE, Ayoub N. HDAC-dependent decrease in histone crotonylation during DNA damage. J Mol Cell Biol. 2019;11(9):804–6.
Yu H, Bu C, Liu Y, Gong T, Liu X, Liu S, et al. Global crotonylome reveals CDYL-regulated RPA1 crotonylation in homologous recombination-mediated DNA repair. Sci Adv. 2020;6(11):eaay4697.
Song X, Yang F, Liu X, Xia P, Yin W, Wang Z, et al. Dynamic crotonylation of EB1 by TIP60 ensures accurate spindle positioning in mitosis. Nat Chem Biol. 2021;17(12):1314–23.
Ruiz-Andres O, Sanchez-Nino MD, Cannata-Ortiz P, Ruiz-Ortega M, Egido J, Ortiz A, et al. Histone lysine crotonylation during acute kidney injury in mice. Dis Model Mech. 2016;9(6):633–45.
Liu Y, Li M, Fan M, Song Y, Yu H, Zhi X, et al. Chromodomain Y-like protein–mediated histone crotonylation regulates stress-induced depressive behaviors. Biol Psychiatry. 2019;85(8):635–49.
Jiang G, Nguyen D, Archin NM, Yukl SA, Mendez-Lagares G, Tang Y, et al. HIV latency is reversed by ACSS2-driven histone crotonylation. J Clin Invest. 2018;128(3):1190–8.
Fu H, Tian CL, Ye X, Sheng X, Wang H, Liu Y, et al. Dynamics of telomere rejuvenation during chemical induction to pluripotent stem cells. Stem Cell Reports. 2018;11(1):70–87.
Fang Y, Xu X, Ding J, Yang L, Doan MT, Karmaus PWF, et al. Histone crotonylation promotes mesoendodermal commitment of human embryonic stem cells. Cell Stem Cell. 2021;28(4):748–63 e7.
Li L, Chen K, Wang T, Wu Y, Xing G, Chen M, et al. Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade. Nat Metab. 2020;2(9):882–92.
Cui H, Xie N, Banerjee S, Ge J, Jiang D, Dey T, et al. Lung myofibroblasts promote macrophage profibrotic activity through lactate-induced histone lactylation. Am J Respir Cell Mol Biol. 2021;64(1):115–25.
Irizarry-Caro RA, McDaniel MM, Overcast GR, Jain VG, Troutman TD, Pasare C. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc Natl Acad Sci U S A. 2020;117(48):30628–38.
Smestad J, Erber L, Chen Y, Maher LJ III. Chromatin succinylation correlates with active gene expression and is perturbed by defective TCA cycle metabolism. Iscience. 2018;2:63–75.
Jing Y, Ding D, Tian G, Kwan KCJ, Liu Z, Ishibashi T, et al. Semisynthesis of site-specifically succinylated histone reveals that succinylation regulates nucleosome unwrapping rate and DNA accessibility. Nucleic Acids Res. 2020;48(17):9538–49.
Wang G, Meyer JG, Cai W, Softic S, Li ME, Verdin E, et al. Regulation of UCP1 and mitochondrial metabolism in Brown adipose tissue by reversible Succinylation. Mol Cell. 2019;74(4):844–57 e7.
Subasinghe S, Unabia S, Barrow CJ, Mok SS, Aguilar MI, Small DH. Cholesterol is necessary both for the toxic effect of Aβ peptides on vascular smooth muscle cells and for Aβ binding to vascular smooth muscle cell membranes. J Neurochem. 2003;84(3):471–9.
Vetri V, Librizzi F, Militello V, Leone M. Effects of succinylation on thermal induced amyloid formation in Concanavalin A. Eur Biophys J. 2007;36(7):733–41.
Iqbal K, Wisniewski HM, Grundke-Iqbal I, Korthals JK, Terry RD. Chemical pathology of neurofibrils. Neurofibrillary tangles of Alzheimer's presenile-senile dementia. J Histochem Cytochem. 1975;23(7):563–9.
Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85(12):4506–10.
Hershberger KA, Abraham DM, Liu J, Locasale JW, Grimsrud PA, Hirschey MD. Ablation of Sirtuin5 in the postnatal mouse heart results in protein succinylation and normal survival in response to chronic pressure overload. J Biol Chem. 2018;293(27):10630–45.
Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z, et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci U S A. 2016;113(16):4320–5.
Zhou B, Du Y, Xue Y, Miao G, Wei T, Zhang P. Identification of Malonylation, Succinylation, and Glutarylation in serum proteins of acute myocardial infarction patients. Proteomics Clin Appl. 2020;14(1):e1900103.
Ali HR, Michel CR, Lin YH, McKinsey TA, Jeong MY, Ambardekar AV, et al. Defining decreased protein succinylation of failing human cardiac myofibrils in ischemic cardiomyopathy. J Mol Cell Cardiol. 2020;138:304–17.
Boylston JA, Sun J, Chen Y, Gucek M, Sack MN, Murphy E. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J Mol Cell Cardiol. 2015;88:73–81.
Jo C, Park S, Oh S, Choi J, Kim E-K, Youn H-D, et al. Histone acylation marks respond to metabolic perturbations and enable cellular adaptation. Exp Mol Med. 2020;52(12):2005–19.
Kebede AF, Nieborak A, Shahidian LZ, Le Gras S, Richter F, Gomez DA, et al. Histone propionylation is a mark of active chromatin. Nat Struct Mol Biol. 2017;24(12):1048–56.
Es-haghi A, Shariatizi S, Ebrahim-Habibi A, Nemat-Gorgani M. Amyloid fibrillation in native and chemically-modified forms of carbonic anhydrase II: role of surface hydrophobicity. Biochim Biophys Acta. 2012;1824(3):468–77.
Liu S, Liu G, Cheng P, Xue C, Zhou Y, Chen X, et al. Genome-wide profiling of histone lysine Butyrylation reveals its role in the positive regulation of gene transcription in Rice. Rice (N Y). 2019;12(1):86.
Goudarzi A, Zhang D, Huang H, Barral S, Kwon OK, Qi S, et al. Dynamic competing histone H4 K5K8 acetylation and Butyrylation are hallmarks of highly active gene promoters. Mol Cell. 2016;62(2):169–80.
Galvan-Pena S, Carroll RG, Newman C, Hinchy EC, Palsson-McDermott E, Robinson EK, et al. Malonylation of GAPDH is an inflammatory signal in macrophages. Nat Commun. 2019;10(1):338.
Pougovkina O, Te Brinke H, Wanders RJ, Houten SM, de Boer VC. Aberrant protein acylation is a common observation in inborn errors of acyl-CoA metabolism. J Inherit Metab Dis. 2014;37(5):709–14.
Colak G, Pougovkina O, Dai L, Tan M, Te Brinke H, Huang H, et al. Proteomic and biochemical studies of lysine Malonylation suggest its Malonic Aciduria-associated regulatory role in mitochondrial function and fatty acid oxidation. Mol Cell Proteomics. 2015;14(11):3056–71.
Zhang Q, Cai T, Xiao Z, Li D, Wan C, Cui X, et al. Identification of histone malonylation in the human fetal brain and implications for diabetes-induced neural tube defects. Mol Genet Genomic Med. 2020;8(9):e1403.
Zhu S, Batushansky A, Jopkiewicz A, Makosa D, Humphries KM, Van Remmen H, et al. Sirt5 Deficiency Causes Posttranslational Protein Malonylation and Dysregulated Cellular Metabolism in Chondrocytes Under Obesity Conditions. CARTILAGE. 2021;13(2_suppl):1185S–99S.
Bao X, Liu Z, Zhang W, Gladysz K, Fung YME, Tian G, et al. Glutarylation of histone H4 lysine 91 regulates chromatin dynamics. Mol Cell. 2019;76(4):660–75 e9.
Schmiesing J, Storch S, Dorfler AC, Schweizer M, Makrypidi-Fraune G, Thelen M, et al. Disease-linked Glutarylation impairs function and interactions of mitochondrial proteins and contributes to mitochondrial heterogeneity. Cell Rep. 2018;24(11):2946–56.
Cheng YM, Hu XN, Peng Z, Pan TT, Wang F, Chen HY, et al. Lysine glutarylation in human sperm is associated with progressive motility. Hum Reprod. 2019;34(7):1186–94.
Zhang H, Tang K, Ma J, Zhou L, Liu J, Zeng L, et al. Ketogenesis-generated β-hydroxybutyrate is an epigenetic regulator of CD8+ T-cell memory development. Nat Cell Biol. 2019;22(1):18–25.
Luo W, Yu Y, Wang H, Liu K, Wang Y, Huang M, et al. Up-regulation of MMP-2 by histone H3K9 beta-hydroxybutyrylation to antagonize glomerulosclerosis in diabetic rat. Acta Diabetol. 2020;57(12):1501–9.
Chen L, Miao Z, Xu X. β-Hydroxybutyrate alleviates depressive behaviors in mice possibly by increasing the histone3-lysine9-β-hydroxybutyrylation. Biochem Biophys Res Commun. 2017;490(2):117–22.
Wei W, Mao A, Tang B, Zeng Q, Gao S, Liu X, et al. Large-scale identification of protein Crotonylation reveals its role in multiple cellular functions. J Proteome Res. 2017;16(4):1743–52.
Liu X, Wei W, Liu Y, Yang X, Wu J, Zhang Y, et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov. 2017;3:17016.
Xu W, Wan J, Zhan J, Li X, He H, Shi Z, et al. Global profiling of crotonylation on non-histone proteins. Cell Res. 2017;27(7):946–9.
Xiao Y, Li W, Yang H, Pan L, Zhang L, Lu L, et al. HBO1 is a versatile histone acyltransferase critical for promoter histone acylations. Nucleic Acids Res. 2021;49(14):8037–59.
Yan KZ, Rousseau J, Machol K, Cross LA, Agre KE, Gibson CF, et al. Deficient histone H3 propionylation by BRPF1-KAT6 complexes in neurodevelopmental disorders and cancer. Sci Adv. 2020;6(4):eaax0021.
Madsen AS, Olsen CA. Profiling of substrates for zinc-dependent lysine Deacylase enzymes: HDAC3 exhibits Decrotonylase activity In Vitro. Angew Chem. 2012;124(36):9217–21.
Feldman JL, Baeza J, Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem. 2013;288(43):31350–6.
Bao X, Wang Y, Li X, Li X-M, Liu Z, Yang T, et al. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. eLife. 2014;3:e02999.
Wei W, Liu X, Chen J, Gao S, Lu L, Zhang H, et al. Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 2017;27(7):898–915.
Zhang Q, Zeng L, Zhao C, Ju Y, Konuma T, Zhou MM. Structural insights into histone Crotonyl-lysine recognition by the AF9 YEATS domain. Structure. 2016;24(9):1606–12.
Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, et al. Molecular coupling of histone Crotonylation and active transcription by AF9 YEATS domain. Mol Cell. 2016;62(2):181–93.
Andrews FH, Shinsky SA, Shanle EK, Bridgers JB, Gest A, Tsun IK, et al. The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol. 2016;12(6):396–8.
Zhao D, Guan H, Zhao S, Mi W, Wen H, Li Y, et al. YEATS2 is a selective histone crotonylation reader. Cell Res. 2016;26(5):629–32.
Xiong X, Panchenko T, Yang S, Zhao S, Yan P, Zhang W, et al. Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat Chem Biol. 2016;12(12):1111–8.
Flynn EM, Huang OW, Poy F, Oppikofer M, Bellon SF, Tang Y, et al. A subset of human Bromodomains recognizes Butyryllysine and Crotonyllysine histone peptide modifications. Structure. 2015;23(10):1801–14.
Ichijima Y, Sin HS, Namekawa SH. Sex chromosome inactivation in germ cells: emerging roles of DNA damage response pathways. Cell Mol Life Sci. 2012;69(15):2559–72.
Turner JM. Meiotic sex chromosome inactivation. Development. 2007;134(10):1823–31.
Abu-Zhayia ER, Awwad SW, Ben-Oz BM, Khoury-Haddad H, Ayoub N. CDYL1 fosters double-strand break-induced transcription silencing and promotes homology-directed repair. J Mol Cell Biol. 2018;10(4):341–57.
Liu Y, Liu S, Yuan S, Yu H, Zhang Y, Yang X, et al. Chromodomain protein CDYL is required for transmission/restoration of repressive histone marks. J Mol Cell Biol. 2017;9(3):178–94.
Xu X, Zhu X, Liu F, Lu W, Wang Y, Yu J. The effects of histone crotonylation and bromodomain protein 4 on prostate cancer cell lines. Transl Androl Urol. 2021;10(2):900–14.
Fellows R, Denizot J, Stellato C, Cuomo A, Jain P, Stoyanova E, et al. Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat Commun. 2018;9(1):105.
Zhu P, Martin E, Mengwasser J, Schlag P, Janssen KP, Gottlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5(5):455–63.
Ashktorab H, Belgrave K, Hosseinkhah F, Brim H, Nouraie M, Takkikto M, et al. Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma. Dig Dis Sci. 2009;54(10):2109–17.
Christott T, Bennett J, Coxon C, Monteiro O, Giroud C, Beke V, et al. Discovery of a selective inhibitor for the YEATS domains of ENL/AF9. SLAS Discov. 2019;24(2):133–41.
Li X, Li XM, Jiang Y, Liu Z, Cui Y, Fung KY, et al. Structure-guided development of YEATS domain inhibitors by targeting pi-pi-pi stacking. Nat Chem Biol. 2018;14(12):1140–9.
Hou JY, Cao J, Gao LJ, Zhang FP, Shen J, Zhou L, et al. Upregulation of alpha enolase (ENO1) crotonylation in colorectal cancer and its promoting effect on cancer cell metastasis. Biochem Biophys Res Commun. 2021;578:77–83.
Wan J, Liu H, Ming L. Lysine crotonylation is involved in hepatocellular carcinoma progression. Biomed Pharmacother. 2019;111:976–82.
Guo Z, Gu M, Huang J, Zhou P-k, Ma T. Global profiling of the crotonylome in Small cell lung Cancer. bioRxiv. 2020:2020.06.29.175877. https://doi.org/10.1101/2020.06.29.175877.
Ge Y, Fuchs E. Stretching the limits: from homeostasis to stem cell plasticity in wound healing and cancer. Nat Rev Genet. 2018;19(5):311–25.
Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer. 2012;12(3):170–80.
Machour FE, Ayoub N. Transcriptional regulation at DSBs: mechanisms and consequences. Trends Genet. 2020;36(12):981–97.
Patel R, Kumar A, Lokhande KB, Swamy K, Sharma P, Kumar N. Molecular Docking and Simulation Studies Predict Lactyl-CoA as the Substrate for P300 Directed Lactylation. ChemRxiv. 2020. Cambridge: Cambridge Open Engage; 2020. This content is a preprint and has not been peer-reviewed.
Gaffney DO, Jennings EQ, Anderson CC, Marentette JO, Shi T, Schou Oxvig AM, et al. Non-enzymatic lysine Lactoylation of glycolytic enzymes. Cell. Chem Biol. 2020;27(2):206–13 e6.
Yu J, Chai P, Xie M, Ge S, Ruan J, Fan X, et al. Histone lactylation drives oncogenesis by facilitating m(6)a reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22(1):85.
Jiang J, Huang D, Jiang Y, Hou J, Tian M, Li J, et al. Lactate modulates cellular metabolism through histone Lactylation-mediated gene expression in non-Small cell lung Cancer. Front Oncol. 2021;11:647559.
San-Millan I, Julian CG, Matarazzo C, Martinez J, Brooks GA. Is lactate an Oncometabolite? Evidence supporting a role for lactate in the regulation of transcriptional activity of Cancer-related genes in MCF7 breast Cancer cells. Front Oncol. 2019;9:1536.
Xie ZY, Dai JBA, Dai LZ, Tan MJ, Cheng ZY, Wu YM, et al. Lysine Succinylation and lysine Malonylation in histones. Mol Cell Proteomics. 2012;11(5):100–7.
Wagner GR, Payne RM. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. 2013;288(40):29036–45.
Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013;4(4):842–51.
Gibson GE, Xu H, Chen H-L, Chen W, Denton TT, Zhang S. Alpha-ketoglutarate dehydrogenase complex-dependent succinylation of proteins in neurons and neuronal cell lines. J Neurochem. 2015;134(1):86–96.
Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, et al. KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature. 2017;552(7684):273–7.
Tong Y, Guo D, Yan D, Ma C, Shao F, Wang Y, et al. KAT2A succinyltransferase activity-mediated 14-3-3zeta upregulation promotes beta-catenin stabilization-dependent glycolysis and proliferation of pancreatic carcinoma cells. Cancer Lett. 2020;469:1–10.
Yuan Y, Yuan H, Yang G, Yun H, Zhao M, Liu Z, et al. IFN-alpha confers epigenetic regulation of HBV cccDNA minichromosome by modulating GCN5-mediated succinylation of histone H3K79 to clear HBV cccDNA. Clin Epigenetics. 2020;12(1):135.
Yang G, Yuan Y, Yuan H, Wang J, Yun H, Geng Y, et al. Histone acetyltransferase 1 is a succinyltransferase for histones and non-histones and promotes tumorigenesis. EMBO Rep. 2021;22(2):e50967.
Kurmi K, Hitosugi S, Wiese EK, Boakye-Agyeman F, Gonsalves WI, Lou Z, et al. Carnitine Palmitoyltransferase 1A has a lysine Succinyltransferase activity. Cell Rep. 2018;22(6):1365–73.
Wang C, Zhang C, Li X, Shen J, Xu Y, Shi H, et al. CPT1A-mediated succinylation of S100A10 increases human gastric cancer invasion. J Cell Mol Med. 2019;23(1):293–305.
Li X, Zhang C, Zhao T, Su ZP, Li MJ, Hu JC, et al. Lysine-222 succinylation reduces lysosomal degradation of lactate dehydrogenase a and is increased in gastric cancer. J Exp Clin Cancer Res. 2020;39(1):172.
Du JT, Zhou YY, Su XY, Yu JJ, Khan S, Jiang H, et al. Sirt5 is a NAD-dependent protein lysine Demalonylase and Desuccinylase. Science. 2011;334(6057):806–9.
Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18(6):920–33.
Li L, Shi L, Yang S, Yan R, Zhang D, Yang J, et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun. 2016;7:12235.
Wang Y, Jin J, Chung MWH, Feng L, Sun HY, Hao Q. Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation. P Natl Acad Sci USA. 2018;115(10):2365–70.
Xie L, Liu W, Li Q, Chen S, Xu M, Huang Q, et al. First succinyl-proteome profiling of extensively drug-resistant mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. J Proteome Res. 2015;14(1):107–19.
Gao Y, Lee H, Kwon OK, Tan M, Kim KT, Lee S. Global proteomic analysis of lysine Succinylation in Zebrafish (Danio rerio). J Proteome Res. 2019;18(10):3762–9.
He D, Wang Q, Li M, Damaris RN, Yi X, Cheng Z, et al. Global proteome analyses of lysine acetylation and Succinylation reveal the widespread involvement of both modification in metabolism in the embryo of germinating Rice seed. J Proteome Res. 2016;15(3):879–90.
Fukushima A, Alrob OA, Zhang L, Wagg CS, Altamimi T, Rawat S, et al. Acetylation and succinylation contribute to maturational alterations in energy metabolism in the newborn heart. Am J Physiol Heart Circ Physiol. 2016;311(2):H347–63.
Park J, Chen Y, Tishkoff DX, Peng C, Tan MJ, Dai LZ, et al. SIRT5-mediated lysine Desuccinylation impacts diverse metabolic pathways. Mol Cell. 2013;50(6):919–30.
Ostergaard E. Disorders caused by deficiency of succinate-CoA ligase. J Inherit Metab Dis. 2008;31(2):226–9.
Gut P, Matilainen S, Meyer JG, Pallijeff P, Richard J, Carroll CJ, et al. SUCLA2 mutations cause global protein succinylation contributing to the pathomechanism of a hereditary mitochondrial disease. Nat Commun. 2020;11(1):5927.
Gibson GE, Hirsch JA, Cirio RT, Jordan BD, Fonzetti P, Elder J. Abnormal thiamine-dependent processes in Alzheimer's disease. Lessons from diabetes. Mol Cell Neurosci. 2013;55:17–25.
Gibson GE, Park LC, Sheu K-FR, Blass JP, Calingasan NY. The α-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochem Int. 2000;36(2):97–112.
Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57(5):695–703.
Lutz MI, Milenkovic I, Regelsberger G, Kovacs GG. Distinct patterns of sirtuin expression during progression of Alzheimer's disease. Neuro Molecular Med. 2014;16(2):405–14.
Ren S, Yang M, Yue Y, Ge F, Li Y, Guo X, et al. Lysine Succinylation contributes to Aflatoxin production and pathogenicity in Aspergillus flavus. Mol Cell Proteomics. 2018;17(3):457–71.
Hu B, Zhao M, Luo D, Yu C, Shao S, Zhao L, et al. Quantitative analysis of the proteome and the Succinylome in the thyroid tissue of high-fat diet-induced Hypothyroxinemia in rats. Int J Endocrinol. 2020;2020:3240198.
Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, et al. SIRT5 Desuccinylates and activates pyruvate kinase M2 to block macrophage IL-1β production and to prevent DSS-induced colitis in mice. Cell Rep. 2017;19(11):2331–44.
Lu X, Yang P, Zhao X, Jiang M, Hu S, Ouyang Y, et al. OGDH mediates the inhibition of SIRT5 on cell proliferation and migration of gastric cancer. Exp Cell Res. 2019;382(2):111483.
Song Y, Wang J, Cheng Z, Gao P, Sun J, Chen X, et al. Quantitative global proteome and lysine succinylome analyses provide insights into metabolic regulation and lymph node metastasis in gastric cancer. Sci Rep. 2017;7:42053.
Tong Y, Guo D, Lin SH, Liang J, Yang D, Ma C, et al. SUCLA2-coupled regulation of GLS succinylation and activity counteracts oxidative stress in tumor cells. Mol Cell. 2021;81(11):2303–16 e8.
Kwon OK, Bang IH, Choi SY, Jeon JM, Na AY, Gao Y, et al. SIRT5 Is the desuccinylase of LDHA as novel cancer metastatic stimulator in aggressive prostate cancer. Genomics Proteomics Bioinformatics. 2022:S1672-0229(22)00018–3. https://doi.org/10.1016/j.gpb.2022.02.004.
Lin ZF, Xu HB, Wang JY, Lin Q, Ruan Z, Liu FB, et al. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem Biophys Res Commun. 2013;441(1):191–5.
Qi H, Ning X, Yu C, Ji X, Jin Y, McNutt MA, et al. Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis. 2019;10(3):170.
Li F, He X, Ye D, Lin Y, Yu H, Yao C, et al. NADP(+)-IDH mutations promote Hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol Cell. 2015;60(4):661–75.
Ma Y, Qi Y, Wang L, Zheng Z, Zhang Y, Zheng J. SIRT5-mediated SDHA desuccinylation promotes clear cell renal cell carcinoma tumorigenesis. Free Radic Biol Med. 2019;134:458–67.
Lukey MJ, Greene KS, Cerione RA. Lysine succinylation and SIRT5 couple nutritional status to glutamine catabolism. Mol Cell Oncol. 2020;7(3):1735284.
Ren M, Yang X, Bie J, Wang Z, Liu M, Li Y, et al. Citrate synthase desuccinylation by SIRT5 promotes colon cancer cell proliferation and migration. Biol Chem. 2020;401(9):1031–9.
Yang X, Wang Z, Li X, Liu B, Liu M, Liu L, et al. SHMT2 Desuccinylation by SIRT5 drives Cancer cell proliferation. Cancer Res. 2018;78(2):372–86.
Xiangyun Y, Xiaomin N. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget. 2017;8(4):6984.
Abril YLN, Fernandez IR, Hong JY, Chiang YL, Kutateladze DA, Zhao Q, et al. Pharmacological and genetic perturbation establish SIRT5 as a promising target in breast cancer. Oncogene. 2021;40(9):1644–58.
Guo Z, Pan F, Peng L, Tian S, Jiao J, Liao L, et al. Systematic proteome and lysine Succinylome analysis reveals enhanced cell migration by Hyposuccinylation in esophageal squamous cell carcinoma. Mol Cell Proteomics. 2021;20:100053.
Zhang N, Gao R, Yang J, Zhu Y, Zhang Z, Xu X, et al. Quantitative global proteome and lysine Succinylome analyses reveal the effects of energy metabolism in renal cell carcinoma. Proteomics. 2018;18(19):e1800001.
Warburg O, Negelein E, Posener K. Versuche an überlebendem Carcinomgewebe. Klin Wochenschr. 1924;3(24):1062–4.
Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–37.
Zhu D, Hou L, Hu B, Zhao H, Sun J, Wang J, et al. Crosstalk among proteome, acetylome and succinylome in colon cancer HCT116 cell treated with sodium dichloroacetate. Sci Rep. 2016;6(1):37478.
Liu B, Lin Y, Darwanto A, Song X, Xu G, Zhang K. Identification and characterization of propionylation at histone H3 lysine 23 in mammalian cells. J Biol Chem. 2009;284(47):32288–95.
Zhang K, Chen Y, Mang ZH, Zhao YM. Identification and verification of lysine Propionylation and Butyrylation in yeast Core histones using PTMap software. J Proteome Res. 2009;8(2):900–6.
Lin H, Su X, He B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem Biol. 2012;7(6):947–60.
Cheng Z, Tang Y, Chen Y, Kim S, Liu H, Li SS, et al. Molecular characterization of propionyllysines in non-histone proteins. Mol Cell Proteomics. 2009;8(1):45–52.
Han Z, Wu H, Kim S, Yang X, Li Q, Huang H, et al. Revealing the protein propionylation activity of the histone acetyltransferase MOF (males absent on the first). J Biol Chem. 2018;293(9):3410–20.
Ringel AE, Wolberger C. Structural basis for acyl-group discrimination by human Gcn5L2. Acta Crystallogr D Struct Biol. 2016;72(Pt 7):841–8.
Nahomi RB, Nandi SK, Rakete S, Michel C, Fritz KS, Nagaraj RH. Lysine malonylation and propionylation are prevalent in human lens proteins. Exp Eye Res. 2020;190:107864.
Vollmuth F, Geyer M. Interaction of propionylated and butyrylated histone H3 lysine marks with Brd4 bromodomains. Angew Chem Int Ed Eng. 2010;49(38):6768–72.
Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630.
Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25(3):393–405.
Singh B, Boopathy S, Somasundaram K, Umapathy S. Fourier transform infrared microspectroscopy identifies protein propionylation in histone deacetylase inhibitor treated glioma cells. J Biophotonics. 2012;5(3):230–9.
Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From dietary Fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165(6):1332–45.
Hogh RI, Moller SH, Jepsen SD, Mellergaard M, Lund A, Pejtersen M, et al. Metabolism of short-chain fatty acid propionate induces surface expression of NKG2D ligands on cancer cells. FASEB J. 2020;34(11):15531–46.
Xu G, Wang J, Wu Z, Qian L, Dai L, Wan X, et al. SAHA regulates histone acetylation, Butyrylation, and protein expression in neuroblastoma. J Proteome Res. 2014;13(10):4211–9.
Zhao YL, Fang XL, Wang Y, Zhang JM, Jiang S, Liu Z, et al. Comprehensive analysis for histone acetylation of human Colon Cancer cells treated with a novel HDAC inhibitor. Curr Pharm Design. 2014;20(11):1866–73.
Souza BK, da Costa Lopez PL, Menegotto PR, Vieira IA, Kersting N, Abujamra AL, et al. Targeting histone Deacetylase activity to arrest cell growth and promote neural differentiation in Ewing sarcoma. Mol Neurobiol. 2018;55(9):7242–58.
Zhang K, Li L, Zhu M, Wang G, Xie J, Zhao Y, et al. Comparative analysis of histone H3 and H4 post-translational modifications of esophageal squamous cell carcinoma with different invasive capabilities. J Proteome. 2015;112:180–9.
Hegedus R, Manea M, Orban E, Szabo I, Kiss E, Sipos E, et al. Enhanced cellular uptake and in vitro antitumor activity of short-chain fatty acid acylated daunorubicin-GnRH-III bioconjugates. Eur J Med Chem. 2012;56:155–65.
Kapuvari B, Hegedus R, Schulcz A, Manea M, Tovari J, Gacs A, et al. Improved in vivo antitumor effect of a daunorubicin - GnRH-III bioconjugate modified by apoptosis inducing agent butyric acid on colorectal carcinoma bearing mice. Investig New Drugs. 2016;34(4):416–23.
Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr. 2008;28:253–72.
Qian L, Nie L, Chen M, Liu P, Zhu J, Zhai L, et al. Global profiling of protein lysine Malonylation in Escherichia coli reveals its role in energy metabolism. J Proteome Res. 2016;15(6):2060–71.
Ishiguro T, Tanabe K, Kobayashi Y, Mizumoto S, Kanai M, Kawashima SA. Malonylation of histone H2A at lysine 119 inhibits Bub1-dependent H2A phosphorylation and chromosomal localization of shugoshin proteins. Sci Rep. 2018;8(1):7671.
Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, et al. SIRT5 regulates both cytosolic and mitochondrial protein Malonylation with glycolysis as a major target. Mol Cell. 2015;59(2):321–32.
Du Z, Liu X, Chen T, Gao W, Wu Z, Hu Z, et al. Targeting a Sirt5-positive subpopulation overcomes multidrug resistance in wild-type Kras colorectal carcinomas. Cell Rep. 2018;22(10):2677–89.
Gao W, Xu Y, Chen T, Du Z, Liu X, Hu Z, et al. Targeting oxidative pentose phosphate pathway prevents recurrence in mutant Kras colorectal carcinomas. PLoS Biol. 2019;17(8):e3000425.
Bruning U, Morales-Rodriguez F, Kalucka J, Goveia J, Taverna F, Queiroz KCS, et al. Impairment of angiogenesis by fatty acid synthase inhibition involves mTOR Malonylation. Cell Metab. 2018;28(6):866–80 e15.
Wang Y-Q, Wang H-L, Xu J, Tan J, Fu L-N, Wang J-L, et al. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nature. Communications. 2018;9(1):545.
Zhou L, Wang F, Sun R, Chen X, Zhang M, Xu Q, et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 2016;17(6):811–22.
Fiaschi T, Chiarugi P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int J Cell Biol. 2012;2012:762825.
Cook JA, Gius D, Wink DA, Krishna MC, Russo A, Mitchell JB. Oxidative stress, redox, and the tumor microenvironment. Semin Radiat Oncol. 2004;14(3):259–66.
Meng X, Xing S, Perez LM, Peng X, Zhao Q, Redoña ED, et al. Proteome-wide analysis of lysine 2-hydroxyisobutyrylation in developing Rice (Oryza sativa) seeds. Sci Rep. 2017;7(1):17486.
Huang H, Luo Z, Qi S, Huang J, Xu P, Wang X, et al. Landscape of the regulatory elements for lysine 2-hydroxyisobutyrylation pathway. Cell Res. 2018;28(1):111–25.
Huang H, Tang S, Ji M, Tang Z, Shimada M, Liu X, et al. p300-mediated lysine 2-Hydroxyisobutyrylation regulates glycolysis. Mol Cell. 2018;70(4):663–78 e6.
Huang J, Luo Z, Ying W, Cao Q, Huang H, Dong J, et al. 2-Hydroxyisobutyrylation on histone H4K8 is regulated by glucose homeostasis in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2017;114(33):8782–7.
Dong H, Zhai G, Chen C, Bai X, Tian S, Hu D, et al. Protein lysine de-2-hydroxyisobutyrylation by CobB in prokaryotes. Sci Adv. 2019;5(7):eaaw6703.
Zhao L-n, Yuan H-f, Wang Y-f, Yun H-l, Zheng W, Yuan Y, et al. IFN-α inhibits HBV transcription and replication by promoting HDAC3-mediated de-2-hydroxyisobutyrylation of histone H4K8 on HBV cccDNA minichromosome in liver. Acta Pharmacol Sin. 2021. https://doi.org/10.1038/s41401-021-00765-7.
Li QQ, Hao JJ, Zhang Z, Krane LS, Hammerich KH, Sanford T, et al. Proteomic analysis of proteome and histone post-translational modifications in heat shock protein 90 inhibition-mediated bladder cancer therapeutics. Sci Rep. 2017;7(1):201.
Bass MV, Armeev GA, Shaitan KV, Shaytan AK. The effect of Oncomutations and posttranslational modifications of histone H1 on Chromatosome structure and stability. Mosc Univ Biol Sci Bull. 2019;74(3):121–6.
Yuan Y, Yuan HF, Geng Y, Zhao LN, Yun HL, Wang YF, et al. Aspirin modulates 2-hydroxyisobutyrylation of ENO1K281 to attenuate the glycolysis and proliferation of hepatoma cells. Biochem Biophys Res Commun. 2021;560:172–8.
Zhang Z, Xie H, Zuo W, Tang J, Zeng Z, Cai W, et al. Lysine 2-hydroxyisobutyrylation proteomics reveals protein modification alteration in the actin cytoskeleton pathway of oral squamous cell carcinoma. J Proteome. 2021;249:104371.
Nie LB, Liang QL, Elsheikha HM, Du R, Zhu XQ, Li FC. Global profiling of lysine 2-hydroxyisobutyrylome in toxoplasma gondii using affinity purification mass spectrometry. Parasitol Res. 2020;119(12):4061–71.
Chriett S, Dabek A, Wojtala M, Vidal H, Balcerczyk A, Pirola L. Prominent action of butyrate over beta-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci Rep. 2019;9(1):742.
Kaczmarska Z, Ortega E, Goudarzi A, Huang H, Kim S, Marquez JA, et al. Structure of p300 in complex with acyl-CoA variants. Nat Chem Biol. 2017;13(1):21–9.
Huang H, Zhang D, Weng Y, Delaney K, Tang Z, Yan C, et al. The regulatory enzymes and protein substrates for the lysine beta-hydroxybutyrylation pathway. Sci Adv. 2021;7(9):eabe2771.
Zhang X, Cao R, Niu J, Yang S, Ma H, Zhao S, et al. Molecular basis for hierarchical histone de-beta-hydroxybutyrylation by SIRT3. Cell Discov. 2019;5:35.
Nishitani S, Fukuhara A, Shin J, Okuno Y, Otsuki M, Shimomura I. Metabolomic and microarray analyses of adipose tissue of dapagliflozin-treated mice, and effects of 3-hydroxybutyrate on induction of adiponectin in adipocytes. Sci Rep. 2018;8(1):8805.
Zhang H, Chang Z, Qin LN, Liang B, Han JX, Qiao KL, et al. MTA2 triggered R-loop trans-regulates BDH1-mediated beta-hydroxybutyrylation and potentiates propagation of hepatocellular carcinoma stem cells. Signal Transduct Target Ther. 2021;6(1):135.
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008.
Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26(15):2157–65.
Liu K, Li F, Sun Q, Lin N, Han H, You K, et al. p53 beta-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019;10(3):243.
Liao P, Bhattarai N, Cao B, Zhou X, Jung JH, Damera K, et al. Crotonylation at serine 46 impairs p53 activity. Biochem Biophys Res Commun. 2020;524(3):730–5.
Liu X, Rong F, Tang J, Zhu C, Chen X, Jia S, et al. Repression of p53 function by SIRT5-mediated desuccinylation at lysine 120 in response to DNA damage. Cell Death Differ. 2022;29(4):722–36.
Simithy J, Sidoli S, Yuan Z-F, Coradin M, Bhanu NV, Marchione DM, et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat Commun. 2017;8(1):1141.
Ducasse M, Brown MA. Epigenetic aberrations and cancer. Mol Cancer. 2006;5:60.
Chen L, Miao Y, Liu M, Zeng Y, Gao Z, Peng D, et al. Pan-Cancer analysis reveals the functional importance of protein lysine modification in Cancer development. Front Genet. 2018;9:254.
Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017;16(1):41.
Lu Y, Chan YT, Tan HY, Li S, Wang N, Feng Y. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol Cancer. 2020;19(1):79.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key Research and Development Plan (2017YFE0196300), the National Natural Science Foundation of China (No. U1932135), the Science and Technology Commission of Shanghai (20DZ2270800 and 19JC1410200).
Author information
Authors and Affiliations
Contributions
SG provided direction and guidance throughout the preparation of this manuscript. YF collected and interpreted studies and was a major contributor to the writing and editing of the manuscript. JY reviewed and made significant revisions to the manuscript. FL assisted in the revision of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fu, Y., Yu, J., Li, F. et al. Oncometabolites drive tumorigenesis by enhancing protein acylation: from chromosomal remodelling to nonhistone modification. J Exp Clin Cancer Res 41, 144 (2022). https://doi.org/10.1186/s13046-022-02338-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02338-w