
Abstract
Background
There has been a recent surge in interest in predicting biological effects associated with genomic alterations in order to implement personalized cancer treatment strategies. However, no reports have yet evaluated the utility of profiling blood-based circulating tumor DNA (ctDNA) in hepatocellular carcinoma (HCC) patients treated with lenvatinib (LEN).
Method
We retrospectively performed ctDNA next-generation sequencing (NGS) analysis in 24 patients with advanced HCC at baseline and 4 weeks after initiation of LEN. Association of the changes in variant allele frequencies (VAFs) during treatment and clinical outcome were evaluated.
Results
In total, 131 single nucleotide variants, 17 indels, and 23 copy number variations were detected as somatic alterations in 28, 6, and 12 genes, respectively in 23 of 24 patients. The most frequently altered genes were TP53 (54%), CTNNB1 (42%), TERT (42%), ATM (25%), and ARID1A (13%). The reduction in the mean frequency of variants (VAFmean) following 4 weeks of LEN treatment was associated with longer progression-free survival. The specificity and sensitivity of the reduction of VAFmean for predicting partial response were 0.67 and 1.0, respectively, which were higher than those of serum α-fetoprotein level (0.10 and 0.93, respectively). No association between the mutation status at baseline and the effectiveness of LEN was observed.
Conclusion
Our study demonstrated that somatic alterations could be detected in the majority of advanced HCC patients by ctDNA profiling and that ctDNA-kinetics during LEN treatment was a useful marker of disease progression. These results suggest that ctDNA profiling is a promising method that provides valuable information in clinical practice.
Similar content being viewed by others


Background
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors and is a leading cause of cancer-related death worldwide [1]. Lenvatinib (LEN) is an oral multikinase inhibitor that targets VEGF receptors 1–3, FGF receptors 1–4, PDGF receptor α, RET, and KIT [2]. Its approval is based on an international, multicenter, randomized, open-label, sorafenib non-inferiority trial (REFLECT; NCT01761266) that demonstrated a statistically significant improvement in progression-free survival (PFS) with LEN over sorafenib in patients with previously untreated, metastatic, or unresectable HCC [3]. More recently, an international, open-label, phase 3 trial (IMbrave; NCT03434379) demonstrated that treatment with atezolizumab plus bevacizumab was associated with significantly better overall survival (OS) and PFS outcome than sorafenib in patients with advanced unresectable HCC not previously treated with systemic therapy [4]. Consequently, atezolizumab plus bevacizumab was approved for the first-line treatment. While the number of drug options is increasing, the availability of biomarkers to predict treatment response to systemic therapies is limited to α-fetoprotein (AFP) for second-line ramucirumab [5]. Recently, associations between mutations in the PI3K/MTOR pathway and resistance to sorafenib as well as between mutations in the WNT pathway and resistance to immune checkpoint inhibitors have been reported [6, 7].
Molecular profiling has been gaining interest as a means to identify genomic alterations in cancer. The genetic landscape of HCC has been studied extensively [8,9,10,11]. However, the application of repeated biopsy in advanced HCC stages is limited for the following reasons: (i) the high specificity of radiological diagnosis; (ii) potential biopsy-related complications, such as bleeding and infection; (iii) and the limitation of single tissue biopsy in assessing tumor heterogeneity [12,13,14]. As an alternative, the use of circulating tumor DNA (ctDNA), also known as “liquid biopsy,” is a rapidly growing area of interest as a non-invasive test for the diagnosis and surveillance of cancer. Several studies have shown that ctDNA contains comprehensive information about tumor genomes, including variants originating from multiple independent tumors [15,16,17].
We have demonstrated that the relative level of ctDNA within a patient with HCC [18] or colorectal cancer [19] reflects the underlying tumor composition and that ctDNA levels change with time under therapeutic pressure. Several studies have shown that major somatic alterations in HCC, such as TP53, CTNNB1, TERT, and ARID1A, could be detected in ctDNA of HCC patients and that ctDNA has high specificity for detecting mutations in matched HCC tissue [20,21,22,23,24].
In various carcinomas, e.g., lung, bladder, and breast cancer, it has been reported that ctDNA kinetics can serve as a marker of therapeutic efficacy and can predict prolonged survival, suggesting that a change in VAF is directly related to antitumor activity and may have clinical significance [25,26,27].
On the other hand, there has been no report evaluating the utility of profiling blood-based ctDNA at baseline and/or during treatment with LEN in advanced HCC patients. The objectives of this study are as follows: 1) to evaluate the possibility of successful identification of ctDNA profiling using next-generation sequencing (NGS) in unresectable HCC patients treated with LEN; 2) to determine the utility of ctDNA in longitudinal monitoring of HCC treatment with LEN; and 3) to investigate the therapeutic implications of alterations that increase in frequency during LEN treatment by developing a search tool to identify therapeutic agents that might be effective against specific somatic alterations.
Methods
Patients
ctDNA was evaluated at the following two time points in 24 HCC patients (referred to as Hiroshima Guardant subjects HG1 through HG24) who had started LEN treatment at Hiroshima University Hospital between April 2018 and October 2019: (i) just before initiation of LEN and (ii) 4 weeks after initiation of LEN. The inclusion criteria were as follows: Child-Pugh liver function class A, an Eastern Cooperative Oncology Group performance status score of 1 or less, TNM stage 3 or higher, and a relative dose intensity > 70% during the initial 4 weeks. Patients were excluded when LEN treatment had been interrupted in the timeframe between the cfDNA profiling assays. Patients who received other treatments, such as transcatheter arterial chemo-embolization (TACE), during LEN treatment were censored. The end of follow-up was June 2020, and the median follow-up period was 14.3 months. Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0.
The study protocol was approved by the Hiroshima University ethical committee (approval numbers E-726-2 and HI-98) in accordance with the Declaration of Helsinki [28]. All patients provided written informed consent.
Treatment regimens
Patients with a body weight of 60 kg or more started at an elevated dose of 12 mg once per day, while the remaining patients started at the standard dose of 8 mg once per day. Treatment interruptions and dose reductions were permitted in the event of adverse drug reactions. Twenty cases of LEN treatment were interrupted because of adverse events (4/20) or progressive disease (PD) (16/20). After discontinuation of LEN, 13 of the 20 patients underwent systemic post-LEN treatments, 2 underwent TACE, and 6 received best supportive care.
Clinical and laboratory assessments
Clinical and laboratory assessments were performed before treatment. Objective response was evaluated by modified Response Evaluation Criteria in Solid Tumors (mRECIST) [29] after 6 weeks (3–15 w) of treatment and every 2 months subsequently. Tumor size was evaluated based on a sum of the diameters (longest for non-nodal lesions, short axis for nodal lesions) for all target lesions, defined in RECIST 1.1 [30]. Plasma collected from each patient at baseline was aliquoted and stored at − 80 °C prior to ctDNA profiling as we previously reported [19]. In brief, 10 milliliters of peripheral venous whole blood were collected using EDTA as an anticoagulant at baseline and a median of 4 weeks (3–6 w) after the start of LEN treatment. Subsequently, the drawn blood was immediately processed to isolate plasma by a two-step centrifugation process: 3500 rpm for 10 min followed by 12,000 rpm for 10 min at 4 °C. Separated plasma was stored at − 80 °C. A timeline of the ctDNA profiling and objective response evaluation is shown in Fig. 1a. The primary endpoint of the study was PFS, and the secondary endpoint was OS.

The timeline of the current study and comprehensive genomic classification. A The timeline of ctDNA profiling and objective response evaluation. ctDNA profiling was performed at baseline and after 4 weeks. Objective response was evaluated by mRECIST after 6 weeks of treatment and every two months subsequently. B Comprehensive genomic classification related to HCC included in the Guardant360 panel
Next-generation sequencing
Cell-free DNA (cfDNA) was extracted from approximately 2 ml of plasma, and genomic alterations were analyzed as described by Lanman et al. [31] using Guardant360 v2.11, an NGS panel of 74 cancer-related genes utilizing Digital Sequencing of cell-free ctDNA. The Guardant360 method detects all four major variant classes (single nucleotide variants [SNVs] in all 74 genes; indels in 74 genes; copy number variations [CNVs] in 18 genes; and fusions in 6 genes). The targeted genes are shown in Supplementary Table 1. ctDNA sequencing, variant calling, and variant filtering were performed as described previously [32, 33] using a proprietary bioinformatics pipeline performed by Guardant Health. Manual review was performed at Guardant Health following variant calling. Minimal PCR amplification was conducted to ensure assay robustness and uniformity, and the limit of detectable VAF of the panel was 0.03% [32]. Single nucleotide polymorphisms (SNPs) and neutral variants registered in gnomAD and dbSNP databases were excluded using their proprietary bioinformatics pipeline [32].
Genomic classification related to HCC
Patients were classified according to the mutations in major aberrant pathways of HCC. The following Gene Ontology and Reactome gene sets were used to determine the pathway component genes.
-
WNT pathway: GO CANONICAL WNT SIGNALING PATHWAY and REACTOME BETA CATENIN INDEPENDENT WNT SIGNALING
-
PI3K/MTOR pathway: GO PHOSPHATIDYLINOSITOL 3 KINASE SIGNALING and REACTOME MTOR SIGNALLING
-
Cell cycle control: GO CELL CYCLE and REACTOME CELL CYCLE
-
Chromatin remodeling: GO CHROMATIN REMODELING and REACTOME CHROMATIN MODIFYING ENZYMES (Fig. 1b).
Definitions
VAFmean: The mean of the VAF(s) of somatic mutated genes in each patient. For example, given the following VAFs at baseline: gene A = 11%, gene B = 4%, gene C = 0%, and gene D = 0% at after 4 weeks: gene A = 12%, gene B = 4%, gene C = 2%, and gene D = 0%. In this case, the VAFmean at baseline is (11 + 4 + 0) / 3 = 5, and after 4 weeks it is (12 + 4 + 2) / 3 = 6. Gene D is not detected at either time point, so it is excluded.
VAFmean-change: The change in VAFmean during the treatment, calculated by subtracting VAFmean at baseline from VAFmean at 4 weeks.
Statistical analysis
Statistical analysis was performed using JMP Pro 14.0.0 (SAS Institute Inc., Cary, NC, USA). Intergroup differences were tested using the Mann-Whitney U test or the Fisher’s exact test for continuous or categorical variables, respectively. Comparison of the VAFmean between baseline and 4 weeks was assessed by paired t test. For continuous values, the median value was used as a threshold if no specific cutoff had been established. Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were calculated for the assessment of the diagnostic utility of the change in VAFmean or tumor markers (serum AFP and des-gamma-carboxy pro-thrombin (DCP)). PFS and OS were estimated using Kaplan–Meier methods, and differences among subgroups were evaluated using the log-rank test. Univariate and multivariate Cox regression analysis was performed for potential biomarkers to predict PFS. All comparisons were considered significant if the P value was < 0.05.
Validation
To address the lack of statistical power due to the small sample size and to reduce overfitting of the Cox regression model, we performed bootstrapping (B = 1000) using the rms 6.1–0 package in R version 4.0.2. We also used the R glmnet 4.1 package to calculate penalized hazard ratios using K-fold cross-validation to determine lambda.
Matching variants with drugs of potential therapeutic effect
Translating ctDNA profiles to potentially actionable therapeutic strategies involves two steps. First, because most variants are expected to be benign or to have uncertain significance, the reported variants must be filtered to select variants of known or likely pathogenic effect. To this end, we compared predictions across several sources, including OncoKB [34], ClinVar [35], COSMIC [36], and CancerVar [37]. Second, because most variants have not been explicitly screened for their response to individual drugs, likely pathogenic variants were weighted based on evidence levels and specificity of the match. We searched for exact variant matches followed by broader partial matches based on the variant class against several databases, including CanDL (https://candl.osu.edu/), CIViC [38], Cancer Genome Interpreter [39], OncoKB [34], and CancerVar [37], filtering annotations based on the level of evidence, associated cancer type, approval status, and the predicted response to the therapy. An evidence report was then prepared based on review of the associated literature.
Results
Clinical characteristics
Patient demographics and baseline characteristics are shown in Table 1. The median PFS and OS were 6.4 months [95% confidence interval (CI), 4.8–8.4] and 17.8 months [95% CI, 13.2–22.4], respectively.
Genomic profiling of ctDNA
In total, 131 somatic SNVs, 17 somatic indels and 23 CNVs were detected in 28, 6, and 12 genes in 23 of 24 patients, respectively (Fig. 2, Supplementary Table 2), suggesting that the method used has the sensitivity required for detecting ctDNA in most patients with advanced HCC. The percentages of patients with the top 5 somatic mutations at baseline were as follows: TP53 (54%), CTNNB1 (42%), TERT (42%), ATM (25%), ARID1A (13%). The distribution of VAFmean is shown in Fig. 3a. VAFmean increased between baseline and 4 weeks in 6/24 (25%) cases. The distribution of VAFmean-change is shown in Fig. 3b. Patient HG-21 was excluded from the analysis of VAFmean-change because no ctDNA was detected at baseline and after 4 weeks. The median value of VAFmean was 0.85% (0–13.86). For the samples analyzed in this study, the depth of uniquely sequenced reads ranged from 952 to 10,459 (median, 4201).

An overview of somatic alterations and changes detected in VAFs. A A heatmap showing the genomic profiling of ctDNA in 24 patients. SNVs or indels are shown in the upper block in a white (0%), yellow (0.5%), and red (> 10%) color scale. CNVs are shown in the lower block in a blue and red color scale. In cases in which multiple variants were detected in the same patient, the greater VAF/CNV is shown

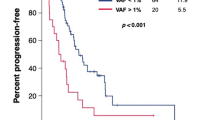
VAFmean kinetics during the 4-week LEN treatment. A The distribution of VAFmean. Blue bars and red bars indicate VAFmean at baseline and at 4 weeks, respectively. B The distribution of VAFmean-change. C Boxplot showing VAFmean at baseline (blue) and at 4 weeks (red) according to the first objective response at 6 weeks. A paired t test was used for the comparison between the VAF at baseline and at 4 weeks. D Kaplan–Meier curve estimates of PFS according to VAFmean at baseline. E Kaplan–Meier curve estimates of PFS according to VAFmean-change
Prior treatments are shown in Supplementary Table 3. Twenty-one patients had a history of prior treatment, and LEN was administrated as a first-line systemic treatment in 18 patients. There was no association between prior treatment and the number of mutations or the comprehensive genomic classification according to the mutated genes (Supplementary Table 4).
Clinical characteristics of the patients according to the baseline VAFmean and VAFmean-change
The patients were divided into a high VAFmean group and a low VAFmean group based on the median value of the VAFmean at baseline. There was no significant difference in the clinical characteristics between the patients in the high and low VAFmean group except for AFP (Table 2). There was no significant difference in clinical characteristics with respect to VAFmean-change.
VAFmean kinetics during the 4-week LEN treatment
Figure 3c shows VAFmean at baseline and at 4 weeks according to the first objective response (6 weeks). In patients with PD or stable disease (SD), there was no significant difference in VAFmean between baseline and 4 weeks (p = 0.78). On the other hand, in patients with partial response (PR) or complete response (CR), VAFmean significantly decreased following 4 weeks of LEN treatment (p = 0.02). The specificity, sensitivity, PPV, and NPV of VAFmean-change < 0 for predicting best response PR and CR was 0.67, 1.0, 0.82, and 1.0, respectively (Table 3).
PFS and OS according to the VAFmean at baseline and VAFmean-change
There was no significant difference in either PFS (Fig. 3d) or OS of patients with high or low VAFmean at baseline, shown in Kaplan–Meier curves; p = 0.63 and p = 0.31, respectively. The median PFS (95%CI) and OS (95%CI) of patients with VAFmean high/low were 7.0 (1.6–14.4) / 6.1 (4.1–12.1) months and 14.1 (7.2-not reached) / 21.5 (7.6-not reached) months, respectively. The median (95%CI) PFS and OS of patients with VAFmean-change < 0 / ≥0 following 4 weeks of LEN treatment were 9.3 (5.7–14.1) / 2.9 (1.2–183) months and 17.8 (13.2–21.2) months / not reached, respectively. Patients with VAFmean-change < 0 showed longer PFS (p < 0.001, Fig. 3e) than patients with VAFmean-change ≥0, but there was no significant difference in OS (p = 0.99).
Correlation of tumor size, VAFmean and tumor markers
Tumor size was evaluated based on the sum of the diameters for all target lesions, defined in RECIST 1.1 [30], and shown in Supplementary Table 5. There was a positive Spearman’s rank correlation between changes in tumor size and VAFmean-change (r = 0.56, p = 0.004). The positive correlation was also observed between VAFmean and tumor size at baseline (r = 0.41, p = 0.05) but not after LEN treatment (r = 0.31, p = 0.14) (Fig. 4a-c).

Correlation of tumor size, VAFmean and tumor markers. A correlation between VAFmean and tumor size at baseline (A) or after LEN treatment (B), and changes in tumor size and VAFmean-change(C) in Spearman’s rank correlation coefficient. A correlation between AFP/DCP and tumor size at baseline (D, G) or after LEN treatment (E, H). A correlation between changes in tumor size and in AFP/DCP before and after LEN treatment (F, I). Because the range of variation is large, the tumor markers are shown on a logarithmic scale (log2), and the change in tumor markers before and after LEN treatment is shown as a logarithmic scale (log2) of the ratio. After 4 weeks of LEN treatment, most cases showed a decrease in AFP (22/24, 92%) and an increase in DCP (19/24, 79%)
On the other hand, there was no significant correlation between tumor size and AFP or DCP at baseline or 4 weeks (Fig. 4d, e, g, h). Regarding the changes during LEN treatment, tumor size was positively correlated with DCP (Fig. 4i) but not with AFP (Fig. 4f).
Comparison with tumor makers
AFP positive HCC was only 11/24 cases (45.8%). In all AFP positive cases (≥20 ng/mL at baseline), AFP was reduced by LEN treatment regardless of the results of the first response evaluation (Table 3). The specificity, sensitivity, PPV and NPV of the decrease of AFP for predicting PR and CR was 0.10, 0.93, 0.59, and 0.50, respectively, inferior to VAFmean-change < 0 (Table 3). There was no significant difference in either PFS (Supplementary Figure 1D) or OS of patients with decrease and increase AFP, shown in Kaplan–Meier curves; p = 0.30 and p = 0.16, respectively. The VAFmean-change may be better as early markers than changes in tumor markers.
Also in DCP, which is one of the important tumor markers in HCC, there was no significant difference in either PFS (Supplementary Figure 1E) or OS of patients with decrease and increase DCP, p = 0.80 and p = 0.24, respectively. The specificity, sensitivity, PPV and NPV of the decrease of DCP for predicting PR and CR was 0.9, 0.29, 0.8, 0.47, respectively (Table 3).
Response to LEN according to specific mutations
Previous reports [6, 7] have shown that mutations in the PI3K/MTOR pathway were associated with resistance to tyrosine kinase inhibitors including sorafenib. We investigated the PFS according to the mutation in pathways frequently mutated in HCC patients, such as PI3K/MTOR pathway, WNT pathway, chromatin remodeling, cell cycle control and telomere maintenance. There were 6 patients with mutations related to PI3K/MTOR pathway, 13 patients with mutations related to WNT pathway, 5 patients with mutations related to chromatin remodeling, 18 patients with mutations related to the cell cycle, and 10 patients with mutations in the TERT promoter. Kaplan-Meier curves were drawn in PFS according to the status of the baseline somatic mutations of each classification (Fig. 5a-e), and no significant difference was found in all cases (p = 0.92, 0.65, 0.09, 0.60 and 0.82, respectively).

PFS according to the mutation in pathways frequently mutated in HCC patients. PFS according to the VAFmean of PI3K/MTOR pathway (A), WNT pathway (B), chromatin remodeling (C), cell cycle control (D), and telomere maintenance (E) at baseline
In the analysis of each of the top 3 genes at baseline, TP53, CTNNB1, and TERT, no significant differences in PFS were found (p = 0.09, 0.66 and 0.82, respectively) (Supplementary Figure 1A-C).
VAFmean-change and response to treatment in different types of mutations
We classified the mutations into missense mutations, nonsense mutations, frame shift mutations, in-frame mutations, mutations in promoter regions, and splice site mutations. Figure 6 shows the VAFmean-change with respect to mutation type. The direction of the VAFmean-change was the same regardless of the mutation type in all but 5 patients. However, there were some mutations that changed in opposite directions. The VAFmean-change of missense mutations in HG-09, nonsense mutations in HG-05, splice site mutations in HG-01 and 19, and promoter region mutations in HG-12 showed the opposite direction from those of the remaining mutations. Each VAFmean-change in the opposite direction represented a VAF-change in a single gene mutation; TP53 c.97-1G > A in HG-01, RB1 R552* in HG-05, TP53 L130F in HG-09, TERT c.-124C > T in HG-12, and TP53 c.993 + 1G > T in HG-19.

VAFmean-change in the different types of mutations. A heatmap showing the VAFmean-change in the different types of mutations including missense mutations, nonsense mutations, frame shift mutations, in-frame mutations, mutations in the promoter region, and splice site mutations. The cases are sorted according to the magnitude of the VAFmean-change in all mutations. Each VAFmean-change is shown in a blue/green color scale (< 0) and a yellow/purple color scale (≥0)
Factors associated with the cumulative incidence of poor PFS
The following parameters listed in Table 1 were analyzed by univariate analysis. AFP level was analyzed in two ways, the value at baseline and the change after 4 weeks of LEN treatment. Factors associated with poor PFS in univariate analysis were then analyzed by multivariable analysis.
Univariate analysis identified the following pretreatment factors associated with poor PFS: DCP (high/low, p = 0.03) and VAFmean-change (≥0/< 0, p < 0.001). Multiple Cox proportional hazard analysis identified only VAFmean-change ≥0/< 0 (HR 8.4, 95% CI = 2.3–31.2, p = 0.002) as an independent factor associated with poor PFS (Table 4). Results of bootstrapping using the rms package indicated a corrected Somer’s D statistic of 0.54 with optimism of 0.02 and a Harrell C index of 0.77. To calculate penalized hazard ratios, we performed 1000 iterations of 10-fold cross-validation with cv.glmnet to determine lambda (0.15). The penalized hazard ratio decreased from 8.4 to 4.8 for VAFmean-change ≥0/< 0 and from 2.3 to 1.6 for DCP (high/low).
Assessment of alterations that increased during LEN treatment
In 10 patients, 22 SNVs in 12 genes and 2 indels in 2 genes were detected after 4 weeks of LEN treatment that had not been detected at baseline. An increase in the VAF of 15 SNVs in 6 genes and 2 indels in 2 genes were observed in 9 and 2 patients, respectively. Predicted associations between these variants and drugs with potential therapeutic efficacy are shown in Supplementary Table 6 (complete list) and Table 5 (extracted list of patients with PD at 6 weeks). In patient HG-06, a novel SNV appeared in DDR2, suggesting that the patient might respond to dasatinib. In patient HG-22, the emergence of ATM G2891D and increases in the VAFs of R2832H and S1905fs were observed, suggesting that drugs such as cisplatin, olaparib, and temozolomide might provide a potential therapeutic response. Patient HG-18 experienced emergence of ESR1 R477*, suggesting that the patient might respond to fulvestrant but show resistance to exemestane. In patient HG-12, the frequency of ARID1A A2027fs increased, suggesting a potential response to ATR inhibitors, EZH2 inhibitors, and PARP inhibitors.
Discussion
We previously reported that detection of ctDNA before surgery could predict microscopic vascular invasion of the portal vein and recurrence, especially extrahepatic metastasis within 2 years in HCC patients who underwent liver resection [18]. Several other studies have also examined the role of ctDNA as a predictive or prognostic marker [40,41,42,43,44]. For molecular-targeted therapies, some reports have shown that ctDNA is useful for evaluation of treatment response and resistance, such as RAS mutation in colorectal cancer or EGFR mutations in lung cancer, known as targeting driver genes [45,46,47,48]. In HCC, Oh et al. reported that the higher amount of cfDNA (total cfDNA) was associated with shorter time to progression and OS in HCC patients treated with sorafenib, but the VEGFA amplification was not significantly associated with treatment outcome [49]. However, to our knowledge, there are no reports related to changes in VAF during HCC treatment. The usefulness of the VAFmean was previously reported by Raja et al., in which the reduction of VAFmean was associated with longer PFS and OS in non–small cell lung cancer and urothelial cancer treated with durvalumab [27]. In our study, we demonstrated that the reduction of VAFmean after initiation of LEN treatment for advanced HCC was associated with longer PFS. On the other hand, it was not associated with OS, possibly due to the effect of post-LEN treatments. In the OS analysis, 11 cases of censoring (survival cases within the follow-up period) were included, which may have affected the results. In the group showing VAFmean-change < 0, 7 out of 17 cases were censored, and in the group showing VAFmean-change ≥0, 4 out of 6 cases were censored. Although von Felden et al. recently reported that patients with mutations in the PI3K/MTOR pathway had significantly shorter PFS than those without these mutations after tyrosine kinase inhibitor treatment [7], no significant difference in PFS according to the presence of mutations in the PI3K/MTOR pathway at baseline was observed in our study. One reason might be that TSC2, which is involved in the PI3K/MTOR pathway and is frequently mutated in HCC, is not included in the Guardant360 panel used in our study. Another possible reason is that the therapeutic effects of LEN and sorafenib are different. In the study reported by von Felden et al., sorafenib was the most frequently used tyrosine kinase inhibitor (n = 18, 75%) [7].
A solid tumor consists of many sub clones with a range of different acquired mutations [12, 50]. It could be said that the response evaluated by the imaging to the treatment is synonymous with the response to the major clones in the tumor. Therefore, we considered that the VAF over a set of genes can reflect the response to the treatment with more precision. Zhang et al. suggested that on-treatment ctDNA kinetics are predictive of benefit with immune checkpoint blockade, which is a larger data set. They also had weighted the somatic SNVs and indels from the Guardant360 report equally when determining mean of VAF [51]. On the other hand, the analysis of the VAFmean-change within a particular mutation type revealed that there were minor populations that had a different direction of VAF change from the other major clones. That difference was considered to reflect the inequality of the responsiveness to LEN.
There was a positive correlation between the kinetics of the VAFmean and the changes in the sum of the tumor diameters of the target lesions during LEN treatment. This finding supports the idea that VAFmean level could serve as a non-invasive surrogate marker reflecting tumor burden. Serum AFP is the most widely used biomarker of HCC both for early diagnosis and evaluation of therapeutic efficacy and prognosis [52]. On the other hand, there was no correlation between tumor size and serum AFP nor between the change in the sum of the tumor diameters and the kinetics of AFP. There were also 6 patients who had discordant kinetics of AFP and VAFmean. For example, although patients HG-18 and HG-22 showed decreases in AFP (from 221 to 168 ng/mL and from 3834 to 1273 ng/mL, respectively) during the 4 weeks of LEN treatment, VAFmean increased (from 1.27 to 1.44% and from 0 to 0.19%, respectively). They were diagnosed as PD at the first response evaluation, which matched the VAFmean kinetics. In the remaining 4 patients, AFP had changed within a normal range (< 20 ng/mL). In addition, the specificity, sensitivity, PPV and NPV of the decrease of AFP or DCP for predicting PR and CR was inferior to those of VAFmean-change < 0. At least in evaluating the early response, the bottleneck in using AFP as a biomarker is that AFP had not been often positive and had tended to decrease after the start of LEN treatment in most cases (22/24, 92%). A systematic review showed that the sensitivity of AFP was 41–65% when using the commonly used positive cutoff value (AFP level ≥ 20 ng/mL) for HCC [53]. Another study investigated early tumor marker response and treatment response in patients with advanced HCC treated with LEN and concluded that the AFP levels of most patients had declined after 2 weeks, and by 4 weeks the group that had achieved a sustained reduction in AFP demonstrated a higher objective response [54]. This suggests that it is difficult to evaluate treatment response using AFP as a biomarker based on only one point. From these findings, it is possible that VAFmean-change might provide additional information to conventional tumor markers.
Although the variable timing of blood collection and imaging evaluation (Supplementary Table 5) is a major limitation of this study, the differences in the correlation between tumor size and VAFmean at baseline and after 4 weeks could reflect the fact that antitumor effects that cannot be evaluated by tumor size alone, which supports the clinical usefulness of mRECIST.
This study suffers from several other important limitations: 1) a relatively modest cohort size, 2) no comparison with DNA from tumor, non-tumor liver and peripheral blood mononuclear cells, 3) non-uniform prior treatment history and variable timing of blood collection and imaging evaluation, and 4) a limited set of cancer-associated genes contained in the panel. Despite these limitations, our cohort represents a substantial effort to interrogate an uncommon but important clinical phenotype of ctDNA kinetics before and after LEN treatment. To compensate, in part, for the limited sample size and lack of a separate validation cohort for our multivariable Cox regression model, we performed internal validation with bootstrapping to estimate model over-optimism using the rms package and calculated penalized hazard ratios using LASSO regularization with the glmnet package. This approach does not obviate the need for independent validation of the results helps to reduce overfitting and suggests that the model may perform comparably with new data. Although the Guardant360 assay is not designed to distinguish between germline and somatic variants, ctDNA genotyping can distinguish germline mutations (present at ~ 50% VAF) from somatic mutations (present but typically at much lower VAF) [55, 56].
We used the definition of somatic mutations derived by Guardant’s analysis platform, in which high frequency mutations close to 50% are defined as germline mutation. Furthermore, the allelic frequency of a germline mutation detected in plasma will not change during treatment. Although that platform is currently widely used, we would like to mention that sequencing DNA from peripheral blood mononuclear cells could support to consider a mutation as somatic. Moreover, the mutations described in this study are in line with the genomic landscape of HCC [11, 57, 58]. The mutations in TP53 were detected more frequently than in previous reports, but it has been demonstrated that advanced-stage HCC is associated with higher frequencies of TP53 mutations [58] (Supplementary Table 7). Several reports have shown that somatic mutations can be observed in background liver [59,60,61]. However, clonal expansions in the case of cirrhosis have been reported to be millimeters in diameter [59], which suggests that the amount of ctDNA harboring the same mutation from background liver is less than that derived from the tumor region. For these reasons, we conclude that the majority of mutations detected in this study were derived from the tumor region. However, the possibility of a germline or background-liver origin cannot be completely excluded.
In our current cohort, prior treatment history was not associated with baseline genomic profiling or number of mutations. However, it is well known that mutation profile could change under therapeutic pressure [19, 62], so the effect should be considered non-negligible. Patient HG-23 (SD case with VAFmean-change ≥0) had a longer PFS of 6.1 months compared to PD cases (median PFS 1.7 months). Although patient HG-23 had been previously treated with durvalumab that had been discontinued due to interstitial pneumonia as an adverse effect, the antitumor effect persisted for a while without treatment. Prior immune checkpoint inhibitor therapy might have affected the PFS of LEN treatment. In patient HG-21, no variant was detected at either time point. A possible reason is that patient HG-21 had a smaller tumor size (Supplementary Table 3), and the amount of ctDNA may have been low. However, we cannot rule out that a mutation that was not included in the panel may have played a role. The Guardant360 panel does not contain several important HCC-associated genes such as AXIN1/2 and TSC2 [11, 57, 58]. More comprehensive platforms such as the 500 gene Guardant OMNI platform, which launched for research use only in 2017, or HCC-dedicated platforms could make it possible to analyze genes that are not yet included in the target gene list [63]. Therefore, it is necessary to balance cost-effectiveness with the selection of a panel that is well-suited to its intended purpose.
At least one somatic alteration in a cancer-related gene was detected in 23 out of 24 patients by ctDNA profiling using Guardant360 v2.11. Moreover, we were able to successfully match the newly emerged or elevated cancer-related variants with drugs that have already been approved for some types of cancer in 3 of the 4 patients with PD at 6 weeks. Although we have not established whether those mutations are associated with resistance to LEN treatment, these results suggest that ctDNA profiling may be useful to search for effective alternative therapies after progressive disease. Ikeda et al. reported that a patient with a CDKN2A-inactivating mutation and a CTNNB1-activating mutation received palbociclib and celecoxib treatment, and low levels of AFP were found at 2 months. Another patient with a PTEN-inactivating mutation and a MET-activating mutation received sirolimus and cabozantinib, and AFP was found to have declined by 63% (8320 to 3045 ng/mL) [23].
Four patients showed intrinsic resistance to LEN on PD after 6 weeks. In these patients, 7 SNVs in 5 genes that had not been detected baseline were increased after 4 weeks of LEN treatment: TP53 R282W and C238S, ESR1 R477*, DDR2 W778L, ATM G2891D, AR R609K, and NFE2L2 D29G. None of these genes is known to be associated with resistance to LEN treatment. ESR1 confers resistance to aromatase inhibitors [64, 65]. Discoidin domain receptors (DDRs), including DDR1 and DDR2, are two members of the collagen receptor family in the tyrosine kinase receptor subgroup. DDR1 activation by p53 induces the MAP kinase pathway and increases resistance to apoptosis [66], and other studies have demonstrated a chemo-resistant role of DDR1 activation in several cancers [67,68,69]. On the other hand, little is known about the role of DDR2 in the acquisition of tumor cell resistance to chemotherapy. Moreover, there is no hot spot for mutations in DDRs, and there is a lack of functional analysis of mutations in these genes [70]. ATM encodes a PI3K-related serine/threonine protein kinase (PIKK) and plays a central role in the repair of DNA double-strand breaks. Once activated, ATM phosphorylates many downstream effectors and causes cell-cycle checkpoint arrest, DNA repair, and apoptosis; hence it is thought that ATM plays a role in suppression of carcinogenesis [71]. Somatic mutations in ATM occur in many tumor types, particularly hematologic malignancies, and generally have been associated with inferior prognosis [71,72,73]. On the other hand, it has been reported that blockade of ATM improves the antitumor effects of sorafenib in HCC cells, with suppression of Akt signaling and significant potentiation of the cytotoxic effects [74, 75]. Involvement of this gene in LEN treatment resistance is unknown, but it may play a role.
On the other hand, in PD cases, RAF1 K171R disappeared after 4 weeks of LEN treatment in patient HG-6, and PIK3CA G1007G disappeared in patient HG-12, who was naive to prior therapy. RAF1 is a kinase best known as the effector linking RAS to MEK/ERK activation. RAF1 has been reported as a negative regulator of hepatocarcinogenesis [76], although it has also been reported that RAF1 acts as an oncogene in HCC and that miR-4510 blocks HCC development through RAF1 targeting and RAS/RAF/MEK/ERK signaling inactivation [77]. Tian et al. reported that highly expressed RAF1 is associated with sorafenib resistance [78]. Interestingly, in the PR case HG-9, RAF1 mutation had increased, and CTNNB1 mutation had decreased following 4 weeks of LEN treatment, while in the PD case HG-6, RAF1 had disappeared and CTNNB1 had increased. Previous papers have revealed different modes of crosstalk between the two signals, WNT/β-catenin and RAS/MAPK, depending on the cellular context [79, 80].
Because most of the patients who require personalized systemic treatment are at advanced stages and their background is similar to that of the cohort of the present study, it could be said that this study was a simulation of clinical cancer genomics-based personalized treatment. Several recent studies have shown that β-catenin pathway activation represented by CTNNB1 mutation was associated with an immune-cold microenvironment and resistance to immune checkpoint inhibitors in HCC patients [5, 81, 82]. On the other hand, our study suggested that the CTNNB1 mutation status at baseline did not influence the effectiveness of LEN treatment. Further study is necessary to make conclusions. Considering our findings in light of previous studies, the assessment of mutations in genes in the β-catenin pathway, including CTNNB1, by ctDNA could be a useful indicator for treatment decisions; e.g., patients harboring such mutations might expect more benefit with LEN than immune checkpoint inhibitors.
Conclusion
Our findings suggest that ctDNA profiling is well-suited for clinical cancer genomics with the following advantages: i) it is non-invasive, ii) it facilitates monitoring of changes in VAF over time, and iii) ctDNA kinetics may be provide additional information over conventional tumor markers.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- ctDNA:
-
Circulating tumor DNA
- HCC:
-
Hepatocellular carcinoma
- LEN:
-
Lenvatinib
- NGS:
-
Next-generation sequencing
- VAF:
-
Variant allele frequency
- SNV:
-
Single nucleotide variant
- CNV:
-
Copy number variation
- PFS:
-
Progression-free survival
- OS:
-
Overall survival
- AFP:
-
α-fetoprotein
- TACE:
-
Transcatheter arterial chemo-embolization
- PPV:
-
Positive predictive value
- NPV:
-
Negative predictive value
- RECIST:
-
Response evaluation criteria in solid tumors
- PD, SD, PR, CR:
-
Progressive disease, Stable disease, Partial response, Complete response
- cfDNA:
-
Cell-free DNA
- SNP:
-
Single nucleotide polymorphism
- 95% CI:
-
95% confidence interval
References
Abubakar II, Tillmann T, Banerjee A. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385(9963):117–71.
Kudo M. Lenvatinib may drastically change the treatment landscape of hepatocellular carcinoma. Liver Cancer. 2018;7(1):1–19. https://doi.org/10.1159/000487148.
Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–73. https://doi.org/10.1016/S0140-6736(18)30207-1.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–905. https://doi.org/10.1056/NEJMoa1915745.
Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(2):282–96. https://doi.org/10.1016/S1470-2045(18)30937-9.
Harding JJ, Nandakumar S, Armenia J, Khalil DN, Albano M, Ly M, et al. Prospective genotyping of hepatocellular carcinoma: clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin Cancer Res. 2019;25(7):2116–26. https://doi.org/10.1158/1078-0432.CCR-18-2293.
von Felden J, Craig AJ, Garcia-Lezana T, Labgaa I. Mutations in circulating tumor DNA predict primary resistance to systemic therapies in advanced hepatocellular carcinoma. Oncogene. 2021;40(1):140–51. https://doi.org/10.1038/s41388-020-01519-1.
Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44(7):760–4. https://doi.org/10.1038/ng.2291.
Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58(5):1693–702. https://doi.org/10.1002/hep.26540.
Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–11. https://doi.org/10.1038/ng.3252.
Schulze K, Nault JC, Villanueva A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J Hepatol. 2016;65(5):1031–42. https://doi.org/10.1016/j.jhep.2016.05.035.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92. https://doi.org/10.1056/NEJMoa1113205.
Russano M, Napolitano A, Ribelli G. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: the potentiality of blood samples. J Exp Clin Cancer Res. 2020;39(1):95. https://doi.org/10.1186/s13046-020-01601-2.
Weng J, Atyah M, Zhou C, Ren N. Prospects and challenges of circulating tumor DNA in precision medicine of hepatocellular carcinoma. Clin Exp Med. 2020;20(3):329–37. https://doi.org/10.1007/s10238-020-00620-9.
Chan KC, Jiang P, Zheng YW, Liao GJ, Sun H, Wong J, et al. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem. 2013;59(1):211–24. https://doi.org/10.1373/clinchem.2012.196014.
Liao H, Li H. Advances in the detection technologies and clinical applications of circulating tumor DNA in metastatic breast Cancer. Cancer Manag Res. 2020;12:3547–60. https://doi.org/10.2147/CMAR.S249041.
Wei J, Liu X, Li T, Xing P, Zhang C, Yang J. The new horizon of liquid biopsy in sarcoma: the potential utility of circulating tumor nucleic acids. J Cancer. 2020;11(18):5293–308. https://doi.org/10.7150/jca.42816.
Ono A, Fujimoto A, Yamamoto Y, Akamatsu S, Hiraga N, Imamura M, et al. Circulating tumor DNA analysis for liver cancers and its usefulness as a liquid biopsy. Cell Mol Gastroenterol Hepatol. 2015;1(5):516–34. https://doi.org/10.1016/j.jcmgh.2015.06.009.
Yamauchi M, Urabe Y, Ono A, Miki D, Ochi H, Chayama K. Serial profiling of circulating tumor DNA for optimization of anti-VEGF chemotherapy in metastatic colorectal cancer patients. Int J Cancer. 2018;142(7):1418–26. https://doi.org/10.1002/ijc.31154.
He G, Chen Y, Zhu C, Zhou J, Xie X, Fei R, et al. Application of plasma circulating cell-free DNA detection to the molecular diagnosis of hepatocellular carcinoma. Am J Transl Res. 2019;11(3):1428–45.
Huang A, Zhang X, Zhou SL, Cao Y, Huang XW, Fan J, et al. Detecting circulating tumor DNA in hepatocellular carcinoma patients using droplet digital PCR is feasible and reflects Intratumoral heterogeneity. J Cancer. 2016;7(13):1907–14. https://doi.org/10.7150/jca.15823.
Ikeda S, Lim JS, Kurzrock R. Analysis of tissue and circulating tumor DNA by next-generation sequencing of hepatocellular carcinoma: implications for targeted therapeutics. Mol Cancer Ther. 2018;17(5):1114–22. https://doi.org/10.1158/1535-7163.MCT-17-0604.
Ikeda S, Tsigelny IF, Skjevik ÅA, Kono Y, Mendler M, Kuo A, et al. Next-generation sequencing of circulating tumor DNA reveals frequent alterations in advanced hepatocellular carcinoma. Oncologist. 2018;23(5):586–93. https://doi.org/10.1634/theoncologist.2017-0479.
Howell J, Atkinson SR, Pinato DJ, Knapp S, Ward C, Minisini R, et al. Identification of mutations in circulating cell-free tumour DNA as a biomarker in hepatocellular carcinoma. Eur J Cancer. 2019;116:56–66. https://doi.org/10.1016/j.ejca.2019.04.014.
Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, et al. Early assessment of lung Cancer immunotherapy response via circulating tumor DNA. Clin Cancer Res. 2018;24(8):1872–80. https://doi.org/10.1158/1078-0432.CCR-17-1341.
O'Leary B, Hrebien S, Morden JP, Beaney M, Fribbens C, Huang X, et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun. 2018;9(1):896. https://doi.org/10.1038/s41467-018-03215-x.
Raja R, Kuziora M, Brohawn PZ, Higgs BW, Gupta A, Dennis PA, et al. Early reduction in ctDNA predicts survival in patients with lung and bladder Cancer treated with Durvalumab. Clin Cancer Res. 2018;24(24):6212–22. https://doi.org/10.1158/1078-0432.CCR-18-0386.
World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–4.
Lencioni R, Llovet JM. Modified RECIST (mRECIST) assessment for hepatocellular carcinoma. Semin Liver Dis. 2010;30(1):52–60. https://doi.org/10.1055/s-0030-1247132.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–47. https://doi.org/10.1016/j.ejca.2008.10.026.
Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One. 2015;10(10):e0140712. https://doi.org/10.1371/journal.pone.0140712.
Zill OA, Banks KC, Fairclough SR, Mortimer SA, Vowles JV, Mokhtari R, et al. The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced Cancer patients. Clin Cancer Res. 2018;24(15):3528–38. https://doi.org/10.1158/1078-0432.CCR-17-3837.
Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, et al. Validation of a plasma-based comprehensive Cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res. 2018;24(15):3539–49. https://doi.org/10.1158/1078-0432.CCR-17-3831.
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017;1:1–6 PO.17.00011.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–5. https://doi.org/10.1093/nar/gkt1113.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in Cancer. Nucleic Acids Res. 2019;47(D1):D941–d7. https://doi.org/10.1093/nar/gky1015.
Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100(2):267–80. https://doi.org/10.1016/j.ajhg.2017.01.004.
Griffith M, Spies NC, Krysiak K, McMichael JF, Coffman AC, Danos AM, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat Genet. 2017;49(2):170–4. https://doi.org/10.1038/ng.3774.
Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, et al. Cancer genome interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018;10(1):25. https://doi.org/10.1186/s13073-018-0531-8.
Wang J, Huang A, Wang YP, Yin Y, Fu PY, Zhang X, et al. Circulating tumor DNA correlates with microvascular invasion and predicts tumor recurrence of hepatocellular carcinoma. Ann Transl Med. 2020;8(5):237. https://doi.org/10.21037/atm.2019.12.154.
Chen VL, Xu D, Wicha MS, Lok AS, Parikh ND. Utility of liquid biopsy analysis in detection of hepatocellular carcinoma, determination of prognosis, and disease monitoring: a systematic review. Clin Gastroenterol Hepatol. 2020;18(13):2879–2902.e9.
Cai J, Chen L. Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut. 2019;68(12):2195–205. https://doi.org/10.1136/gutjnl-2019-318882.
An Y, Guan Y, Xu Y, Han Y, Wu C, Bao C, et al. The diagnostic and prognostic usage of circulating tumor DNA in operable hepatocellular carcinoma. Am J Transl Res. 2019;11(10):6462–74.
Long G, Fang T, Su W, Mi X, Zhou L. The prognostic value of postoperative circulating cell-free DNA in operable hepatocellular carcinoma. Scand J Gastroenterol. 2020;55(12):1441–6. https://doi.org/10.1080/00365521.2020.1839127.
Bulbul A, Leal A, Husain H. Applications of cell-free circulating tumor DNA detection in EGFR mutant lung cancer. J Thorac Dis. 2020;12(5):2877–82. https://doi.org/10.21037/jtd.2020.01.66.
Klein-Scory S, Wahner I, Maslova M, Al-Sewaidi Y, Pohl M, Mika T, et al. Evolution of RAS mutational status in liquid biopsies during first-line chemotherapy for metastatic colorectal Cancer. Front Oncol. 2020;10:1115. https://doi.org/10.3389/fonc.2020.01115.
van Helden EJ, Angus L. RAS and BRAF mutations in cell-free DNA are predictive for outcome of cetuximab monotherapy in patients with tissue-tested RAS wild-type advanced colorectal cancer. Mol Oncol. 2019;13(11):2361–74. https://doi.org/10.1002/1878-0261.12550.
Yamada T, Matsuda A, Takahashi G, Iwai T, Takeda K, Ueda K, et al. Emerging RAS, BRAF, and EGFR mutations in cell-free DNA of metastatic colorectal patients are associated with both primary and secondary resistance to first-line anti-EGFR therapy. Int J Clin Oncol. 2020;25(8):1523–32. https://doi.org/10.1007/s10147-020-01691-0.
Oh CR, Kong SY, Im HS, Kim HJ, Kim MK, Yoon KA, et al. Genome-wide copy number alteration and VEGFA amplification of circulating cell-free DNA as a biomarker in advanced hepatocellular carcinoma patients treated with Sorafenib. BMC Cancer. 2019;19(1):292. https://doi.org/10.1186/s12885-019-5483-x.
Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–4. https://doi.org/10.1038/nature09807.
Zhang Q, Luo J, Wu S, Si H, Gao C, Xu W, et al. Prognostic and predictive impact of circulating tumor DNA in patients with advanced cancers treated with immune checkpoint blockade. Cancer Discov. 2020;10(12):1842–53. https://doi.org/10.1158/2159-8290.CD-20-0047.
Wang T, Zhang KH. New blood biomarkers for the diagnosis of AFP-negative hepatocellular carcinoma. Front Oncol. 2020;10:1316. https://doi.org/10.3389/fonc.2020.01316.
Gupta S, Bent S, Kohlwes J. Test characteristics of alpha-fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C. a systematic review and critical analysis. Ann Intern Med. 2003;139(1):46–50. https://doi.org/10.7326/0003-4819-139-1-200307010-00012.
Kodama K, Kawaoka T, Namba M, Uchikawa S, Ohya K, Morio K, et al. Correlation between early tumor marker response and imaging response in patients with advanced hepatocellular carcinoma treated with Lenvatinib. Oncology. 2019;97(2):75–81. https://doi.org/10.1159/000499715.
Nance T, Helman E, Artieri C, Yen J, Slavin TP, Chudova D, et al. Abstract 4272: A novel approach to differentiation of somatic vs. germline variants in liquid biopsies using a betabinomial model. Cancer Res. 2018;78(13 Supplement):4272.
Nakamura Y, Taniguchi H. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat Med. 2020;26(12):1859–64. https://doi.org/10.1038/s41591-020-1063-5.
Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46(12):1267–73. https://doi.org/10.1038/ng.3126.
Nault JC, Martin Y, Caruso S, Hirsch TZ, Bayard Q, Calderaro J. Clinical impact of genomic diversity from early to advanced hepatocellular carcinoma. Hepatology. 2020;71(1):164–82. https://doi.org/10.1002/hep.30811.
Brunner SF, Roberts ND, Wylie LA, Moore L, Aitken SJ, Davies SE, et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature. 2019;574(7779):538–42. https://doi.org/10.1038/s41586-019-1670-9.
Lombardo D, Saitta C, Giosa D, Di Tocco FC, Musolino C, Caminiti G, et al. Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from southern Italy. Oncol Lett. 2020;19(3):2368–74. https://doi.org/10.3892/ol.2020.11332.
Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4(1):2218. https://doi.org/10.1038/ncomms3218.
Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. 2017;7(8):805–17. https://doi.org/10.1158/2159-8290.CD-17-0343.
Si H, Kuziora M, Quinn KJ, Helman E, Ye J, Liu F, et al. A blood-based assay for assessment of tumor mutational burden in first-line metastatic NSCLC treatment: results from the MYSTIC study. Clin Cancer Res. 2021;27(6):1631–40. https://doi.org/10.1158/1078-0432.CCR-20-3771.
Carausu M, Melaabi S. ESR1 mutation detection and dynamics in meningeal Carcinomatosis in breast Cancer. J Breast Cancer. 2020;23(2):218–23. https://doi.org/10.4048/jbc.2020.23.e4.
Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446–51. https://doi.org/10.1038/ng.2823.
Ongusaha PP, Kim JI, Fang L, Wong TW, Yancopoulos GD, Aaronson SA, et al. p53 induction and activation of DDR1 kinase counteract p53-mediated apoptosis and influence p53 regulation through a positive feedback loop. EMBO J. 2003;22(6):1289–301. https://doi.org/10.1093/emboj/cdg129.
Ambrogio C, Gómez-López G, Falcone M, Vidal A, Nadal E, Crosetto N, et al. Combined inhibition of DDR1 and notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med. 2016;22(3):270–7. https://doi.org/10.1038/nm.4041.
Ambrogio C, Nadal E, Villanueva A, Gómez-López G, Cash TP, Barbacid M, et al. KRAS-driven lung adenocarcinoma: combined DDR1/notch inhibition as an effective therapy. ESMO Open. 2016;1(5):e000076. https://doi.org/10.1136/esmoopen-2016-000076.
Das S, Ongusaha PP, Yang YS, Park JM, Aaronson SA, Lee SW. Discoidin domain receptor 1 receptor tyrosine kinase induces cyclooxygenase-2 and promotes chemoresistance through nuclear factor-kappaB pathway activation. Cancer Res. 2006;66(16):8123–30. https://doi.org/10.1158/0008-5472.CAN-06-1215.
Henriet E, Sala M, Abou Hammoud A, Tuariihionoa A, Di Martino J, Ros M, et al. Multitasking discoidin domain receptors are involved in several and specific hallmarks of cancer. Cell Adhes Migr. 2018;12(4):363–77. https://doi.org/10.1080/19336918.2018.1465156.
Choi M, Kipps T, Kurzrock R. ATM mutations in Cancer: therapeutic implications. Mol Cancer Ther. 2016;15(8):1781–91. https://doi.org/10.1158/1535-7163.MCT-15-0945.
Beggs AD, Domingo E, McGregor M, Presz M, Johnstone E, Midgley R, et al. Loss of expression of the double strand break repair protein ATM is associated with worse prognosis in colorectal cancer and loss of Ku70 expression is associated with CIN. Oncotarget. 2012;3(11):1348–55. https://doi.org/10.18632/oncotarget.694.
Lin K, Adamson J, Johnson GG, Carter A, Oates M, Wade R, et al. Functional analysis of the ATM-p53-p21 pathway in the LRF CLL4 trial: blockade at the level of p21 is associated with short response duration. Clin Cancer Res. 2012;18(15):4191–200. https://doi.org/10.1158/1078-0432.CCR-11-2936.
Fujimaki S, Matsuda Y, Wakai T, Sanpei A, Kubota M, Takamura M, et al. Blockade of ataxia telangiectasia mutated sensitizes hepatoma cell lines to sorafenib by interfering with Akt signaling. Cancer Lett. 2012;319(1):98–108. https://doi.org/10.1016/j.canlet.2011.12.043.
Liu J, Liu Y, Meng L, Ji B, Yang D. Synergistic antitumor effect of Sorafenib in combination with ATM inhibitor in hepatocellular carcinoma cells. Int J Med Sci. 2017;14(6):523–9. https://doi.org/10.7150/ijms.19033.
Jeric I, Maurer G, Cavallo AL, Raguz J, Desideri E, Tarkowski B, et al. A cell-autonomous tumour suppressor role of RAF1 in hepatocarcinogenesis. Nat Commun. 2016;7(1):13781. https://doi.org/10.1038/ncomms13781.
Ghousein A, Mosca N, Cartier F, Charpentier J, Dupuy JW, Raymond AA. miR-4510 blocks hepatocellular carcinoma development through RAF1 targeting and RAS/RAF/MEK/ERK signalling inactivation. Liver Int. 2020;40(1):240–51. https://doi.org/10.1111/liv.14276.
Tian N, Wu D, Tang M, Sun H, Ji Y, Huang C, et al. RAF1 expression is correlated with HAF, a parameter of liver computed tomographic perfusion, and may predict the early therapeutic response to Sorafenib in advanced hepatocellular carcinoma patients. Open Med. 2020;15(1):167–74. https://doi.org/10.1515/med-2020-0024.
Jeong WJ, Yoon J, Park JC, Lee SH, Lee SH, Kaduwal S, et al. Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Sci Signal. 2012;5(219):ra30.
Biechele TL, Kulikauskas RM, Toroni RA, Lucero OM, Swift RD, James RG, et al. Wnt/β-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci Signal. 2012;5(206):ra3.
Hu ZQ, Xin HY, Luo CB, Li J, Zhou ZJ, Zou JX, et al. Associations among the mutational landscape, immune microenvironment, and prognosis in Chinese patients with hepatocellular carcinoma. Cancer Immunol Immunother. 2021;70(2):377–89. https://doi.org/10.1007/s00262-020-02685-7.
Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C. Castro de Moura M, et al. identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology. 2017;153(3):812–26. https://doi.org/10.1053/j.gastro.2017.06.007.
Acknowledgements
We would like to thank the study participants. Also, we gratefully acknowledge the technicians Chikako Kanehara and Emi Nishio for technical assistance.
Funding
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Number 17 K15948).
Author information
Authors and Affiliations
Contributions
YF and AO performed the analysis and wrote the paper. NH performed Cox bootstrapping analysis, matched variants with drugs of potential therapeutic effect, and edited the manuscript. SU, KK, YT, HF, TN, EM, DM, TK, MY, WO and MT were in charge of clinical data collection and recruited and monitored patients during follow up. MI, HA and KC designed the study, supervised the experiments and edited the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Hiroshima University ethical committee (approval number were E-726-2 and HI-98) in accordance with the Declaration of Helsinki (27). All patients provided written informed consent.
Consent for publication
Not applicable.
Competing interests
Potential competing interests: KC received lecture fees from Bristol-Myers Squibb, MSD, AbbVie, Gilead, and Sumitomo Dainippon Pharma and received grants from Bristol-Myers Squibb, Sumitomo Dainippon Pharma, MSD, AbbVie, TORAY, Eisai, Janssen Pharmaceutical, Daiichi-Sankyo, Roche, Otsuka Pharmaceutical, and Mitsubishi-Tanabe Pharma during the conduct of the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fujii, Y., Ono, A., Hayes, C.N. et al. Identification and monitoring of mutations in circulating cell-free tumor DNA in hepatocellular carcinoma treated with lenvatinib. J Exp Clin Cancer Res 40, 215 (2021). https://doi.org/10.1186/s13046-021-02016-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-02016-3