
Abstract
STAT proteins represent an important family of evolutionarily conserved transcription factors that play key roles in diverse biological processes, notably including blood and immune cell development and function. Classically, STAT proteins have been viewed as inducible activators of transcription that mediate cellular responses to extracellular signals, particularly cytokines. In this ‘canonical’ paradigm, latent STAT proteins become tyrosine phosphorylated following receptor activation, typically via downstream JAK proteins, facilitating their dimerization and translocation into the nucleus where they bind to specific sequences in the regulatory region of target genes to activate transcription. However, growing evidence has challenged this paradigm and identified alternate ‘non-canonical’ functions, such as transcriptional repression and roles outside the nucleus, with both phosphorylated and unphosphorylated STATs involved. This review provides a revised framework for understanding the diverse kaleidoscope of STAT protein functional modalities. It further discusses the implications of this framework for our understanding of STAT proteins in normal blood and immune cell biology and diseases such as cancer, and also provides an evolutionary context to place the origins of these alternative functional modalities.
Similar content being viewed by others
Background
The signal transducer and activators of transcription (STAT) family of proteins were initially identified as transcription factors that could facilitate the rapid induction of target genes in response to specific extracellular stimuli, including cytokines, growth factors and other agents, through tyrosine phosphorylation-mediated activation, much of it mediated by Janus kinases (JAKs) [1,2,3,4,5]. These genes impact key cellular processes, including differentiation, proliferation, survival and functional activation [6]. Gene ablation studies of the various STATs have identified essential roles particularly in blood and immune cell development and function (Table 1), but also in mammopoiesis, lactation, postnatal growth and a variety of homeostatic processes.
More recent studies, however, have uncovered alternative STAT functions that lie outside this ‘canonical’ paradigm of transcriptional activation including gene repression [16] and non-nuclear roles [17], as well as functions not requiring tyrosine phosphorylation [18], collectively termed ‘non-canonical’ signaling. This review describes the various STAT protein functionalities and places these in a robust framework that captures the diversity of STAT roles in the normal biology of blood and immune cells and relevant diseases, placing this diversity in an evolutionary context.
Structure and function of STAT protein family
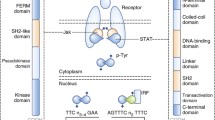
The STAT protein family consists seven members: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6 [19, 20]. Each is approximately 750–900 amino acid residues [21] comprising six conserved domains that mediate different aspects of STAT function [22] (Fig. 1).

Structure of STAT proteins. Schematic illustration of a representative STAT protein showing its six conserved functional domains: N-terminal, coiled-coil, DNA-binding, linker, Src-homology 2 (SH2) and C-terminal. The positions of a nuclear localization signal (NLS) and tyrosine (Y) and serine (S) residues phosphorylated in response to extracellular stimuli are shown, along with the sites of interaction of various transcriptional co-activators (green) and co-repressors (red)
N-terminal domain
Consisting of multiple alpha-helices that form a hook-like structure, the N-terminal domain (NTD) mediates interactions between STAT molecules that facilitate dimerization even in the absence of phosphorylation [23, 24].
Coiled-coil domain
Containing several alpha-helices in a ropelike structure, the coiled-coil domain (CCD) facilitates binding to other transcription factors and co-activators such as p48/Interferon regulatory factor 9 (IRF9) and N-Myc and STAT interactor (Nmi) [25], but also to co-repressors such as Silencing mediator for retinoic acid receptor and thyroid hormone receptor (SMRT)/nuclear receptor co-repressor (N-CoR) [26]. This domain is also implicated in nuclear translocation via a nuclear localization signal (NLS) motif that interacts with importin proteins to facilitate nuclear entry [22, 27].
DNA-binding domain
The DNA-binding domain (DBD) possess an immunoglobin-like structure that mediates recognition and binding to specific DNA target sequences [22, 28]. Many of these conform to the so-called gamma-activated sequence (GAS), a palindromic TTCN3-4GAA motif present within the promoter region of STAT-responsive target genes [6, 29].
Linker domain
The short linker domain (LD) is crucial for providing structural support during activation and DNA binding, and also serves as a contact point during the formation of transcriptional complexes [22, 30].
Src homology 2 domain
The Src homology2 (SH2) domain is a highly conserved structural module found in a myriad of signaling proteins that is able to bind to specific phosphotyrosine-containing motifs on other signaling components to mediate protein–protein interactions [31]. In the case of STATs, this includes phosphotyrosines found on activated receptor complexes as well as those found adjacent to the SH2 domain on STAT proteins that facilitate dimerization [1, 20, 22].
C-terminal domain
A diverse and poorly defined sequence located at the C terminus, this domain is often referred to as the transactivation domain (TAD), since it interacts with numerous transcriptional co-activators such as histone modifying acetyl transferases p300/CREB-binding protein (CBP) [32] and general control of amino acid synthesis protein-5 (GCN5) [33], and the chromatin remodeling factor Brahma [34] to activate gene transcription [35]. A serine residue present within the C-terminal domain undergoes phosphorylation independent of tyrosine phosphorylation and can enhance transcriptional activation [19].
Canonical STAT function
In the well-established ‘canonical’ STAT functional mode (Fig. 2), unphosphorylated STATs (uSTATs) reside in an inactive state in the cytoplasm and require tyrosine phosphorylation to become active.

Canonical STAT mode of action. Schematic representation of the archetypal ‘canonical’ STAT functional modality and its control. In this paradigm, STAT proteins (orange/yellow) exist in the cytoplasm as latent, unphosphorylated STAT (uSTAT) molecules. In response to binding of their cognate extracellular ligands (light pink), transmembrane receptors (dark blue) undergo conformational changes that results in the activation of kinases such as the receptor-associated JAKs (light brown), which subsequently mediate phosphorylation (P, green) of tyrosine residues within the intracellular receptor complex, thereby creating docking sites for signaling molecules, including uSTATs. These in turn become tyrosine phosphorylated, with the phosphorylated (pSTAT) molecules able to form dimers that can translocate into the nucleus and bind to specific DNA sequences (blue) to activate the transcription of responsive genes. These encode effector proteins (brown) responsible for cell differentiation, proliferation, survival and activation, as well as SOCS proteins (blue). These mediate a negative feedback loop by blocking STAT activation through interfering with STAT docking, inhibiting JAKs and/or mediating degradation of receptor signaling components. Other negative regulators include PIAS proteins (grey blue) that act via blocking STAT dimerization and nuclear entry and Protein tyrosine phosphatase (PTP) proteins (orange) that can dephosphorylate receptor complex components in the cytoplasm as well as pSTAT molecules in the nucleus to regenerate uSTAT molecules that return to the cytoplasm
Receptors
Extracellular regulators, such as cytokines and growth factors, bind to specific transmembrane receptors present on the plasma membrane of responsive cells. As a result, the intrinsic tyrosine kinase domains found in growth factor receptors and associated non-receptor tyrosine kinases, such as Janus kinases (JAKs), in the case of cytokine receptors become activated.
JAKs
JAKs associate with the membrane proximal region of cytokine receptors via interaction with conserved sequence motifs [36]. Cytokine-induced conformational changes in the receptor complex facilitate JAK activation through autophosphorylation, with activated JAKs able to phosphorylate tyrosine residues within the cytoplasmic domain of the receptor and associated molecules [37]. This creates docking sites for downstream signaling proteins containing SH2 domains, including uSTAT molecules [2, 31, 38].
STATs
Once docked to the receptor complex the uSTATs in turn become tyrosine phosphorylated, with the resultant phospho-STAT (pSTAT) able to form a dimer with another pSTAT via reciprocal SH2 domain–phosphotyrosine interactions, which can rapidly translocate into the nucleus by direct interaction with importin complexes in the nuclear membrane mediated by GTPases such as Rac1 [7, 39, 40] Within the nucleus the pSTAT dimers bind to specific DNA sequences in the promoter region of target genes typically based on variations of a core palindromic TTCN3-4GAA motif [6, 19]. This results in the transcriptional activation of these genes via associated co-activators [35]. The target genes encode proteins that are associated with various cellular activities such as proliferation, differentiation, survival and activation [6].
Negative regulators
STATs subsequently undergo inactivation by dephosphorylation via nuclear protein tyrosine phosphatases (PTPs) and are exported back to the cytoplasm [18, 41]. Canonical STAT function is also negatively regulated by protein inhibitors of activated STAT (PIAS) protein through direct binding to STAT proteins to suppress nuclear entry and DNA-binding activity, cytoplasmic PTPs that dephosphorylate various receptor components to inhibit STAT activation [42], and by members of the SOCS family of negative feedback regulators, which are induced by STAT signaling and then serve to inhibit further signaling via a number of mechanisms [43, 44].
Variations
Even within this ‘canonical’ mode, there are variations, such as the formation of a STAT1/STAT2/IRF9 hetero-trimeric complex in response to type I interferons [45], which targets an alternative sequence, the interferon-sensitive response element (ISRE: YAGTTC(A/T)TTTYCC) [46]. There is also evidence of other heterodimeric combinations, such as STAT1/IRF9, STAT1/STAT3, STAT3/STAT5 and STAT5A/STAT5B [47,48,49,50]. In addition, multimeric sites comprising more than one STAT dimer binding cooperatively to enhance the impact on transcription have been described [51, 52].
A new framework to describe alternative STAT functional modalities
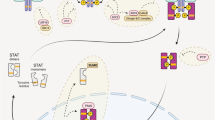
There is growing evidence that STATs can exert their effects via additional modalities distinct from the canonical mode. Collectively these alternative modalities, termed ‘non-canonical’ [16], utilize a variety of different mechanisms, including both gene repression and activation, mediated by both tyrosine phosphorylated and unphosphorylated STAT proteins, and involving roles both inside and outside the nucleus. This review provides an overarching framework to describe these modalities to facilitate a greater coherence to the literature and as a necessary prelude to defining the specific function(s) of each modality (Fig. 3).

Alternative STAT functional modalities. Schematic depiction of alternate modes by which STATs can impact on cellular functions: A inducible transcriptional activation (‘canonical’ signaling), B inducible transcriptional repression, C basal transcriptional activation, D basal transcriptional repression, E inducible non-nuclear function, F basal non-nuclear function. Shown are unphosphorylated STAT (uSTAT) molecules and their conversion into phosphorylated STAT (pSTAT) molecules and their dimerization where appropriate, as well as their movement between the cytoplasm, nucleus and other cellular compartments, along with the molecular function(s) that they exert in each case
Nuclear functions
The predominant function for STAT proteins is still regarded to be as a transcriptional regulator in the nucleus, but several variations of this have now been described. In addition to transcriptional activation, pSTATs have also been shown to mediate repression of target genes, for example STAT5 during embryonic erythropoiesis [53]. Moreover, it is now well established that uSTATs are capable of entering the nucleus to regulate gene expression through either activation or repression [16]. These can represent cytoplasmic uSTAT dimers that translocate into the nucleus, which for uSTAT3 involves importins [54], but for uSTAT1 is mediated by direct interaction with nucleoporins [55]. Alternatively, they may be uSTATs that result from dephosphorylation of nuclear pSTAT molecules [56]. Such uSTATs can bind similar, overlapping or totally distinct sites to the corresponding pSTAT [57, 58]. For example, uSTAT3 has been shown to preferentially bind with AT-rich DNA sequences and specific DNA structures, leading to the heterochromatin formation resulting in the gene silencing [59].
Inducible transcriptional activation
This represents the classical ‘canonical’ mode of signaling utilized by all STATs (Fig. 3A, Table 2), in which uSTAT proteins are dormant in the cytoplasm and then become activated by tyrosine phosphorylation in response to cytokines [1], growth factors [3], G-protein-coupled receptors [60] or other stimuli such as osmotic stress [61]. Tyrosine phosphorylation allows dimerization and subsequent translocation of pSTAT dimers into the nucleus to activate target genes via interaction with co-activators such as CREB-binding protein (CBP), p300 and NCoA-1/SRC-1 [35, 62], as well as chromatin remodelers such as BRG1 [63] until pSTAT molecules are inactivated by dephosphorylation [18, 41].
Numerous cytokines have been shown to utilize STATs in this manner leading to induction of a raft of genes involved in specific cellular functions along with proliferation, differentiation and survival [6]. For example, IFNγ signaling leads to the phosphorylation, dimerization and nuclear translocation specifically of pSTAT1 to activate various genes such as various IRF, CXCL and ISG genes and micro RNAs essential for immune responses to pathogen and tumors via so-called γ-activated sites (GAS) motifs within their promoters [18, 80, 81]. Alternatively, IL-6 induces the transcription of IL21 via pSTAT3 in T cell precursors, with the encoded IL-21 in turn inducing IL17 that is essential for Th17 differentiation also via pSTAT3 activation [66]. Similarly, IL-12 induces pSTAT4 in CD3+ T cells that induces genes such as IFNγ that impact on cell differentiation and function [82]. In addition, in the erythroid lineage erythropoietin (EPO) to induce genes such as BCL2l1 and TRAF5 via pSTAT5 in concert with KLF-1 and GATA1 to regulate erythropoiesis [68].
Inducible transcriptional repression
In this alternative modality, dormant cytoplasmic uSTAT proteins are also tyrosine phosphorylated in response to external stimuli and form pSTAT dimers that move to the nucleus and bind to specific DNA sequences, but instead actively repress transcription of certain target genes (Fig. 3B, Table 2). This is facilitated via interaction with co-repressors such as Silencing mediator for retinoic acid receptor and thyroid hormone receptor/nuclear co-repressor 2 (SMRT/Ncor2) that induce histone modification [26]. The pSTAT dimers involved would also be inactivated over time to remove the repressive effect. Numerous examples of this modality have been described. For example, IL-4-induced pSTAT6 mediates the repression of various genes involved in the alternative macrophage polarization, such as NLRP3 and IL1B, thereby inhibiting inflammasome stimulation and pyroptosis [71]. Similarly, IL-7-induced pSTAT5 binds to intronic tetrameric STAT sites in B cells to repress the transcription of the IGK gene to suppress immunoglobin gene rearrangement [70], while IL-2-induced pSTAT5 suppresses a Th17-like program during Th9 cell differentiation [83].
Basal transcriptional activation
In this functional mode, nuclear uSTAT molecules mediate the transcriptional activation of certain target genes, often through the formation of novel complexes with other transcription factors [35]. In response to external signals, the levels of pSTAT become acutely increased, which serves to indirectly deplete levels of uSTAT that results in an acute deactivation of uSTAT-activated genes (Fig. 3C, Table 2). For example, uSTAT1 can form a heterodimeric complex with interferon regulatory factor-1 (IRF1) that can bind GAS motifs to induce genes such as LMP2 that encodes a subunit of the 20S proteosome [57]. In addition, uSTAT1 has been shown to activate expression of pro-apoptotic genes, such as caspases CPP22, ICE and ICH1 necessary for TNFα-mediated apoptosis [84]. In a similar vein, uSTAT3 mediates induction of ion channels and neurotransmitter receptors in the brain [85] and can also augment the expression of select STAT1- and STAT2-responsive genes by increasing promoter accessibility [86]. However, in several cases ‘canonical’ signaling can also increase the transcription of the respective STAT gene, thereby elevating uSTAT levels that leads to sustained basal transcriptional activation over a longer time frame. For example, via a complex of uSTAT1, IRF9 and uSTAT2 resulting in prolonged expression of a subset of IFN-induced genes [18], or in the case of uSTAT3 to induce a second wave of alternate genes [72].
Basal transcriptional repression
In this modality, uSTAT molecules can repress transcription of specific target genes via interaction with co-repressors and/or chromatin modifiers, including heterochromatin protein (HP)1α that mediates heterochromatin formation [87]. In response to external signals, acute levels of these uSTAT molecules tend to decrease leading to derepression of these genes (Fig. 3D, Table 2). For example, uSTAT5 can act as a direct repressor of genes via the transcription repressor CTCF to restrain megakaryocytic differentiation. However, thrombopoietin (TPO)-stimulated STAT5 phosphorylation results in decreased levels of uSTAT5 thereby abrogating the repression of these genes [58, 74].
Non-nuclear roles
STATs have also been implicated in various non-nuclear roles, including in mitochondria, endoplasmic reticulum (ER), Golgi apparatus and cytoplasm in both unphosphorylated and phosphorylated states, the majority not involving gene regulation [17, 88].
Inducible non-nuclear functions
In this mode, dormant uSTAT is activated in response to external stimuli and moves into non-nuclear organelles to exert various biological roles (Fig. 3E, Table 2). For example, IL-6-mediated mitochondrial translocation of STAT3 has been shown to mediate cytokine expression in CD4 + T cells [75], while mitochondrial STAT3 induced by IL-21 contributed to the generation of CD8 + memory T cells and antibody production in B cells [89]. IL-2 has also been shown to induce translocation of pSTAT5 into mitochondria, where it was able to associate with the D-loop regulatory region of mtDNA, suggesting it may also participate in the regulation of mitochondrial DNA transcription [76].
Basal non-nuclear functions
In this remaining modality, uSTAT molecules exert a function outside of the nucleus (Fig. 3F, Table 2), with the impact of external signals currently unclear. For example, uSTAT1 functions at immunological synapses in NK cells to contribute to cytotoxicity [77]. Meanwhile uSTAT3 sequesters the FoxO transcription factors in the cytoplasm to prolong T cell activation, which is released by pro-inflammatory cytokines that convert it to pSTAT3 [78]. uSTAT3 has also been shown to be involved in the regulation of microtubule structural integrity in murine embryonic fibroblast cells through its antagonistic association with the cytoplasmic microtubule destabilizing protein stathmin [90]. Alternatively, uSTAT5A and uSTAT5B have been identified in the ER and Golgi apparatus in human pulmonary arterial endothelial and smooth muscle cells, where they contribute to the anterograde vesicular secretory pathway. Knockdown of STAT5A/B resulted in dilation and/or fragmentation of the ER, Golgi and mitochondria, along with effects on other organelles [79].
Implications of this framework
This framework provides a powerful lens with which to view the various studies that address STAT functions, allowing a fresh and more complete perspective of STATs in both normal biology and disease.
Reassessment of the current literature
The majority of studies to date examining STAT function have utilized ‘global’ knockout/knockdown strategies, which provide excellent information about the overall function of the individual STAT proteins [91]. However, the key limitation of such approaches is that they do not provide detail about which specific STAT signaling modality is responsible for particular phenotypic changes. More problematic, however, has been the inherent assumption in many cases that the alterations observed relate exclusively to the loss of the canonical/inducible transcriptional activation mode. Therefore, the potential role of the various non-canonical modalities should also be directly considered. This will require alternative methodological approaches using more specific gene modifications that selectively target particular modalities, as already performed in a number of breakout studies [73, 86, 92].
More comprehensive insight into STAT biology
This framework also helps understand the full gamut of responses to cytokines and growth factors, rather than just the acute effects. Perhaps the best documented examples relate to interferon signaling where our understanding is unparalleled [93]. Thus, it is known that in the absence of IFNs, uSTAT-containing complexes facilitate low basal expression of a core set of genes [94]. IFN signaling then induces high level, but transient canonical pSTAT-mediated expression of various target genes that serve to inhibit growth and promote apoptosis and immune surveillance genes [94]. However, the targets also include STAT1, STAT2 and IRF9 [95]. A few hours after IFN stimulation, pSTAT levels decline, but those of uSTAT become increased as a combined consequence of pSTAT de-phosphorylation and increased STAT gene expression, allowing for a sustained impact on the transcription of a different set of immune genes that elicit long-lived antiviral and immune responses [18]. Similarly, it has been shown that IL-6 can increase STAT3 expression in fibroblasts (through canonical signaling via pSTAT3), which ultimately results in increased levels of uSTAT3 that drives the expression of late phase genes such as CDC2, CCNB1 and E2F11 [72, 73]. Finally, with respect to megakaryocyte differentiation, it has been demonstrated that in the absence of TPO uSTAT5 blocks differentiation by repressing the genes responsible for this process, including MPL and FTR. However, TPO-stimulated pSTAT5 phosphorylation results in decreased levels of uSTAT5 and so the repression of these genes is removed in concert with the induction of genes by pSTAT5 promoting proliferation and survival [58, 74].
More nuanced understanding of relevant human diseases and their potential treatment
STAT proteins have been widely implicated in human diseases, particularly immune deficiencies, inflammatory diseases, cancers and other proliferative disorders [96,97,98]. However, the current understanding of disease etiology has primarily focused on impacts to canonical signaling. This framework allows a recalibration of STAT-mediated diseases such that the full gamut of non-canonical functionalities can also be considered (Table 3).
This is not to say that canonical STAT signaling resulting in inducible gene activation is not important in disease. Indeed in numerous cancers and proliferative disorders, STAT proteins have been observed to be constitutively phosphorylated as a result of mutation of upstream activators, or through excessive signaling associated with inflammation, infection or other pathologies [117,118,119,120]. This is typically associated with an increase in the expression of genes usually induced through the canonical pathway. For example, in various human cancers STAT3 induces genes that permit sustained proliferation, such as those encoding cyclin D1 (CCND1) [103] and telomerase (TERT) [104]. However, there is also evidence for inducible transcriptional repression. For example, IL-2-mediated pSTAT3 represses p53 to enhance survival of chronic lymphocytic leukemia cells [111]. Similarly, in mammary epithelial cells prolactin-mediated pSTAT5A results in repression of the BCL6A gene through binding to tetrameric STAT sites within the 5’ untranslated exon of this gene to decrease breast cancer tumorigenesis [121].
Unphosphorylated STATs also play a role in disease. This can involve basal transcriptional activation. For example, in non-small cell lung cancer uSTAT6 upregulates the cyclooxygenase 2 gene (PTGS2) through a consensus STAT6 site to provide protection against apoptosis [114]. One of the targets of canonical STAT3 signaling is the STAT3 gene itself, which serves to increase the levels of uSTAT3 as well over the longer term, resulting in chronic pSTAT3-mediated transcriptional effects in concert with additional uSTAT3-mediated regulation. In lung, head and neck cancers, this includes genes involved in cell cycle progression, such as CCNB1, E2F11 and CDC7 [73]. Importantly, genes not typically regulated by pSTAT3 can be affected, such as those encoding cytokines (IL6, IL8, RANTES) and oncoproteins (MRAS, MET) in hTERT-HME1 cells. Expression of these genes was shown to be mediated by a novel transcription complex formed when uSTAT3 binds unphosphorylated NFκβ (uNFκβ) [72]. Similarly, in colon cancer, Jun activation domain-binding protein 1 (Jab1) regulates uSTAT3 DNA binding and expression of VEGF, MDR1 and NANOG [122]. However, the basal repression modality of uSTATs is also important. For example, uSTAT3 and uSTAT5A have been shown to interact with HP-1α, promoting the formation of heterochromatin which contributes to gene silencing that suppresses growth of colon and lung cancer cells, respectively [123, 124].
In addition, non-nuclear roles have been implicated in disease, including cancer, which can be inducible or basal. For example, pSTAT5 induced by BCR-ABL, JAK2V617F or KITD816V in leukemic cells has been shown to interact with the scaffold adaptor Gab2 in the cytoplasm to mediate activation of the phosphatidylinositol-3 kinases (PI3K)/AKT pathway to facilitate cell survival [116]. Alternatively, mitochondrial STAT5 and has been found to interact with the E2 subunit of the pyruvate dehydrogenase complex to regulate metabolism in leukemic T cells [76]. In leukemic pre-B cells uSTAT5 protects against oxidative damage independent of transcriptional changes [51]. The pathogenesis of pulmonary arterial hypertension has also been proposed to be mediated by non-nuclear functions of STAT3 [125] and STAT5 [17], which appear to impact on the stability of the ER and Golgi body [79].
These examples clearly indicate that relevant diseases involve both canonical and non-canonical modalities. This knowledge provides critical insights for the design and application of therapeutic agents targeting STAT proteins. This is particularly important since most of the developed pharmacological inhibitors target either upstream canonical signaling components such as the kinase activity of JAKs or the SH2 domains of STATs [121] (Table 4). However, these drugs may not be effective on the non-canonical pathways. Moreover, there is a possibility for negative consequences, with inhibition of pSTAT potentially resulting in accumulation of uSTAT that may exert unintended and potentially deleterious effects.
Additional STAT modifications
The framework presented has focused solely on tyrosine phosphorylation and in the context of full-length STAT proteins. However, a range of other modifications of STAT proteins are possible. These are best viewed as mechanisms to further fine tune the STAT response, generally serving to modulate a particular functional modality, for example, phosphorylation of a critical serine residue in the C-terminal domain of STAT1, STAT3, STAT5A and STAT5B, such as STAT5 by IL-2 [126]. This has been shown to positively impact on canonical STAT-mediated gene transcription, such as that induced by hypothalamic STAT3 [127] and mammary STAT5 [128]. For STAT1, this has been shown to be through increased association with histone acetylase complexes [129]. Serine phosphorylation plays a particularly important role in oncogenic STAT signaling [6, 130]. For example, serine phosphorylation of STAT5 was required for BCR-ABL-induced leukemogenesis [123], and phosphorylation of S727 on STAT3 augmented the induction of genes involve in the cellular growth and survival that contributed to progression of chronic lymphocytic leukemia [131]. However, serine phosphorylation can also mediate translocation of STAT3 molecules into mitochondria, during RAS-dependent oncogenic transformation, where it served to negatively regulate the activity of electron transport chain components complex I and complex II by a mechanism that did not require DNA binding or tyrosine phosphorylation [132]. In mammary tumors, pSTAT3 has also been shown to control ER Ca2+ flux via interacting inositol 1,4,5-triphosphate receptor (IP3R3) results proteasomal degradation of IP3R3 and inhibits oxidative/ER stress and apoptosis [133].
In addition, acetylation of lysine residues in STAT3 (K685) and STAT5 (K694) by histone acetylases has been described, such in response to cytokines such as IL-6 and prolactin, respectively [134]. Acetylation differentially regulates STATs, impacting on transcriptional activation and protein stability [135]. Acetylation at K685 in uSTAT3 is important in the formation of a stable dimer and its accumulation in the nucleus to regulate target genes [136], with STAT3 shown to be constitutively acetylated at this site in CLL [137]. Contrastingly, SUMOylation of alternative lysine resides, such as K696 in the STAT5A, can antagonize the effect of acetylation and negatively impact canonical STAT signaling through decreased tyrosine phosphorylation [18, 138, 139]. Other chemical modifications have also been reported, including methylation, oxidation and glutathionylation [140,141,142], but their role in normal STAT biology remains speculative. Finally, truncated versions of several STATs have been described, from either alternative splicing or proteolysis. In general, these are phosphorylated, dimerize, translocate into the nucleus and bind to the putative DNA-binding site in response to cytokines and growth factors. However, instead of transcriptional activation, they often exert a dominant-negative effect through blocking access of transactivation competent STATs and have been also implicated in disease [143,144,145].
Evolutionary origins of multiple STAT functional modalities
A key question regarding the multiple STAT functionalities is whether they represent specific ad hoc innovations or instead have deeper evolutionary origins. This is particularly relevant since STATs have a long evolutionary history, predating cytokine receptors and JAKs [146]. Moreover, investigations of STATs in extant primitive species have identified both canonical and non-canonical signaling modalities [147].
Evidence of ‘canonical’ inducible transcriptional activation is evident throughout metazoans, including invertebrates such as Drosophila melanogaster. This organism possesses a single STAT known as Stat92E (or Marelle), with a similar structure to mammalian STAT proteins [148, 149]. This is activated downstream of the single Drosophila cytokine receptor (Dome) and JAK (Hopscotch) [90], with pStat92E molecules inducing genes associated with immunity and development [150].
However, STAT proteins evolved prior to cytokine receptors and JAKs [146]. For example, the nematode C. elegans possesses two STAT-like proteins, STA-1 and STA-2, but no upstream cytokine receptor signaling components [116]. Indeed, STA-2 lacks the tyrosine phosphorylation motif as well as the coiled-coil domain but can act as an inducible transcriptional activator [116, 151]. This is achieved by an alternative mechanism in which epidermal injuries mediates its release from hemidesmosomes allowing it to move to the nucleus to induce the transcription of genes associated with innate immunity [152]. More primitive eukaryotes, such as the slime mold Dictyostelium discoideum, also possess STAT-like proteins—again in the absence of upstream cytokine receptor signaling components [147]. D. discoideum has four STAT proteins, STATa, STATb, STATc and STATd, composed of coiled-coil, DNA-binding and SH2 domains, but without transactivation domain and N-terminal sequences. STATa is tyrosine phosphorylated in response to extracellular cyclic AMP (cAMP) through the cyclic AMP receptor (cAR1) and translocate into the nucleus to activate genes such as cudA, thereby acting as an inducible transcriptional activator [153]. STATc can also become tyrosine phosphorylated and activate expression of genes such as gapA and rtoA in response to hyperosmotic and other stressors [154]. Finally, plants have also been shown to possess STAT-related GRAS proteins, with highly similar SH2 and DNA-binding domains to other STATs [155], which can act as transcriptional activators to regulate plant development [156, 157].
There is considerable evidence of non-canonical signaling in these organisms as well. For example, Drosophila Stat92E has been shown to participate in basal transcriptional repression, with uStat92E able to enter the nucleus and target alternate DNA sites, particularly those involving metabolic and stress-related pathways [150]. This is achieved via an evolutionarily conserved association with HP1 that induces histone methylation and heterochromatin stabilization resulting in gene repression that can enhance genome stability [79] and suppress tumor growth [158]. Dome signaling is able to mediate derepression of these target genes [125]. C. elegans STA-1, which shares a similar structure to mammalian STATs but lacks a TAD, also acts as a basal repressor, inhibiting transcription of antiviral genes in the absence of the viral infection [159]. D. discoideum STATa similarly functions as an transcriptional repressor of alternative genes such as ecmB [153], and serine phosphorylation is also important in the regulation of this STAT, being able to increase its nuclear export [160]. D. discoideum STATc can also repress other genes such as ecmA, by preventing activator binding [161]. Finally, plant GRAS proteins can function as transcriptional repressors [156, 157].
Collectively these studies imply that both canonical and non-canonical STATs signaling modalities were present early in evolution and have simply been maintained (and further diversified) along the evolutionary path to higher vertebrates, including mammals. Therefore, the framework provided is applicable across all STAT proteins. It should be noted, however, that the classical ‘canonical’ signaling paradigm can best be viewed as a relatively recent innovation within metazoans, coincident with the coalescence of a functional cytokine receptor signaling module in the common precursor of vertebrates and invertebrates [162].
Conclusion
Our knowledge of STAT proteins has progressed from a simple view of them mediating acute cytokine-mediated transcriptional responses to one where they are involved in a veritable kaleidoscope of functions, many of which impact on immunity and cancer. Notably, many of these modalities are evident in STAT proteins of divergent organisms, suggesting they have early evolutionary origins. However, there remains a gap in knowledge regarding the full breadth of roles mediated by the non-canonical modalities in physiology and disease. This review provides a framework to view these functional modalities that should contribute to filling this gap and provide new insights that will better inform precise therapeutic interventions that target the appropriate modality in disease states.
Availability of data and materials
Not applicable.
Abbreviations
- CBP:
-
CREB-binding protein
- CCD:
-
Coiled-coil domain
- DBD:
-
DNA-binding domain
- EPO:
-
Erythropoietin
- ER:
-
Endoplasmic reticulum
- GAS:
-
Gamma-activated sequence
- GCN:
-
General control of amino acid synthesis protein
- HP:
-
Heterochromatin protein
- IRF:
-
Interferon regulatory factor
- JAKs:
-
Janus kinase
- LD:
-
Linker domain
- N-CoR:
-
Nuclear receptor co-repressor
- NLS:
-
Nuclear localization signal
- Nmi:
-
N-Myc and STAT interactor
- NTD:
-
N-terminal domain (NTD)
- PAK:
-
P21-activated kinase
- PDC:
-
Pyruvate dehydrogenase complex
- PIAS:
-
Protein inhibitor of activated STAT
- pSTAT:
-
Phosphorylated STAT
- PTP:
-
Protein tyrosine phosphatases
- SH2:
-
Src homology-2
- SMRT:
-
Silencing mediator for retinoic acid receptor and thyroid hormone receptor
- SOCS:
-
Suppressor of cytokine signaling
- STAT:
-
Signal transducer and activators of transcription
- TAD:
-
Transactivation domain
- TPO:
-
Thrombopoietin
- uSTAT:
-
Unphosphorylated STAT
References
Shuai K, Stark GR, Kerr IM, Darnell JE. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science. 1993;261(5129):1744–6.
Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21.
Zhong Z, Wen Z, Darnell JE. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264(5155):95–8.
Ram PA, Park S-H, Choi HK, Waxman DJ. Growth hormone activation of Stat1, Stat3, and Stat5 in rat liver: differential kinetics of hormone desensitization and growth hormone stimulation of both tyrosine phosphorylation and serine/threonine phoshorylation. J Biol Chem. 1996;271(10):5929–40.
Amiri F, Shaw S, Wang X, Tang J, Waller JL, Eaton DC, et al. Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int. 2002;61(5):1605–16.
Levy DE, Darnell JE. STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–62.
Au-Yeung N, Mandhana R, Horvath CM. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. JAKSTAT. 2013;2(3):e23931-1-8.
Rauch I, Muller M, Decker T. The regulation of inflammation by interferons and their STATs. JAKSTAT. 2013;2:e23820.
Hillmer EJ, Zhang H, Li HS, Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016;31:1–15.
Hirano T. IL-6 in inflammation, autoimmunity and cancer. Int Immunol. 2020;33(3):127–48.
Kaplan MH. STAT4. Immunol Res. 2005;31(3):231–41.
Keeter WC, Moriarty A, Butcher MJ, Ma KW, Nadler JL, Galkina E. IL-12 induced STAT4 activation plays a role in pro-inflammatory neutrophil functions. J Immunol. 2018;200:166–259.
Rani A, Murphy JJ. STAT5 in cancer and immunity. J Interferon Cytokine Res. 2016;36(4):226–37.
Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunol Res. 2011;50(1):87–96.
Wang W, Wang L, Zha B. The roles of STAT6 in regulating B cell fate, activation, and function. Immunol Lett. 2021;233:87–91.
Affolter M, Pyrowolakis G, Weiss A, Basler K. Signal-induced repression: the exception or the rule in developmental signaling? Dev Cell. 2008;15(1):11–22.
Sehgal PB. Non-genomic STAT5-dependent effects at the endoplasmic reticulum and Golgi apparatus and STAT6-GFP in mitochondria. JAKSTAT. 2013;2(4):e24860.
Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc Natl Acad Sci U S A. 2009;106(23):9373–8.
Copeland NG, Gilbert DJ, Schindler C, Zhong Z, Wen Z, Darnell JE, et al. Distribution of the mammalian Stat gene family in mouse chromosomes. Genomics. 1995;29(1):225–8.
Heim MH. The STAT protein family. In: Sehgal PB, Levy DE, Hirano T, editors. Signal transducers and activators of transcription (STATs). Berlin: Springer; 2003. p. 11–26.
Kisseleva T, Bhattacharya S, Braunstein J, Schindler C. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285(1–2):1–24.
Lim CP, Cao X. Structure, function, and regulation of STAT proteins. Mol Biosyst. 2006;2(11):536–50.
Strehlow I, Schindler C. Amino-terminal signal transducer and activator of transcription (STAT) domains regulate nuclear translocation and STAT deactivation. J Biol Chem. 1998;273(43):28049–56.
Vinkemeier U, Moarefi I, Darnell JE, Kuriyan J. Structure of the amino-terminal protein interaction domain of STAT-4. Science. 1998;279(5353):1048–52.
Zhu M-h, John S, Berg M, Leonard WJ. Functional association of Nmi with Stat5 and Stat1 in IL-2-and IFN γ-mediated signaling. Cell. 1999;96(1):121–30.
Nakajima H, Brindle PK, Handa M, Ihle JN. Functional interaction of STAT5 and nuclear receptor co-repressor SMRT: implications in negative regulation of STAT5-dependent transcription. EMBO J. 2001;20(23):6836–44.
Brown S, Hu N, Hombría JC-G. Novel level of signalling control in the JAK/STAT pathway revealed by in situ visualisation of protein-protein interaction during Drosophila development. Development. 2003;130(14):3077–84.
Horvath CM, Wen Z, Darnell J. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 1995;9(8):984–94.
Decker T, Kovarik P, Meinke A. GAS elements: A few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res. 1997;17(3):121–34.
Yang E, Wen Z, Haspel RL, Zhang JJ, Darnell JE Jr. The linker domain of Stat1 is required for gamma interferon-driven transcription. Mol Cell Biol. 1999;19(7):5106–12.
Liu KD, Gaffen SL, Goldsmith MA. JAK/STAT signaling by cytokine receptors. Curr Opin Immunol. 1998;10(3):271–8.
Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D’Andrea A, et al. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-α. Nature. 1996;383(6598):344–7.
Paulson M, Press C, Smith E, Tanese N, Levy DE. IFN-Stimulated transcription through a TBP-free acetyltransferase complex escapes viral shutoff. Nat Cell Biol. 2002;4(2):140–7.
Huang M, Qian F, Hu Y, Ang C, Li Z, Wen Z. Chromatin-remodelling factor BRG1 selectively activates a subset of interferon-α-inducible genes. Nat Cell Biol. 2002;4(10):774–81.
Shuai K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene. 2000;19(21):2638–44.
Yamaoka K, Saharinen P, Pesu M, Holt VET, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks). Genome Biol. 2004;5(12):253–9.
Babon JJ, Lucet IS, Murphy JM, Nicola NA, Varghese LN. The molecular regulation of Janus kinase (JAK) activation. Biochem J. 2014;462(1):1–13.
Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178(5):2623–9.
Kawashima T, Bao YC, Nomura Y, Moon Y, Tonozuka Y, Minoshima Y, et al. Rac1 and a GTPase-activating protein, MgcRacGAP, are required for nuclear translocation of STAT transcription factors. Mol Cell Biol. 2006;175(6):937–46.
Köster M, Hauser H. Dynamic redistribution of STAT1 protein in IFN signaling visualized by GFP fusion proteins. Eur J Biochem. 1999;260(1):137–44.
Haspel RL, Darnell JE. A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc Natl Acad Sci USA. 1999;96(18):10188–93.
Kashiwada M, Giallourakis CC, Pan P-Y, Rothman PB. Immunoreceptor tyrosine-based inhibitory motif of the IL-4 receptor associates with SH2-containing phosphatases and regulates IL-4-induced proliferation. J Immunol. 2001;167(11):6382–7.
Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci. 2000;113(16):2813–9.
Sobah ML, Liongue C, Ward AC. SOCS proteins in immunity, inflammatory diseases, and immune-related cancer. Front Med. 2021;8:1532.
Qureshi SA, Salditt-Georgieff M, Darnell JE. Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3. Proc Natl Acad Sci USA. 1995;92(9):3829–33.
Levy DE, Kessler D, Pine R, Darnell J. Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. 1989;3(9):1362–71.
Finbloom DS, Winestock KD. IL-10 induces the tyrosine phosphorylation of Tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J Immunol. 1995;155(3):1079–90.
Novak U, Mui A, Miyajima A, Paradiso L. Formation of STAT5-containing DNA binding complexes in response to colony-stimulating factor-1 and platelet-derived growth factor. J Biol Chem. 1996;271(31):18350–4.
Delgoffe GM, Vignali DAA. STAT heterodimers in immunity: A mixed message or a unique signal? JAKSTAT. 2013;2(1):e23060.
Bluyssen HA, Muzaffar R, Vlieststra RJ, van der Made AC, Leung S, Stark GR, et al. Combinatorial association and abundance of components of interferon-stimulated gene factor 3 dictate the selectivity of interferon responses. Proc Natl Acad Sci USA. 1995;92(12):5645–9.
Cholez E, Debuysscher V, Bourgeais J, Boudot C, Leprince J, Tron F, et al. Evidence for a protective role of the STAT5 transcription factor against oxidative stress in human leukemic pre-B cells. Leukemia. 2012;26(11):2390–7.
Soldaini E, John S, Moro S, Bollenbacher J, Schindler U, Leonard WJ. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol. 2000;20(1):389–401.
Schmerer M, Torregroza I, Pascal A, Umbhauer M, Evans T. STAT5 acts as a repressor to regulate early embryonic erythropoiesis. Blood. 2006;108(9):2989–97.
Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-α3. Proc Natl Acad Sci USA. 2005;102(23):8150–5.
Marg A, Shan Y, Meyer T, Meissner T, Brandenburg M, Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J Cell Biol. 2004;165(6):823–33.
Meyer T, Marg A, Lemke P, Wiesner B, Vinkemeier U. DNA binding controls inactivation and nuclear accumulation of the transcription factor Stat1. Genes Dev. 2003;17(16):1992–2005.
Chatterjee-Kishore M, Wright KL, Ting JP-Y, Stark GR. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000;19(15):4111–22.
Park HJ, Li J, Hannah R, Biddie S, Leal-Cervantes AI, Kirschner K, et al. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2016;35(6):580–94.
Timofeeva OA, Chasovskikh S, Lonskaya I, Tarasova NI, Khavrutskii L, Tarasov SG, et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J Biol Chem. 2012;287(17):14192–200.
Pelletier S, Duhamel F, Coulombe P, Popoff MR, Meloche S. Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol Cell Biol. 2003;23(4):1316–33.
Gatsios P, Terstegen L, Schliess F, Häussinger D, Kerr IM, Heinrich PC, et al. Activation of the Janus kinase/signal transducer and activator of transcription pathway by osmotic shock. J Biol Chem. 1998;273(36):22962–8.
Litterst CM, Kliem S, Marilley D, Pfitzner E. NCoA-1/SRC-1 is an essential coactivator of STAT5 that binds to the FDL motif in the alpha-helical region of the STAT5 transactivation domain. J Biol Chem. 2003;278(46):45340–51.
Christova R, Jones T, Wu P-J, Bolzer A, Costa-Pereira AP, Watling D, et al. P-STAT1 mediates higher-order chromatin remodelling of the human MHC in response to IFNγ. J Cell Sci. 2007;120(18):3262–70.
Jung SR, Ashhurst TM, West PK, Viengkhou B, King NJ, Campbell IL, et al. Contribution of STAT1 to innate and adaptive immunity during type I interferon-mediated lethal virus infection. PLoS Pathog. 2020;16(4):e1008525.
Hambleton S, Goodbourn S, Young DF, Dickinson P, Mohamad SMB, Valappil M, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci USA. 2013;110(8):3053–8.
Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8(9):967–74.
Dong X, Antao OQ, Song W, Sanchez GM, Zembrzuski K, Koumpouras F, et al. Type I interferon–activated STAT4 regulation of follicular helper T cell–dependent cytokine and immunoglobulin production in Lupus. Arthritis Rheum. 2021;73(3):478–89.
Gillinder KR, Tuckey H, Bell CC, Magor GW, Huang S, Ilsley MD, et al. Direct targets of pSTAT5 signalling in erythropoiesis. PLoS ONE. 2017;12(7):e0180922.
Chen Z, Lund R, Aittokallio T, Kosonen M, Nevalainen O, Lahesmaa R. Identification of novel IL-4/Stat6-regulated genes in T lymphocytes. J Immunol. 2003;171(7):3627–35.
Mandal M, Powers SE, Maienschein-Cline M, Bartom ET, Hamel KM, Kee BL, et al. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat Immunol. 2011;12(12):1212–20.
Czimmerer Z, Daniel B, Horvath A, Rückerl D, Nagy G, Kiss M, et al. The transcription factor STAT6 mediates direct repression of inflammatory enhancers and limits activation of alternatively polarized macrophages. Immunity. 2018;48(1):75–90.
Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007;21(11):1396–408.
Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65(3):939–47.
Decker T. Emancipation from transcriptional latency: unphosphorylated STAT5 as guardian of hematopoietic differentiation. EMBO J. 2016;35(6):555–7.
Yang R, Lirussi D, Thornton TM, Jelley-Gibbs DM, Diehl SA, Case LK, et al. Mitochondrial Ca2+ and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. Elife. 2015;4:e06376.
Chueh F-Y, Leong K-F, Yu C-L. Mitochondrial translocation of signal transducer and activator of transcription 5 (STAT5) in leukemic T cells and cytokine-stimulated cells. Biochem Biophys Res Commun. 2010;402(4):778–83.
Putz EM, Majoros A, Gotthardt D, Prchal-Murphy M, Zebedin-Brandl EM, Fux DA, et al. Novel non-canonical role of STAT1 in natural killer cell cytotoxicity. Oncoimmunology. 2016;5(9):e1186314–24.
Oh H-M, Yu C-R, Dambuza I, Marrero B, Egwuagu CE. STAT3 protein interacts with Class O Forkhead transcription factors in the cytoplasm and regulates nuclear/cytoplasmic localization of FoxO1 and FoxO3a proteins in CD4+ T cells. J Biol Chem. 2012;287(36):30436–43.
Lee JE, Yang Y-M, Liang F-X, Gough DJ, Levy DE, Sehgal PB. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol. 2012;302(5):C804–20.
Schmitt MJ, Philippidou D, Reinsbach SE, Margue C, Wienecke-Baldacchino A, Nashan D, et al. Interferon-γ-induced activation of signal transducer and activator of transcription 1 (STAT1) up-regulates the tumor suppressing microRNA-29 family in melanoma cells. Cell Commun Signal. 2012;10(1):1–14.
Satoh J-i, Kawana N, Yamamoto Y. Pathway analysis of ChIP-Seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul Syst Biol. 2013;7:GRSB.S13204.
Bacon CM, Petricoin EF, Ortaldo JR, Rees RC, Larner AC, Johnston JA, et al. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci USA. 1995;92(16):7307–11.
Canaria DA, Yan B, Clare MG, Zhang Z, Taylor GA, Boone DL, et al. STAT5 Represses a STAT3-independent Th17-like program during Th9 cell differentiation. J Immunol. 2021;207(5):1265–74.
Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR. Defective TNF-α-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science. 1997;278(5343):1630–2.
Hixson KM, Cogswell M, Brooks-Kayal AR, Russek SJ. Evidence for a non-canonical JAK/STAT signaling pathway in the synthesis of the brain’s major ion channels and neurotransmitter receptors. BMC Genomics. 2019;20(1):1–16.
Pfeffer SR, Fan M, Du Z, Yang CH, Pfeffer LM. Unphosphorylated STAT3 regulates the antiproliferative, antiviral, and gene-inducing actions of type I interferons. Biochem Biophys Res Commun. 2017;490(3):739–45.
Hu X, Dutta P, Tsurumi A, Li J, Wang J, Land H, et al. Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc Natl Acad Sci USA. 2013;110(25):10213–8.
Meier JA, Larner AC. Toward a new STATe: The role of STATs in mitochondrial function. Semin Immunol. 2014;26(1):20–8.
Rincon M, Pereira FV. A new perspective: mitochondrial Stat3 as a regulator for lymphocyte function. Int J Mol Sci. 2018;19(6):1656-1-24.
Arbouzova NI, Zeidler MP. JAK/STAT signalling in Drosophila: insights into conserved regulatory and cellular functions. Development. 2006;133(14):2605–16.
Akira S. Functional roles of STAT family proteins: lessons from knockout mice. Stem Cells. 1999;17(3):138–46.
Majoros A, Platanitis E, Szappanos D, Cheon H, Vogl C, Shukla P, et al. Response to interferons and antibacterial innate immunity in the absence of tyrosine-phosphorylated STAT1. EMBO Rep. 2016;17(3):367–82.
Majoros A, Platanitis E, Kernbauer-Hölzl E, Rosebrock F, Müller M, Decker T. Canonical and non-canonical aspects of JAK–STAT signaling: lessons from lnterferons for cytokine responses. Front Immunol. 2017;8:29.
Michalska A, Blaszczyk K, Wesoly J, Bluyssen HAR. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front Immunol. 2018;9:1135.
Lehtonen A, Matikainen S, Julkunen I. Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J Immunol. 1997;159(2):794–803.
Mogensen TH. IRF and STAT transcription factors—from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front Immunol. 2019;9(3047):1–13.
Recio C, Guerra B, Guerra-Rodriguez M, Aranda-Tavio H, Martin-Rodriguez P, de Mirecki-Garrido M, et al. Signal transducer and activator of transcription (STAT)-5: An opportunity for drug development in oncohematology. Oncogene. 2019;38(24):4657–68.
Salas A, Hernandez-Rocha C, Duijvestein M, Faubion W, McGovern D, Vermeire S, et al. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17(6):323–37.
Lorenzini T, Dotta L, Giacomelli M, Vairo D, Badolato R. STAT mutations as program switchers: turning primary immunodeficiencies into autoimmune diseases. J Leuk Biol. 2017;101(1):29–38.
Consonni F, Dotta L, Todaro F, Vairo D, Badolato R. Signal transducer and activator of transcription gain-of-function primary immunodeficiency/immunodysregulation disorders. Curr Opin Pediatr. 2017;29(6):711–7.
Duncan CJA, Hambleton S. Human disease phenotypes associated with loss and gain of function mutations in STAT2: viral susceptibility and type I interferonopathy. J Clin Immunol. 2021;41(7):1446–56.
Shahmarvand N, Nagy A, Shahryari J, Ohgami RS. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 2018;109(4):926–33.
Leslie K, Lang C, Devgan G, Azare J, Berishaj M, Gerald W, et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated Signal Transducer and Activator of Transcription 3. Cancer Res. 2006;66(5):2544.
Konnikova L, Simeone MC, Kruger MM, Kotecki M, Cochran BH. Signal Transducer and Activator of Transcription 3 (STAT3) regulates human telomerase reverse transcriptase (hTERT) expression in human cancer and primary cells. Cancer Res. 2005;65(15):6516.
Polak KL, Chernosky NM, Smigiel JM, Tamagno I, Jackson MW. Balancing STAT activity as a therapeutic strategy. Cancers. 2019;11(11):1716.
Gallucci S, Meka S, Gamero AM. Abnormalities of the type I interferon signaling pathway in lupus autoimmunity. Cytokine. 2021;146:155633.
Barbara M, Harini N, Bettina W, Ha Thi Thanh P, Michaela S, Tobias S, et al. High activation of STAT5A drives peripheral T-cell lymphoma and leukemia. Haematologica. 2020;105(2):435–47.
Maurer B, Kollmann S, Pickem J, Hoelbl-Kovacic A, Sexl V. STAT5A and STAT5B—twins with different personalities in hematopoiesis and leukemia. Cancers. 2019;11(11):1726.
Zhang Y, Molavi O, Su M, Lai R. The clinical and biological significance of STAT1 in esophageal squamous cell carcinoma. BMC Cancer. 2014;14(1):1–14.
Zhang Y, Liu Z. STAT1 in cancer: friend or foe? Discov Med. 2017;24(130):19–29.
Sainz-Perez A, Gary-Gouy H, Gaudin F, Maarof G, Marfaing-Koka A, de Revel T, et al. IL-24 induces apoptosis of chronic lymphocytic leukemia B cells engaged into the cell cycle through dephosphorylation of STAT3 and stabilization of p53 expression. J Immunol. 2008;181(9):6051–60.
Walker SR, Nelson EA, Frank DA. STAT5 represses BCL6 expression by binding to a regulatory region frequently mutated in lymphomas. Oncogene. 2007;26(2):224–33.
Pasqualucci L, Migliazza A, Basso K, Houldsworth J, Chaganti RSK, Dalla-Favera R. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood. 2003;101(8):2914–23.
Cui X, Zhang L, Luo J, Rajasekaran A, Hazra S, Cacalano N, et al. Unphosphorylated STAT6 contributes to constitutive cyclooxygenase-2 expression in human non-small cell lung cancer. Oncogene. 2007;26(29):4253–60.
Shiota G, Okubo M, Noumi T, Noguchi N, Oyama K, Takano Y, et al. Cyclooxygenase-2 expression in hepatocellular carcinoma. Hepatogastroenterology. 1999;46(25):407–12.
Harir N, Pecquet C, Kerenyi M, Sonneck K, Kovacic B, Nyga R, et al. Constitutive activation of Stat5 promotes its cytoplasmic localization and association with PI3-kinase in myeloid leukemias. Blood. 2006;109(4):1678–86.
Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77(5):521–46.
Lu R, Zhang Y-g, Sun J. STAT3 activation in infection and infection-associated cancer. Mol Cell Endocrinol. 2017;451:80–7.
Olbrich P, Freeman AF. STAT1 and STAT3 mutations: important lessons for clinical immunologists. Expert Rev Clin Immunol. 2018;14(12):1029–41.
Hammarén HM, Virtanen AT, Raivola J, Silvennoinen O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine. 2019;118:48–63.
Brachet-Botineau M, Polomski M, Neubauer HA, Juen L, Hédou D, Viaud-Massuard MC, et al. Pharmacological inhibition of oncogenic STAT3 and STAT5 signaling in hematopoietic cancers. Cancers (Basel). 2020;12(1):240.
Nishimoto A, Kugimiya N, Hosoyama T, Enoki T, Li T-S, Hamano K. JAB1 regulates unphosphorylated STAT3 DNA-binding activity through protein–protein interaction in human colon cancer cells. Biochem Biophys Res Commun. 2013;438(3):513–8.
Berger A, Hoelbl-Kovacic A, Bourgeais J, Hoefling L, Warsch W, Grundschober E, et al. PAK-dependent STAT5 serine phosphorylation is required for BCR-ABL-induced leukemogenesis. Leukemia. 2014;28(3):629–41.
Dutta P, Zhang L, Zhang H, Peng Q, Montgrain PR, Wang Y, et al. Unphosphorylated STAT3 in heterochromatin formation and tumor suppression in lung cancer. BMC Cancer. 2020;20(1):1–10.
Mukhopadhyay S, Shah M, Xu F, Patel K, Tuder RM, Sehgal PB. Cytoplasmic provenance of STAT3 and PY-STAT3 in the endolysosomal compartments in pulmonary arterial endothelial and smooth muscle cells: implications in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;294(3):L449–68.
Xue HH, Fink DW Jr, Zhang X, Qin J, Turck CW, Leonard WJ. Serine phosphorylation of Stat5 proteins in lymphocytes stimulated with IL-2. Int Immunol. 2002;14(11):1263–71.
Breit A, Besik V, Solinski HJ, Muehlich S, Glas E, Yarwood SJ, et al. Serine-727 phosphorylation activates hypothalamic STAT-3 independently from tyrosine-705 phosphorylation. J Mol Endocrinol. 2015;29(3):445–59.
Beuvink I, Hess D, Flotow H, Hofsteenge J, Groner B, Hynes NE. Stat5a serine phosphorylation. Serine 779 is constitutively phosphorylated in the mammary gland, and serine 725 phosphorylation influences prolactin-stimulated in vitro DNA binding activity. J Biol Chem. 2000;275(14):10247–55.
Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Müller M, et al. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-gamma-dependent innate immunity. Immunity. 2003;19(6):793–802.
Friedbichler K, Kerenyi MA, Kovacic B, Li G, Hoelbl A, Yahiaoui S, et al. Stat5a serine 725 and 779 phosphorylation is a prerequisite for hematopoietic transformation. Blood. 2010;116(9):1548–58.
Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, et al. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood. 2010;115(14):2852–63.
Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324(5935):1713–6.
Avalle L, Camporeale A, Morciano G, Caroccia N, Ghetti E, Orecchia V, et al. STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca2+ fluxes and apoptotic responses. Cell Death Differ. 2019;26(5):932–42.
Wieczorek M, Ginter T, Brand P, Heinzel T, Krämer OH. Acetylation modulates the STAT signaling code. Cytokine Growth Factor Rev. 2012;23(6):293–305.
Zhuang S. Regulation of STAT signaling by acetylation. Cell Signal. 2013;25(9):1924–31.
Dasgupta M, Unal H, Willard B, Yang J, Karnik SS, Stark GR. Critical role for lysine 685 in gene expression mediated by transcription factor unphosphorylated STAT3. J Biol Chem. 2014;289(44):30763–71.
Rozovski U, Harris DM, Li P, Liu Z, Jain P, Ferrajoli A, et al. STAT3 is constitutively acetylated on lysine 685 residues in chronic lymphocytic leukemia cells. Oncotarget. 2018;9(72):33710–8.
Krämer OH, Moriggl R. Acetylation and sumoylation control STAT5 activation antagonistically. JAKSTAT. 2012;1(3):203–7.
Begitt A, Droescher M, Knobeloch K-P, Vinkemeier U. SUMO conjugation of STAT1 protects cells from hyperresponsiveness to IFNγ. Blood. 2011;118(4):1002–7.
Li L, Cheung S-h, Evans EL, Shaw PE. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res. 2010;70(20):8222.
Xie Y, Kole S, Precht P, Pazin MJ, Bernier M. S-Glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology. 2009;150(3):1122–31.
Kim E, Kim M, Woo D-H, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23(6):839–52.
Moriggl R, Gouilleux-Gruart V, Jähne R, Berchtold S, Gartmann C, Liu X, et al. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol Cell Biol. 1996;16(10):5691–700.
Shao H, Quintero AJ, Tweardy DJ. Identification and characterization of cis elements in the STAT3 gene regulating STAT3α and STAT3β messenger RNA splicing. Blood. 2001;98(13):3853–6.
Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JAM, Lammers J-WJ, et al. STAT3β; a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription? J Biol Chem. 1996;271(22):13221–27.
Liongue C, Sertori R, Ward AC. Evolution of cytokine receptor signaling. J Immunol. 2016;197(1):11–8.
Kawata T. STAT signaling in Dictyostelium development. Dev Growth Differ. 2011;53(4):548–57.
Hou XS, Melnick MB, Perrimon N. Marelle acts downstream of the Drosophila HOP/JAK kinase and encodes a protein similar to the mammalian STATs. Cell. 1996;84(3):411–9.
Yan R, Small S, Desplan C, Dearolf CR, Darnell JE Jr. Identification of a Stat gene that functions in Drosophila development. Cell. 1996;84(3):421–30.
Tsurumi A, Zhao C, Li WX. Canonical and non-canonical JAK/STAT transcriptional targets may be involved in distinct and overlapping cellular processes. BMC Genomics. 2017;18(1):718–28.
Dierking K, Polanowska J, Omi S, Engelmann I, Gut M, Lembo F, et al. Unusual regulation of a STAT protein by an SLC6 family transporter in C. elegans epidermal innate immunity. Cell Host Microbe. 2011;9(5):425–35.
Zhang Y, Li W, Li L, Li Y, Fu R, Zhu Y, et al. Structural damage in the C. elegans epidermis causes release of STA-2 and induction of an innate immune response. Immunity. 2015;42(2):309–20.
Mohanty S, Jermyn KA, Early A, Kawata T, Aubry L, Ceccarelli A, et al. Evidence that the Dictyostelium Dd-STATa protein is a repressor that regulates commitment to stalk cell differentiation and is also required for efficient chemotaxis. Development. 1999;126(15):3391–405.
Araki T, Tsujioka M, Abe T, Fukuzawa M, Meima M, Schaap P, et al. A STAT-regulated, stress-induced signalling pathway in Dictyostelium. J Cell Sci. 2003;116(14):2907–15.
Tian C, Wan P, Sun S, Li J, Chen M. Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol Biol. 2004;54(4):519–32.
Dill A, Sun T. Synergistic derepression of gibberellin signaling by removing RGA and GAI function in Arabidopsis thaliana. Genetics. 2001;159(2):777–85.
Hirsch S, Oldroyd GE. GRAS-domain transcription factors that regulate plant development. Plant Signal Behav. 2009;4(8):698–700.
Shi S, Calhoun HC, Xia F, Li J, Le L, Li WX. JAK signaling globally counteracts heterochromatic gene silencing. Nat Genet. 2006;38(9):1071–6.
Tanguy M, Véron L, Stempor P, Ahringer J, Sarkies P, Miska EA. An alternative STAT signaling pathway acts in viral immunity in Caenorhabditis elegans. mBio. 2017;8(5):e00924-17.
Ginger RS, Dalton EC, Ryves WJ, Fukuzawa M, Williams JG, Harwood AJ. Glycogen synthase kinase-3 enhances nuclear export of a Dictyostelium STAT protein. EMBO J. 2000;19(20):5483–91.
Fukuzawa M, Araki T, Adrian I, Williams JG. Tyrosine phosphorylation-independent nuclear translocation of a Dictyostelium STAT in response to DIF signaling. Mol Cell. 2001;7(4):779–88.
Liongue C, O’Sullivan LA, Trengove MC, Ward AC. Evolution of JAK-STAT pathway components: mechanisms and role in immune system development. PLoS ONE. 2012;7(3):e32777.
Acknowledgements
Not applicable.
Funding
N.A. is supported by a Deakin University Postgraduate Research Scholarship.
Author information
Authors and Affiliations
Contributions
N.A., C.L. and A.C.W. drafted the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Awasthi, N., Liongue, C. & Ward, A.C. STAT proteins: a kaleidoscope of canonical and non-canonical functions in immunity and cancer. J Hematol Oncol 14, 198 (2021). https://doi.org/10.1186/s13045-021-01214-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-021-01214-y