
Abstract
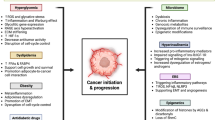
Malignant tumors are often multifactorial. Epidemiological studies have shown that hyperglycemia raises the prevalence and mortality of certain malignancies, like breast, liver, bladder, pancreatic, colorectal, endometrial cancers. Hyperglycemia can promote the proliferation, invasion and migration, induce the apoptotic resistance and enhance the chemoresistance of tumor cells. This review focuses on the new findings in the relationship between hyperglycemia and tumor development.
Similar content being viewed by others

Background
Recent studies have recognized hyperglycemia as a factor for cancer development in patients with diabetes. (The diabetes mentioned in this article is mainly type 2 diabetes). Hyperglycemia increases the prevalence and mortality (either short- or long-term) of many malignancies [1,2,3,4,5]. According to the WHO data, the number of patients with diabetes will increase from 382 million in 2015 to 592 million in 2035 [6]. Patients with diabetes face an increased risk of developing cancers, mainly including breast, liver, bladder, pancreatic, colorectal, endometrial cancers [7] (Table.1). This risk may arouse from special diabetic pathology, such as hyperglycemia, hyperinsulinemia, insulin resistance, distorted insulin-like growth factor-1 (IGF-1) pathway, oxidative stress, enhanced inflammatory processes, and aberrant sex hormone production [8, 9]. Studies have shown that hyperglycemia is one of the key factors in the hypothesis that diabetes is at increased risk of cancer [10,11,12]. Warburg O. first proposed that elevated blood glucose was associated with tumorigenesis [13]. Since then, many researchers have found that hyperglycemia can promote tumor development [1,2,3].
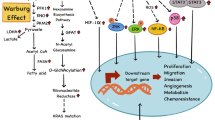
The glucose metabolism in tumor cells is characterized by the “Warburg” effect. Under aerobic or anoxic conditions, the cells initiate glycolysis to convert glucose into lactic acid, a process in which energy is produced [14]. Because of the deficiency of adenosine triphosphate (ATP) produced by glycolysis, the tumor cells increase the intake of glucose to boost energy-providing glycolysis. High glucose level supports tumor progression through a variety of mechanisms, including promoting tumor cell proliferation, invasion and migration and inducing apoptotic resistance and chemoresistance. However, more mechanisms may also be involved. This review aims to explore the mechanisms engaging hyperglycemia with tumor cell behavior, which we hope to benefit the treatment for cancer patients with diabetes.
Effect of hyperglycemia on tumor cell proliferation
Joshi et al. [15] pointed out that hyperglycemia could provide nutrients for the rapid proliferation of malignant tumor cells, thereby accelerating the process of tumor cells. Hou et al. [16] reported that high-concentration glucose (25 mM) significantly increased the proliferation of breast cancer cells (such as MDAMB231) compared to low-concentration glucose (5 mM). The mechanism may be that epidermal growth factor receptor (EGFR) is activated by guanosine triphosphatases (GTPases) Rac1 and Cdc42 to accelerate cell cycle progression and promote breast cancer cell proliferation. Han et al. [17] revealed that the proliferation of pancreatic cancer cells (such as BxPC-3 and Panc-1 cells) was affected by glucose concentration: high glucose (25, 50 mM) significantly increased pancreatic cancer cell proliferation compared to low glucose (5.5 mM). High glucose-induced epidermal growth factor (EGF) expression and EGFR transactivation may increase pancreatic cancer cell proliferation.
Long-term hyperglycemia leads to the production of a wide range of pro-inflammatory factors, such as interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), cyclooxygenase-2 (COX-2). These factors may be closely related to the development of tumors. Pothiwala et al. [18] pointed out that cytokines such as IL-6, TNF-α and COX-2 could stimulate oncogene expression, regulate cell cycle, promote tumor cell proliferation, inhibit apoptosis, and even induce epithelial-to-mesenchymal transition (EMT). EMT is widely recognized in cancer progression by enhancing cell invasion and anti-apoptosis [19, 20]. In EMT, polarized epithelial cells interact with the basement membrane through their basal surface and biochemically differentiate into interstitial phenotypes, a process through which invasive and anti-apoptotic properties are endowed and extracellular matrix generated.
Flores et al. [21] showed that high glucose (30 mM) increased the proliferation of breast cancer cells (MDA-MB-231) compared with low glucose (5.6 mM), and increased insulin further enhanced the proliferative effect of high glucose. High glucose (or high glucose and insulin)-induced cell proliferation may be mediated, at least in part, by oxidative stress, in which plasminogen activation is regulated by reactive oxygen species (ROS) production. Li et al. [22] found that hyperglycemia could induce miR-301a expression in prostate cancer cells in rat models and that miR-301a expression could inhibit the expression of p21 and Smad4, thus promoting the cells cycle from G1 and S phase, tumor cell proliferation and xenograft growth in nude mice. p21 is a cyclin-dependent kinase (CDK) inhibitor that blocks the cell cycle from G1 to S phase [23], and Smad4 can also induce G1/S cell cycle arrest [24]. Other researchers also pointed out that miR-301a promoted human tumor progression [25, 26], confirming the finding of Li et al.
Wang et al. [27] found that hyperglycemia could induce angiogenesis and tumor growth through hypoxia-inducible factor-1/vascular endothelial growth factor-dependent (HIF-1/VEGF) pathway. The mechanism may be that hyperglycemia, by impairing the function of HIF-1 inhibitors, attenuating the resistance of HIF-1 inhibitors against tumor chemotherapy or radiotherapy, increases tumor microvascular formation and tumor growth. They also pointed out that the prognosis of patients with hyperglycemia treated with HIF-1 inhibitor might be worse than those with low blood glucose. Other investigators also found that hyperglycemia promoted the proliferation of malignant breast cancer epithelial cells by increasing leptin/insulin-like growth factor-1 receptor (IGF-1R) signaling and activating the Protein Kinase B/mechanistic target of rapamycin (AKT/mTOR) pathway [28].
Effect of hyperglycemia on tumor cell invasion
Matrix metalloproteinase-2 (MMP-2), a member of the MMPs family, is involved in the breakdown of extracellular matrices, a process promoting tumor invasion [29]. Compared with cholangiocarcinoma cells cultured in low glucose, those cultured with high-concentration glucose showed stronger activation of signal transducer and activator of transcription3 (STAT3) and higher expression of MMP2 in the downstream of STAT3. Lowering blood glucose or using STAT3 inhibitors reduced the invasion of cholangiocarcinoma cells, so Saengboonmee et al. [30] pointed out that hyperglycemia might increase the invasive ability of biliary tumor cells by activating STAT3. Resveratrol could inhibit the proliferation and invasion of liver cancer cells by inhibiting the expression of STAT3 gene in high glucose environment [31].
Kang et al. [32] treated human lung epithelial cells (A549) with high-concentration glucose, finding that the expression of heme oxygenase-1 (HO-1) in cells increased. Moreover, cluster of differentiation 147 (CD147) and MMP-9, two strains of HO-l mediated protein associated with tumor cell invasion and metastasis, also showed increased expression that as a consequence enhanced tumor cell invasiveness. If HO-1 expression was silenced, high glucose-induced protein expression was reduced and tumor cell invasiveness attenuated. The mechanism may be that HO-1 expression is increased by hyperglycemia mediated by up-regulation of ROS or the TGF-β1/PI3K/Akt signaling pathway. In malignant tumors, such as lung cancer and bladder cancer, up-regulation of HO-1 is a factor for cancer poor prognosis [33, 34].
Alisson et al. [35] found that hyperglycemia (25 mM) induced TGF-β secretion in human lung cancer cell A549 compared with low blood glucose concentration group (5 mM). TGF-β is an important inducer of EMT and TGF-β signal conduction can lead to EMT [36] that enhances cell invasion and anti-apoptosis in cancer progression [19, 20]. Both Flores and Viedma have shown that high glucose can promote breast cancer cell invasion by inducing EMT [21, 37].
Sun et al. [38] demonstrated by transwell experiments that compared to those cultured in low glucose (5.56 mM), the breast cancer cells 7 cells (MCF-7) cultured in high glucose (25 mM) medium had stronger invasive ability, while the mRNA and protein expression of Glut1, MMP2 and MMP9 were significantly increased; besides, down-regulating Glut1 inhibited the invasion of MCF-7 cells and inhibited the expression of MMP2 and MMP9. Flores et al. [21] pointed out that hyperglycemia also increased the expression of serine protease urinary plasminogen activator (uPA) in tumor cells through ROS. uPA can proteolyze extracellular matrix components and the basement membrane around the primary tumor, thereby promoting tumor cell invasion. In summary, hyperglycemia can increase the expression of MMPs and uPA, the hydrolysis of extracellular matrix components, the invasion of tumor cells into adjacent normal tissues (Fig. 1).

Mechanisms underlying hyperglycemia-promoted cancer progression
Effect of hyperglycemia on tumor cell migration
Li et al. [39, 40] reported that hyperglycemia could promote the migration and invasion of pancreatic cancer cells (such as BxPC-3 and Panc-1 cells). The possible mechanism is that hyperglycemia can increase the concentration of H2O2 by up-regulating the expression of manganese superoxide dismutase (SOD2), and then activate the extracellular signal-regulated kinase (ERK) and protein 38 mitogen-activated protein kinases (p38 MAPK) pathways. H2O2 is a key factor mediating hyperglycemia-induced pancreatic cancer cell migration and invasion. After the addition of SOD2 inhibitor and polyethylene glycol-conjugated catalase (PEG-CAT), the migration was effectively inhibited. In vitro and in vivo studies showed that H2O2 increased the invasive and migratory ability of pancreatic cancer cells, and their invasion and migration were terminated after being treated with PEG-CAT.
Rahn et al. [41] studied precancerous H6c7-kras pancreatic cells with epithelial features, finding that hyperglycemia activated TGF-β1 signaling by increasing TGF-β1 expression and secretion, leading to a decrease in the expression of downstream Smad-dependent E-cadherin, which was more likely to break away from the mass and invade surrounding tissues, thereby promoting tumor cell metastasis. Takatani et al. [42] found that MCF-7 cultured in high glucose (25 mM) showed increased motility compared those cultured with low glucose (5.5 mM). The difference may be achieved by Zn2+ transported by Zin transporter 6 (ZIP6) and Zin transporter 10 (ZIP10). Zn2+ plays a crucial role in glucose-induced cell migration. Lack of Zn2+ significantly weakens the migrative activity of breast cancer cells in hyperglycemic conditions.
Together, it can be seen that the hyperglycemia promotes the migration of tumor cells, and the migration of tumor cells determines the quality of life and survival time of patients with advanced cancer to some extent. In general, highly migrative tumor cells are always highly invasive. Therefore, in the hyperglycemic environment, invasion always synchronizes migration, both deciding the prognosis of cancer patients.
Effect of hyperglycemia on tumor cell apoptotic resistance
p53 can suppress the canceration of cells and activate tumor cells’ response to anticancer drugs [43]. Garufi et al. [43] pointed out that hyperglycemia could inhibit p53 pro-apoptotic properties by reducing p53 phosphorylation of serine 46 (Ser46). Homeodomain-interacting protein kinase 2 (HIPK2) is a nuclear serine/threonine kinase that regulates the p53-dependent apoptotic pathway and tumor cell apoptosis [44]. Baldari et al. [45] found that hyperglycemia could trigger the degradation of HIPK2 protein, consequently inhibiting p53-induced apoptosis and promoting tumor progression. But in the hypoglycemic environment, the degradation of HIPK2 can be attenuated. Lowering blood glucose level maintains the HIPK2/p53 apoptotic axis function. Studies have shown that chronic inflammation markers (like IL-6, TNF-α, COX-2) produced under hyperglycemic conditions can exert anti-apoptotic activity to cells and induce EMT [46, 47]. Proto-oncogenes and tumor suppressor genes in humans are mutually restricted in controlling cell growth, but when mutated or inactivated, these genes can lead to tumor progression. The effect of hyperglycemia on p53 and inflammatory factors is shown in Fig. 1.
Effect of hyperglycemia on the resistance of tumor cells to chemotherapeutic drugs
Studies showed that [48,49,50,51] elevated blood glucose during chemotherapy increased the chemoresistance of tumor cells. Ma et al. [48] revealed that hyperglycemia attenuated the antiproliferative effect of 5-Fluorouracil (5-FU) on colon cancer cells. Patients with colorectal cancer accompanied by hyperglycemia need a higher dose of 5-FU and longer chemotherapy to adequately inhibit tumor cell growth. Zhao et al. [49] pointed out that hyperglycemia attenuated the chemosensitivity of gastric cancer cells to 5-FU. Hyperglycemia increases the expression of Nampt and Sirt1 in gastric cancer tissues and the expression of mutant p53 (compared with wild-type p53, the overexpression of mutant p53 in tumor cells is positively correlated with the high level expression of P-gp), resulting in the up-regulation of P-glycoprotein (P-gp) and down-regulation of Topoisomerase IIα (Topo-IIα). P-gp is a typical chemoresistance-resistant protein marker, and Topo-IIα a target marker for anticancer drugs. Up-regulation of P-gp and down-regulation of Topo-IIα mean that hyperglycemia leads to drug resistance in gastric cancer cells.
Biernacka et al. [50] reported that high glucose inhibited the apoptosis of prostate cancer cells induced by docetaxel, which may be related to the increased expression of IGFBP2. After IGFBP2 was silenced with small interfering RNA (siRNA), hyperglycemia no longer conferred tumor cells the resistance to chemotherapy drugs. This result was consistent with other studies on esophageal cancer and breast cancer cells [51]. IGFBP2 inactivates the tumor suppressor gene phosphatase and tensin homolog (PTEN) deleted on chromosome ten, leading to chemoresistance [52]. The expression level of IGFBP-2 is positively correlated with the progression of breast, prostate, lung, and colon cancer [51]. Zeng et al. [53] found that in the hyperglycemic environment, the sensitivity of breast cancer cells to chemotherapeutic drugs (such as 5-FU, doxorubicin or paclitaxel) might be related to fatty acid synthase (FAS), since inhibiting fatty acid synthase restored the sensitivity and accelerated the apoptosis of breast cancer cells. Therefore, strict control of glucose in cancer patients may enhance the effectiveness of chemotherapy.
Metformin is the most common hypoglycemic agent that exerts a hypoglycemic effect by reducing hepatic gluconeogenesis and increasing peripheral glucose utilization. A cohort study by Libby et al. [54] found that metformin use in patients with type 2 diabetes reduced the cancer-related overall mortality and mortality. Cancer was diagnosed among 7.3% of 4,085 metformin users compared with 11.6% of 4,085 comparators. After adjusting gender, age, BMI, smoking and other factors, a significantly reduced risk of cancer was found to be associated with metformin: 0.63 (0.53–0.75). Studies have shown that metformin has proliferation-inhibiting and apoptosis-promoting effects on tumor cells [55, 56]. There are a number of mechanisms by which metformin has been reported to act and these include: (1) LKB1-dependent mechanism activated protein kinase (AMPK-mTOR) to inhibit tumor cell proliferation [57, 58]; (2) Significant activation of AMPK in MDA-MB-231 cells on normal blood glucose level [55]. When metformin was used to treat prostate cancer cells, the resistance of prostate cancer cells to docetaxel was inhibited under hyperglycemic conditions, indicating that metformin can restore the sensitivity of prostate cancer cells to docetaxel through decreasing IGFBP-2 levels [59].
There are conflicting views. Lee et al. [60] reported that the risk of prostate cancer was reduced in patients with diabetes. Some metabolic and hormonal factors, including blood glucose and insulin, may involve. However, Betancourt et al. [61] showed the reduced risk of prostate cancer in patients with diabetes might be attributed to the decline in testosterone levels in patients with diabetes. However, Xu et al. [62] revealed that pre-existing high-risk factors such as hyperglycemia or obesity were associated with poor prognosis of prostate cancer; Li et al. [22] pointed out that hyperglycemia increased the expression of miR-301a in prostate cancer cells, thereby promoting G1/S cell cycle transition in vivo and accelerating cell proliferation; Biernacka et al. [50, 51] found that high glucose inhibited the efficacy of docetaxel-induced apoptosis in prostate cancer cells, which may be associated with hyperglycemia-mediated IGFBP2 overproduction. This is the controversy about the relationship between hyperglycemia and prostate cancer. Some researchers believe that patients with diabetes have a lower risk of prostate cancer. However, others believe that diabetes or hyperglycemia may promote the progression of prostate cancer through promoting the proliferation of tumor cells and inhibiting the apoptosis of tumor cells. The latter view is consistent with the impact of diabetes or hyperglycemia on other types of cancer (such as breast, liver, pancreatic, colorectal, bladder, endometrial cancer, etc.).
Conclusion
In summary, hyperglycemia accelerates the progression of tumor through enhancing the proliferation, migration, and invasion of tumor cells. However, the underlying mechanisms vary and still require more in-depth studies.
Availability of data and materials
Not Applicable.
Abbreviations
- 5-FU:
-
5-Fluorouracil
- AKT:
-
Protein Kinase B
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- ATP:
-
Adenosine triphosphate
- CD147:
-
Cluster of differentiation 147
- CDK:
-
Cyclin-dependent kinase
- COX-2:
-
Cyclooxygenase-2
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-to-mesenchymal transition
- ERK:
-
Extracellular signal-regulated kinase
- FAS:
-
Fatty acid synthase
- FDG-PET:
-
Fluorodeoxyglucose positron emission tomography
- Glut1:
-
Glucose transporter 1
- GTPases:
-
Guanosine triphosphatases
- HIF-1:
-
Hypoxia-inducible factor-1
- HIPK2:
-
Homologous domain-interacting protein kinase-2
- HO-1:
-
Heme oxygenase-1
- IGF-1:
-
Insulin-like growth factor-1
- IGF-1R:
-
Insulin-like growth factor-1 receptor
- IGFBP2:
-
Insulin Like Growth Factor Binding Protein 2
- IL-6:
-
Interleukin-6
- MMP:
-
Matrix metalloproteinase
- mTOR:
-
mechanistic target of rapamycin
- p38 MAPK:
-
p38 mitogen-activated protein kinases
- PEG-CAT:
-
polyethylene glycol-conjugated catalase
- P-gp:
-
P-glycoprotein
- PI3K:
-
Phosphoinositide 3-kinase
- PTEN:
-
Phosphatase and tensin homolog deleted on chromosome ten
- ROS:
-
Reactive oxygen species
- siRNA:
-
Small interfering RNA
- SOD2:
-
Superoxide dismutase
- STAT3:
-
Signal transducer and activator of transcription 3
- TGF-β1:
-
Transforming growth factor-β1
- TNF-α:
-
Tumor necrosis factor-α
- Topo-IIα:
-
Topoisomerase IIα
- uPA:
-
urinary plasminogen activator
- VEGF:
-
Vascular endothelial growth factor
- ZIP10:
-
Zin transporter 10
- ZIP6:
-
Zin transporter 6
References
Scappaticcio L, Maiorino MI, Bellastella G, Giugliano D, Esposito K. Insights into the relationships between diabetes, prediabetes, and cancer. ENDOCRINE. 2017;56:231–9.
Pandey A, Forte V, Abdallah M, et al. Diabetes mellitus and the risk of cancer. Minerva Endocrinol. 2011;36:187–209.
Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr Relat Cancer. 2009;16:1103–23.
Huang Y, Cai X, Qiu M, et al. Prediabetes and the risk of cancer: a meta-analysis. Diabetologia. 2014;57:2261–9.
Zhou XH, Qiao Q, Zethelius B, et al. Diabetes, prediabetes and cancer mortality. Diabetologia. 2010;53:1867–76.
Popkin BM. Nutrition transition and the global diabetes epidemic. Curr Diab Rep. 2015;15:64.
Garg SK, Maurer H, Reed K, Selagamsetty R. Diabetes and cancer: two diseases with obesity as a common risk factor. Diabetes Obes Metab. 2014;16:97–110.
Dabrowski M, Szymanska-Garbacz E, Miszczyszyn Z, Derezinski T, Czupryniak L. Risk factors for cancer development in type 2 diabetes: a retrospective case-control study. BMC Cancer. 2016;16:785.
Tseng CH. Type 2 diabetes mellitus and kidney Cancer risk: a retrospective cohort analysis of the National Health Insurance. PLoS One. 2015;10:e142480.
De Lorenzo MS, Baljinnyam E, Vatner DE, et al. Caloric restriction reduces growth of mammary tumors and metastases. Carcinogenesis. 2011;32:1381–7.
Lee C, Longo VD. Fasting vs dietary restriction in cellular protection and cancer treatment: from model organisms to patients. Oncogene. 2011;30:3305–16.
Seyfried TN, Kiebish MA, Marsh J, et al. Metabolic management of brain cancer. Biochim Biophys Acta. 2011;1807:577–94.
WARBURG O. On the origin of cancer cells. Science. 1956;123:309–14.
Westley RL, May FE. A twenty-first century cancer epidemic caused by obesity: the involvement of insulin, diabetes, and insulin-like growth factors. Int J Endocrinol 2013;2013:632461.
Joshi S, Liu M, Turner N. Diabetes and its link with cancer: providing the fuel and spark to launch an aggressive growth regime. Biomed Res Int. 2015;2015:390863.
Hou Y, Zhou M, Xie J, et al. High glucose levels promote the proliferation of breast cancer cells through GTPases. Breast Cancer (Dove Med Press). 2017;9:429–36.
Han L, Ma Q, Li J, et al. High glucose promotes pancreatic cancer cell proliferation via the induction of EGF expression and transactivation of EGFR. PLoS One. 2011;6:e27074.
Pothiwala P, Jain SK, Yaturu S. Metabolic syndrome and cancer. Metab Syndr Relat Disord. 2009;7:279–88.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90.
Kalluri R, Weinberg RA. The basics of epithelialmesenchymal transition. Journal of Clinical Investigation2009, 119 (6) :1420–1428.
Flores-Lopez LA, Martinez-Hernandez MG, Viedma-Rodriguez R, Diaz-Flores M, Baiza-Gutman LA. High glucose and insulin enhance uPA expression, ROS formation and invasiveness in breast cancer-derived cells. Cell Oncol (Dordr). 2016;39:365–78.
Li X, Li J, Cai Y, et al. Hyperglycaemia-induced miR-301a promotes cell proliferation by repressing p21 and Smad4 in prostate cancer. Cancer Lett. 2018;418:211–20.
Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–14.
Ding Z, Wu CJ, Chu GC, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470:269–73.
Zhou P, Jiang W, Wu L, et al. miR-301a is a candidate oncogene that targets the homeobox gene Gax in human hepatocellular carcinoma. Dig Dis Sci. 2012;57:1171–80.
Zhang W, Zhang T, Jin R, et al. MicroRNA-301a promotes migration and invasion by targeting TGFBR2 in human colorectal cancer. J Exp Clin Cancer Res. 2014;33:113.
Wang Y, Zhu YD, Gui Q, Wang XD, Zhu YX. Glucagon-induced angiogenesis and tumor growth through the HIF-1-VEGF-dependent pathway in hyperglycemic nude mice. Genet Mol Res. 2014;13:7173–83.
Lopez R, Arumugam A, Joseph R, et al. Hyperglycemia enhances the proliferation of non-tumorigenic and malignant mammary epithelial cells through increased leptin/IGF1R signaling and activation of AKT/mTOR. PLoS One. 2013;8:e79708.
Kapoor C, Vaidya S, Wadhwan V, Kaur G, Pathak A. Seesaw of matrix metalloproteinases (MMPs). J Cancer Res Ther. 2016;12:28–35.
Saengboonmee C, Seubwai W, Pairojkul C, Wongkham S. High glucose enhances progression of cholangiocarcinoma cells via STAT3 activation. Sci Rep. 2016;6:18995.
Li Y, Zhu W, Li J, Liu M, Wei M. Resveratrol suppresses the STAT3 signaling pathway and inhibits proliferation of high glucose-exposed HepG2 cells partly through SIRT1. Oncol Rep. 2013;30:2820–8.
Kang X, Kong F, Wu X, et al. High glucose promotes tumor invasion and increases metastasis-associated protein expression in human lung epithelial cells by upregulating heme oxygenase-1 via reactive oxygen species or the TGF-beta1/PI3K/Akt signaling pathway. Cell Physiol Biochem. 2015;35:1008–22.
Tsai JR, Wang HM, Liu PL, et al. High expression of heme oxygenase-1 is associated with tumor invasiveness and poor clinical outcome in non-small cell lung cancer patients. Cell Oncol (Dordr). 2012;35:461–71.
Miyata Y, Kanda S, Mitsunari K, Asai A, Sakai H. Heme oxygenase-1 expression is associated with tumor aggressiveness and outcomes in patients with bladder cancer: a correlation with smoking intensity. Transl Res. 2014;164:468–76.
Alisson-Silva F, Freire-de-Lima L, Donadio JL, et al. Increase of O-glycosylated oncofetal fibronectin in high glucose-induced epithelial-mesenchymal transition of cultured human epithelial cells. PLoS One. 2013;8:e60471.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Viedma-Rodriguez R, Martinez-Hernandez MG, Flores-Lopez LA, Baiza-Gutman LA. Epsilon-aminocaproic acid prevents high glucose and insulin induced-invasiveness in MDA-MB-231 breast cancer cells, modulating the plasminogen activator system. Mol Cell Biochem. 2018;437:65–80.
Sun XF, Shao YB, Liu MG, et al. High-concentration glucose enhances invasion in invasive ductal breast carcinoma by promoting Glut1/MMP2/MMP9 axis expression. Oncol Lett. 2017;13:2989–95.
Li W, Ma Z, Ma J, et al. Hydrogen peroxide mediates hyperglycemia-induced invasive activity via ERK and p38 MAPK in human pancreatic cancer. Oncotarget. 2015;6:31119–33.
Cao L, Chen X, Xiao X, et al. Resveratrol inhibits hyperglycemia-driven ROS-induced invasion and migration of pancreatic cancer cells via suppression of the ERK and p38 MAPK signaling pathways [J]. Int J Oncol. 2016;49(2):735–43.
Rahn S, Zimmermann V, Viol F, et al. Diabetes as risk factor for pancreatic cancer: hyperglycemia promotes epithelial-mesenchymal-transition and stem cell properties in pancreatic ductal epithelial cells. Cancer Lett. 2018;415:129–50.
Takatani-Nakase T, Matsui C, Maeda S, et al. High glucose level promotes migration behavior of breast cancer cells through zinc and its transporters. PLoS One. 2014;9:e90136.
Garufi A, D'Orazi G. High glucose dephosphorylates serine 46 and inhibits p53 apoptotic activity. J Exp Clin Cancer Res. 2014;33:79.
D'Orazi G, Rinaldo C, Soddu S. Updates on HIPK2: a resourceful oncosuppressor for clearing cancer. J Exp Clin Cancer Res. 2012;31:63.
Baldari S, Garufi A, Granato M, et al. Hyperglycemia triggers HIPK2 protein degradation. Oncotarget. 2017;8:1190–203.
Jiang GM, Xie WY, Wang HS, et al. Curcumin combined with FAPalphac vaccine elicits effective antitumor response by targeting indolamine-2,3-dioxygenase and inhibiting EMT induced by TNF-αlpha in melanoma. Oncotarget. 2015;6:25932–42.
Guan J, Zhang H, Wen Z, et al. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014;345:132–9.
Ma YS, Yang IP, Tsai HL, et al. High glucose modulates antiproliferative effect and cytotoxicity of 5-fluorouracil in human colon cancer cells. DNA Cell Biol. 2014;33:64–72.
Zhao W, Chen R, Zhao M, et al. High glucose promotes gastric cancer chemoresistance in vivo and in vitro. Mol Med Rep. 2015;12:843–50.
Biernacka KM, Uzoh CC, Zeng L, et al. Hyperglycaemia-induced chemoresistance of prostate cancer cells due to IGFBP2. Endocr Relat Cancer. 2013;20:741–51.
Al QA, Holly J, Perks C. Hypoxia negates hyperglycaemia-induced chemo-resistance in breast cancer cells: the role of insulin-like growth factor binding protein 2. Oncotarget. 2017;8:74635–48.
Uzoh CC, Perks CM, Bahl A, et al. PTEN-mediated pathways and their association with treatment-resistant prostate cancer. BJU Int. 2009;104:556–61.
Zeng L, Biernacka KM, Holly JM, et al. Hyperglycaemia confers resistance to chemotherapy on breast cancer cells: the role of fatty acid synthase. Endocr Relat Cancer. 2010;17:539–51.
Libby G, Donnelly LA, Donnan PT, et al, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2diabetes. Diabetes Care 2009;32(9):1620–1625. PubMed PMID: 19564453.
Zordoky BN, Bark D, Soltys CL, et al. The anti-proliferative effect of metformin in triple-negative MDA-MB-231 breast cancer cells is highly dependent on glucose concentration: implications for cancer therapy and prevention. Biochim Biophys Acta. 2014;1840:1943–57.
Takahashi A, Kimura F, Yamanaka A, et al. Metformin impairs growth of endometrial cancer cells via cell cycle arrest and concomitant autophagy and apoptosis. Cancer Cell Int. 2014;14:53.
Storozhuk Y, Hopmans SN, Sanli T, et al. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. 2013;108:2021–32.
Li B, Takeda T, Tsuiji K, et al. The antidiabetic drug metformin inhibits uterine leiomyoma cell proliferation via an AMP-activated protein kinase signaling pathway. Gynecol Endocrinol. 2013;29:87–90.
Biernacka KM, Persad RA, Bahl A, et al. Hyperglycaemia-induced resistance to docetaxel is negated by metformin: a role for IGFBP-2. Endocr Relat Cancer. 2017;24:17–30.
Lee C, An D, Park J. Hyperglycemic memory in metabolism and cancer. Horm Mol Biol Clin Investig. 2016;26:77–85.
Betancourt-Albrecht M, Cunningham GR. Hypogonadism and diabetes. Int J Impot Res. 2003;15(Suppl 4):S14–20.
Xu H, Zhang LM, Liu J, et al. The association between overall survival of prostate cancer patients and hypertension, hyperglycemia, and overweight in southern China: a prospective cohort study. J Cancer Res Clin Oncol. 2013;139:943–51.
Acknowledgements
This project was sponsored by the grants from the Priority Academic Program Development of Jiangsu Higher Education Institutions, National Natural Sciences Foundation of China (81870436, 81673515), Natural Science Foundation of Jiangsu Province (BK20161591), Jiangsu Provincial Medical Youth Talent (QNRC2016215), Suzhou science and education Youth Project (KJXW2016067), Suzhou industrial technology innovation (SYSD2016046), Jiangsu Province 333 High-level Talent Training Project (LGY2016010), Nanjing Science and Technology Development Plan (201715003) and Jiangsu Province Six Talent Peaks (WSN-030).
Funding
Not Applicable.
Author information
Authors and Affiliations
Contributions
WJ L, HZ and YQ W conceptualized the manuscript. WJ L collected the literature, wrote the manuscript and made the Figs. HZ and YQ W edited and made significant revisions to the manuscript. XH Z, HS, YZ and CY S retrieved the relevant literatures and revised the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not applicable.
Competing interests
The author states that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Wenjie Li and Xuehui Zhang are the first authors and did equal contribution.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, W., Zhang, X., Sang, H. et al. Effects of hyperglycemia on the progression of tumor diseases. J Exp Clin Cancer Res 38, 327 (2019). https://doi.org/10.1186/s13046-019-1309-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-019-1309-6