
Abstract
Cyclin-dependent kinase 4/6 (CDK4/6) inhibitors, which block the transition from the G1 to S phase of the cell cycle by interfering with Rb phosphorylation and E2F release, have shown potent antitumor activity and manageable toxicity in HR+/HER2− breast cancer patients. Some clinical trials involving CDK4/6 inhibitors in other tumors have achieved preliminary impressive efficacy. Whether CDK4/6 inhibitors possess great potential as broad-spectrum antitumor drugs and how to maximize their clinical benefits remain uncertain. TCGA database analysis showed that CDK4/6 genes and related genes are widely expressed among various tumors, and high or moderate expression of CDK4/6 genes commonly indicates poor survival. CDK4/6 gene expression is significantly higher in COAD, ESCA, STAD, LIHC, and HNSC, suggesting that CDK4/6 inhibitors could be more efficacious in those tumors. Moreover, network analysis with the STRING database demonstrated that CDK4/6-related proteins were co-expressed or co-occurred with the classical tumor signaling pathways, such as the cell cycle pathway, RAS pathway, PI3K pathway, Myc pathway, and p53 pathway. The extensive antitumor effects of CDK4/6 inhibitors may be achieved by synergizing or antagonizing with other signaling molecule inhibitors, and combination therapy might be the most effective treatment strategy. This article analyzed the feasibility of expanding the application of CDK4/6 inhibitors at the genetic level and further summarized the associated clinical/preclinical studies to collect supportive evidence. This is the first study that presents a theoretical foundation for CDK4/6 inhibitor precision therapy via combined analysis of comprehensive gene information and clinical research results.
Similar content being viewed by others
Tumorigenesis is a complicated process involving multiple links, multiple factors, and multiple stages, among which the cell cycle plays an essential regulatory role. Dysregulation of the cell cycle is considered to be related to the imbalance of proto-oncogenes, tumor suppressors, and cell cycle-related proteins [1]. Cell cycle inhibitors block cell cycle progression in tumor cells, hence inhibiting tumor cell proliferation and promoting tumor cell apoptosis [2, 3]. To date, cyclin-dependent kinase 4/6 inhibitors (CDK4/6 inhibitors) have occupied the leading position in cell cycle therapy. The successful application of CDK4/6 inhibitors in HR+/HER2− breast cancers brings great clinical benefits to patients and gives more encouragement to physicians and researchers. Since the dysregulation of the cell cycle is one of the crucial characteristics of malignant tumors, we aimed to investigate whether CDK4/6 inhibitors can be applied to various tumors and whether they can be as effective as traditional chemotherapy drugs in most tumors [4].
The expression of CDK4/6 in tumors
CDK4/6-related signaling pathways
Cyclin-dependent kinases (CDKs), part of the serine/threonine protein kinase family, are a group of key kinases that regulate the cell cycle; CDKs are activated by cyclins in a time-dependent manner. Twenty kinds of CDKs have been found, and these CDKs bind with their corresponding regulatory subunits (i.e., cyclins) to form active heterodimers [5]. According to their specialized functions, CDKs can be divided into two main categories: CDKs involved in cell cycle regulation, including CDK 1/2/4/6 and CDKs involved in transcriptional regulation, including CDK 7/8/9/11 [6].
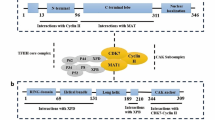
CDK4/6 is the main driving factor in cell cycle regulation and plays a key role in the occurrence and progression of various malignant tumors. Among the four cell cycle phases—G1 phase (prophase of DNA synthesis), S phase (DNA synthesis), G2 phase (prophase of mitosis), and M phase (mitosis)—cyclin D-CDK4/6-retinoblastoma (cyclin D-CDK4/6-Rb) signaling pathway is mainly responsible for regulating the G1-S transition [7]. CDK4 and CDK6 share 71% amino acid homology, and both can bind to cyclin D1/2/3. Under the induction of pro-mitosis signal, cyclin D binds to CDK4/6 and promotes retinoblastoma (Rb) phosphorylation, thus separating transcription factor E2F from the Rb-E2F complex, which causes cells to enter S phase and initiates DNA replication [8, 9] (Fig. 1). Changes in the cyclin D-CDK4/6-Rb pathway have been observed in the tumorigenesis processes of many tumors, such as breast cancer, pancreatic cancer, kidney cancer, liver cancer, and hematologic system tumors [10,11,12,13,14,15,16,17]. Gene amplification, gene mutation, and abnormalities in upstream and downstream regulators of cyclin D, CDK4, and CDK6 can all lead to abnormal activation of the cyclin D-CDK4/6-Rb pathway [8, 18]. The core regulatory effect of CDK4/6 in the cell cycle illustrates its vital role as a target in the treatment of malignant tumors.

The role of CDK4/6 in the cell cycle
The expression of CDK4/6 in different tumors
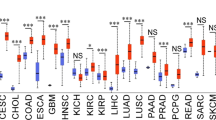
The cdk4, cdk6, rb1, and e2f1 genes are expressed in a wide range of cancers according to analysis of the TCGA PANCAN database, which is composed of 12,839 samples. cdk4 has the highest expression in ACC and the lowest expression in KIRC, cdk6 has the highest expression in LAML and the lowest expression in THCA, and e2f1 has the highest expression in DLBC and the lowest expression in PRAD. The expression of rb1 was the highest in KIRC and the lowest in TGCT. Intriguingly, the expression of cdk4, cdk6, e2f1, and rb1 in breast cancer was moderate or low compared to that in all tumors. The expression of the four genes in other tumors is shown in Fig. 2a. UALCAN cancer database analysis indicated that the expression of cdk4 and e2f1 in breast cancer was significantly higher than that in normal tissues (p < 0.01), while the expression of cdk6 and rb1 was not higher than that in normal tissues. We ultimately observed that the expression levels of cdk4, cdk6, e2f1, and rb1 in digestive system tumors such as COAD, ESCA, STAD, LIHC, and HNSC were significantly higher than those in normal tissues (p < 0.01), suggesting that CDK4/CDK6-E2F1/Rb1 signaling may be involved in the occurrence and progression of these tumors and that CDK4/6 inhibitors may have better efficacy in these tumors; the expression of cdk4 and cdk6 in CESC, PAAD, and THYM was not significantly different compared with that in normal tissues (p > 0.05), suggesting that CDK4/6 inhibitors may have poor or no therapeutic effect in these tumors. However, the expression trends of cdk4, cdk6, e2f1, and rb1 in many other cancers were inconsistent, indicating that this signaling pathway may regulate the cell cycle by crossing with other signaling pathways, and CDK4/6 inhibitor combination therapies may lead to considerable antitumor effects in these cancers (Fig. 2b). Moreover, the cdk4, cdk6, and e2f1 genes show extensive gene amplification and gene mutation in various tumors; deep deletion and gene mutation of the rb1 gene were observed in various tumors, while gene fusion and multiple alterations in these four genes are rare (Fig. 2c). These gene changes may explain the difference in the clinical efficacy of and drug resistance to CDK4/6 inhibitors in different tumors.

CDK4/6-related gene information among various cancers. a Expression of CDK4/6 and related genes. b Comparison of the expression of CDK4/6-related genes between tumor and normal tissues. c Alteration frequency of CDK4/6-related genes
Role of the CDK4/6 gene in tumor progression and prognosis
Based on the analysis from the UALCAN cancer database, moderate expression of the cdk4 gene in KIRC, LGG, KIRP, MESO, KICH, and SKCM was significantly negatively related to overall survival (p < 0.05); high expression of the cdk4 gene in LIHC was closely related to worse overall survival than low expression and may be a sensitive marker for predicting the prognosis of LIHC. The cdk6 gene was expressed at low levels in UCEC and moderately expressed in BLCA, LUAD, PAAD, LGG, SARC, ACC, and MESO, and cdk6 expression was significantly negatively correlated with the overall survival of patients with these tumors. e2f1 was moderately expressed in PAAD, LGG, ACC, and MESO, and highly expressed in KIRC, HNSC, KIRP, ESCA, CHOL, and KICH, which was closely related to the overall survival of those patients. Moreover, in KIRC and LGG, the rb1 gene was moderately expressed, and its upregulation predicted a significant decrease in overall survival (Fig. 3). Thus, cdk4, cdk6, e2f1, and rb1 not only participate in the progression of cancer cells but might also be useful biomarkers for predicting prognosis in some tumors.

Expression levels of CDK4/6-related genes and its corresponding survival
Clinical application of CDK 4/6 inhibitors in breast cancer and other tumors
Application of CDK 4/6 inhibitors in breast cancer
Based on the essential regulatory role of CDK4/6 in the cell cycle, CDK4/6 inhibitors have emerged as antitumor drugs. CDK4/6 inhibitors hinder the transition from G1 phase to S phase by inhibiting Rb phosphorylation and E2F release and induce tumor cycle arrest at G1 phase, which can inhibit tumor cell growth and cause tumor regression [18]. Since CDK inhibitors were developed 20 years ago, CDK4/6 inhibitors have achieved great success in breast cancer [19, 20], and cell cycle therapy has gradually matured. Approximately, 75–80% of patients with breast cancer are hormone receptor (HR) positive, and the proliferation of breast cancer cells depends on the activation of estrogen [21]. Endocrine therapy is the main treatment for HR positive breast cancer [22]; however, drug resistance is inevitable in the course of treatment [20, 23, 24]. Endocrine therapy combined with chemotherapy is not as effective as expected due to the limited survival benefits and higher toxicity in HR-positive breast cancer patients. Therefore, emphasis should be placed on improving endocrine therapy efficacy [25]. The cyclin D-CDK4/6 complex is usually highly expressed or abnormally activated in breast cancer [19]. The mutation rate of cell cycle-related genes in breast cancer is as high as 38% [26]. Increased expression of cyclin D causes continuous phosphorylation of Rb and leads to continuous proliferation of breast cancer cells; blocking CDK4/6 exerts a lethal effect on breast cancer cells. Moreover, Finn’s study confirmed that palbociclib combined with tamoxifen sensitizes endocrine-resistant estrogen receptor-positive breast cancer cell lines in vitro [27]. Thus, investigators turned their attention to CDK4/6 inhibitors.
Alvociclib, the first-generation CDK inhibitor, lacks specificity and blocks CDK1/2/4/6/7/9, causing serious adverse effects and limiting its clinical application [20]. As medical science has advanced, three selective CDK4/6 inhibitors (palbociclib, ribociclib, and abemaciclib) have achieved fairly good curative benefits in breast cancer [28]. Palbociclib is the first FDA-approved CDK4/6 inhibitor [29]. The PALOMA-1 study showed that palbociclib combined with letrozole increased the PFS of HR+/HER2− advanced breast cancer patients from 10.2 to 20.2 months compared to letrozole alone, and its side effects were controllable [25]. Based on this study, the FDA accelerated the approval of palbociclib in combination with letrozole for first-line treatment of postmenopausal HR+/HER2− metastatic breast cancer patients in February 2015 [25]. The PALOMA-3 study demonstrated that the mOS of the palbociclib combined fulvestrant group was higher than that of the fulvestrant group (34.9 m vs 28.0 m). Accordingly, the FDA approved palbociclib in combination with fulvestrant for HR+/HER2− postmenopausal women with advanced breast cancer who had failed previous endocrine therapy in February 2016 [30]. The phase III study MONALEESA-2 showed that ribociclib plus letrozole significantly improved PFS in patients with HR+/HER2− advanced breast cancer compared with placebo plus letrozole (NR vs 14.7 m; HR 0.56, 95% CI 0.43–0.72, p < 0.001) [31]. Based on this trial, ribociclib was approved for first-line treatment of postmenopausal women with HR+/HER2− advanced breast cancer in March 2017 [32]. Palbociclib and ribociclib are breakthroughs in the treatment of advanced HR+/HER2− postmenopausal breast cancer, overturning the old pattern of single endocrine therapy for advanced postmenopausal breast cancer in the past decades, laying the foundation of CDK4/6 inhibitors combined with aromatase inhibitors in first-line treatment for postmenopausal women with HR+/HER2− advanced breast cancer. Abemaciclib is the third FDA-approved CDK4/6 inhibitor. In MONARCH-3 trial, the HR+/HER2− postmenopausal advanced breast cancer patients who did not receive systemic therapy as the aimed population, the 18-month interim results indicated that the PFS of abemaciclib group had not been reached, and the median PFS was 14.7 months in the placebo group; the objective response rates were 59% and 44%, respectively (p = 0.004) [33]. According to this study, the FDA approved abemaciclib combined with aromatase inhibitors for the first-line treatment of postmenopausal HR+/HER2− advanced or metastatic breast cancer in February 2018 [34]. Moreover, in the MONARCH-1 trial, the objective response rate of abemaciclib monotherapy reached 19.7% [35]. Compared with palbociclib and ribociclib, abemaciclib can be administered alone, without serious neutropenia toxicity, which provides a new choice for breast cancer patients and highlights its advantages among the highly competitive CDK-targeted drugs. More clinical trial information on the application of CDK4/6 inhibitors in various breast cancers [36,37,38,39,40] is demonstrated in Table 1.
The major adverse effects of CDK4/6 inhibitors are leukopenia and neutropenia, mainly caused by palbociclib and ribociclib [41]. CDK4/6 inhibitors can also cause gastrointestinal side effects such as diarrhea, nausea, and vomiting. It is worth noting that once neutropenia occurs simultaneously with diarrhea, the risk of infection is greatly increased [42]. Some patients have prolonged QTc intervals, elevated transaminases, thromboembolism, and others [43]. However, these side effects are reversible and can be controlled by dose interruption, dose reduction, and symptomatic supportive treatment [44].
Application and expansion of CDK4/6 inhibitors in other solid tumors
The cdk4/6 genes are generally expressed among various cancers, and their expression is higher or lower than that of normal tissues in most cancer types. High or moderate expression of CDK4/6-related genes often indicates poor survival. The CDK4/6 genes participate in tumorigenesis by synergistically regulating multiple genes in many signaling pathways. CDK4/6 inhibitors may be more effective when combined with other signaling pathway inhibitors, which is consistent with the complexity and intersectionality of tumor signaling pathway interactions. Although the cdk4/6 gene expression level in breast cancer is relatively low, a marked antitumor effect of CDK4/6 inhibitors has been observed in breast cancer, so we speculate that it may also work in other malignancies, especially in those cancers with high cdk4/6-related gene expression. CDK4/6 inhibitors may have the potential to be widely applied in multiple cancers, similar to traditional chemotherapy drugs such as cisplatin or paclitaxel.
Although main battlefield in HR+/HER2− breast cancer, CDK4/6 inhibitors have also been actively explored in other malignancies. Yang’s study showed that CDK4/6 inhibitors increased the sensitivity of acute myeloid leukemia cells to cytotoxic drugs [17]. Patnaik and Taylor’s study showed that CDK4/6 inhibitors achieved disease control rates of 49% and 44%, respectively, in non-small-cell lung cancer patients (n = 68) and melanoma patients (n = 18) [44, 45]. Adkins’ study indicated an objective response rate of 39% for CDK4/6 inhibitors in patients with head and neck squamous cell carcinoma (n = 62) [46]. It is not difficult to conclude that the exploration of CDK4/6 inhibitors in other cancers except breast cancer, such as liposarcoma, lymphoma melanoma, and many other advanced cancers [47,48,49,50], is still at an early stage and is mostly limited to basic experiments and stage I or II clinical trials (See Table 2).
Correlations between CDK4/6-related proteins and classical tumor signaling pathway molecules
The occurrence and progression of tumors involves the interaction of multiple signaling pathways. We used the STRING database and the STRING tool to map the relationship between CDK4/6-Rb1/E2F1 pathway proteins and classical signaling pathway-associated proteins. Network analysis showed that CDK4/6-related proteins were directly or indirectly associated with key molecules of classical signaling pathways, such as the cell cycle pathway, RAS pathway, PI3K pathway, Myc pathway, and p53 pathway. In addition, CDK4/6 protein is co-expressed with EGFR/ERBB2, BRAF/KRAS, PIK3CA/MTOR, MYC/MYCN, NOTCH, MDM4, MSH2, and other proteins (Fig. 4, black line), suggesting that CDK4/6 may have synergistic effects with these co-expressed signaling molecules. Moreover, CDK4/6-related signaling molecules are downstream of most signaling pathways, such as the RAS pathway/PI3K pathway/TGF-β pathway/p53 pathway/Notch pathway/Myc pathway [51], suggesting that CDK4/6 blockade may inhibit these related signaling pathways to some extent. There were also protein correlations derived from experimental evidence (Fig. 4, green line) and some derived from in-depth analysis of scientific papers (Fig. 4, yellow line). CDK4/6 inhibitors may play a role in inhibiting tumor growth by synergizing or antagonizing certain signaling molecule inhibitors and immune checkpoint inhibitors, which indirectly reflects their extensive antitumor effect.

Functional protein association networks of CDK4/6 and classical tumor signaling pathways
CDK4/6 inhibitors combined with endocrine therapy
At present, endocrine therapy combined with CDK4/6 inhibitors has achieved significant therapeutic effects and controllable toxicity in many clinical trials, as mentioned above, and combined therapy has become the most promising therapeutic strategy for HR+/HER2− breast cancer patients.
CDK4/6 inhibitors combined with immunotherapy
CDK4/6 inhibitors not only arrest the tumor cell cycle, but also trigger antitumor immunity. First, CDK4/6 inhibitors downregulate E2F transcription factor-related gene expression and upregulate major histocompatibility complex class I molecule expression in breast cancer cell lines [52]; CDK4/6 inhibitors activate endogenous retroviral components in tumor cells, stimulating the production of type III interferons to promote tumor antigen presentation [52, 53]. Second, CDK4/6 inhibitors inhibit the proliferation of regulatory T (Treg) cells and DNA methyltransferase 1 expression in Treg cells, resulting in promoter hypomethylation and suppression of E2F release [52]; other studies demonstrated that the expression of CDK6 in Treg cells was higher than that of other T cell subtypes. CDK4/6 inhibitors can downregulate Treg cell proliferation by inhibiting CDK6 [54]. Furthermore, CDK4/6 inhibitors promote tumor cell clearance by enhancing cytotoxic T cells (CTLs). CDK6 is an upstream regulatory element of nuclear factor of activated T cells (NFAT), and CDK4/6 inhibitors suppress NFAT phosphorylation, the activation of CTLs, and its ability to kill tumor cells [55]. Finally, the Cyclin D1-CDK4 complex directly phosphorylates speckle-type POZ protein (SPOP), and CDK4/6 inhibitors can enhance the immune escape of tumors by decreasing the ubiquitination of SPOP and reducing the degradation of PD-L1 [56, 57]. These synergistic mechanisms provide a theoretical basis for the combination therapy of CDK4/6 inhibitors plus immune checkpoint inhibitors [52, 58]. In Zhang’s study, CDK4/6 inhibitor combined with PD-1 antibody significantly reduced the proliferation of tumor cells and improved the survival rate of carcinogenic mic e[57], illustrating the synergistic antitumor effect of the CDK4/6 inhibitor and immune checkpoint-related inhibitor. The synergism, antagonism, and interactions of those related genes deserve further exploration (Fig. 4, red line).
CDK4/6 inhibitors combined with targeted therapy
The PI3K-AKT-mTOR pathway proteins and the CDK-Rb-E2F pathway proteins are widely co-expressed in different tumor types, and the two pathways are the relationship between the upstream and downstream, signifying that the combined targeting inhibition of CDK4/6 and PI3K-AKT-mTOR should further improve the efficacy [59]. Cortes’ research indicated that the combination of PI3K inhibitors or mTOR inhibitors might increase the sensitivity of CDK4/6 inhibitors [60]. Teo’s study showed that CDK4/6 inhibitors combined with PI3K inhibitors increased the apoptosis of triple-negative breast cancer cell lines and induced persistent tumor regression in vivo [61]. Moreover, CDK4/6 inhibitors can restore sensitivity to EGFR/HER2 inhibitors by reducing the activity of mTOR [62]. EGFR/HER2 inhibitors combined with CDK4/6 inhibitors may increase the sensitivity to EGFR inhibitor-resistant lung cancer cells [63]. Goel’s study demonstrated that CDK4/6 inhibitors augmented the efficacy of EGFR inhibitors in esophageal squamous cell carcinoma and reversed drug resistance [64]. A phase II clinical trial (NCT02101034) involving patients with HPV-unrelated head and neck squamous-cell carcinomas showed that palbociclib plus cetuximab resulted in an objective response rate of 39% in patients with disease progression on platinum but cetuximab-naive and 19% in patients with disease progression on cetuximab [46]. Teh’s study demonstrated that CDK4/6 inhibitors in combination with BRAF inhibitors or MEK inhibitors are an effective treatment strategy for melanoma, but continued administration increases toxicity. The mechanism of acquired resistance of CDK4/6 inhibitors combined with BRAF inhibitors or MEK inhibitors may be due to mTOR pathway activation, so adding mTOR inhibitors may overcome the resistance of foresaid combined therapy [65]. Chen’s study showed that RAF inhibitors combined with CDK4/6 inhibitors improve the therapeutic effects of RAS or BRAF mutant tumors [66]. Small’s study confirmed that abemaciclib combined with sunitinib significantly decreased tumor size in a preclinical mouse model of renal cell carcinoma without serious adverse effects [67]. Pek’s study demonstrated that in KRAS-dependent and BRAF-mutant metastatic colorectal cancer cell lines, palbociclib combined with MEK inhibitors is an effective treatment strategy [68]. Ribociclib combined with the ALK inhibitor ceritinib showed excellent therapeutic effects in ALK mutant neuroblastoma [69]. However, large-scale clinical trials are needed to confirm the efficacy of the abovementioned combination therapies.
CDK4/6 inhibitor combined with other treatments
Cyclin D3-CDK6 can phosphorylate two key enzymes (6-phosphofructokinase and pyruvate kinase M2) in the glucose metabolism pathway and restrain its metabolic activity, which consumes the antioxidants NADPH and glutathione, therefore increasing the level of reactive oxygen species and leading to apoptosis of tumor cells. Palbociclib combined with low-dose pentose phosphate pathway inhibitors may exert synergistic antitumor effects [70]. Vijayaraghavan’s research showed that CDK4/6 inhibitors combined with autophagy inhibitors can maintain the integrity of the G1/S checkpoint and may be a new therapeutic pattern for multiple solid tumors [71]. Francis’s study confirmed that CDK4/6 inhibitors resensitize Rb-positive sarcoma cells to the Weel kinase inhibitor AZD1775 [72].
The resistance and efficacy prediction of CDK4/6 inhibitors
CDK4/6 inhibitor resistance inevitably occurs in the course of treatment, and overcoming resistance is a challenge for clinicians. Some studies have pointed out that the acquired resistance of CDK4/6 inhibitors mainly results from CDK6 amplification, which usually occurs after prolonged administration of the potent selective CDK4/6 inhibitor [73]. Some researchers believe that CCNE1 amplification causes acquired resistance to CDK4/6 inhibitors, and that sensitivity can be restored by targeting CDK2 [74]. In addition, activating cyclin D gene mutations may enhance sensitivity to CDK4/6 inhibitors, while cyclin D deficiency is associated with CDK4/6 inhibitor resistance [75]. In addition, the role of CDK4/6 inhibitors in arresting the cell cycle depends on Rb status [24]. When Rb function is absent, the G1-S phase transition is no longer dependent on CDK4/6, and CDK4/6 inhibitors may have off-target effects in Rb inactivation models [19]. By influencing the Hippo pathway, FAT atypical cadherin 1 (FAT1) loss leads to CDK6 elevation, which promotes resistance to CDK4/6 inhibitors [76]. A recent study revealed that amplification of fibroblast growth factor receptor 1 (FGFR1) might cause resistance to CDK4/6 inhibitors [77]. We can preliminarily conclude that the increase in cyclin-CDK complex expression and the regulation of the cyclin D-CDK4/6-Rb pathway may be important resistance mechanisms of CDK4/6 inhibitors, as shown in Table 3. Overcoming resistance is a major concern in research, and exploring efficacy predictors and selecting the population that should obtain the most benefit is another research hot spot. Studies have shown that deletion of p16 decreases the endogenous inhibition of CDK4/6 and that low levels of p16 may suggest that cells are sensitive to CDK4/6 inhibitors [78]. High expression of cyclin D/Rb and low expression of p16 are thought to be biomarkers for predicting CDK4/6 inhibitor sensitivity [27]. However, Wang’s study showed that approximately 85% of breast cancer cells have normal Rb status, but due to the rare Rb deletion in ER+ breast cancer, it is less sensitive as a predictive marker [79]. CDK activation requires interferon β expression, suggesting that interferon β may be a predictor of CDK4/6 inhibitor efficacy [80]. Other efficacy predictors include CDK4 phosphorylation and tumor cloning kinetics [75, 81, 82]. However, so far, there is still a long way to go in combatting CDK4/6 inhibitor resistance and identifying sensitive predictive markers.
Prospects and conclusions
CDK4/6-related genes are widely expressed and are closely associated with the prognosis of different cancers. CDK4/6 inhibitors achieve striking antitumor effects by regulating the cell cycle. Clinical trials have confirmed that CDK4/6 inhibitors alone or combined with endocrine therapy have brought great clinical benefit to HR+/HER2− breast cancer patients. Extensive cross-linking of CDK4/6 genes and key genes in classical tumor signaling pathways may exert synergistic or antagonistic effects, given the theoretical basis for CDK4/6 inhibitor combination therapy. CDK4/6 inhibitors have great potential to become broad-spectrum antitumor drugs. CDK4/6 inhibitor combination therapy may become a new strategy for the precise treatment of malignancies. Future research should focus on exploring biomarkers that predict CDK4/6 inhibitor efficacy and screening sensitive populations. More large-scale prospective clinical trials are needed to validate the curative effect of CDK4/6 inhibitors in more cancer types and more combination models.
Availability of data and materials
All data generated or analyzed during this study are included.
Abbreviations
- TCGA:
-
The Cancer Genome Atlas
- ACC:
-
Adrenocortical carcinoma
- BLCA:
-
Bladder urothelial carcinoma
- BRCA:
-
Breast invasive carcinoma
- CESC:
-
Cervical squamous cell carcinoma and endocervical adenocarcinoma
- CHOL:
-
Cholangiocarcinoma
- COAD:
-
Colon adenocarcinoma
- DLBC:
-
Lymphoid neoplasm diffuse large B-cell lymphoma
- ESCA:
-
Esophageal carcinoma
- GBM:
-
Glioblastoma multiforme
- HNSC:
-
Head and neck squamous cell carcinoma
- KICH:
-
Kidney chromophobe
- KIRC:
-
Kidney renal clear cell carcinoma
- KIRP:
-
Kidney renal papillary cell carcinoma
- LAML:
-
Acute myeloid leukemia
- LGG:
-
Brain lower grade glioma
- LIHC:
-
Liver hepatocellular carcinoma
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell carcinoma
- MESO:
-
Mesothelioma
- OV:
-
Ovarian serous cystadenocarcinoma
- PAAD:
-
Pancreatic adenocarcinoma
- PCPG:
-
Pheochromocytoma and paraganglioma
- PRAD:
-
Prostate adenocarcinoma
- READ:
-
Rectum adenocarcinoma
- SARC:
-
Sarcoma
- SKCM:
-
Skin cutaneous melanoma
- STAD:
-
Stomach adenocarcinoma
- TGCT:
-
Testicular germ cell tumors
- THCA:
-
Thyroid carcinoma
- THYM:
-
Thymoma
- UCEC:
-
Uterine corpus endometrial carcinoma
- UCS:
-
Uterine carcinosarcoma
- UVM:
-
Uveal melanoma
- STES:
-
Stomach and esophageal carcinoma
- EGFR:
-
Epidermal growth factor receptor
- PI3K:
-
Phosphatidylinositol 3 kinase
- TGF-β:
-
Transforming growth factor beta
- MTOR:
-
Mammalian target of rapamycin
- MAPK:
-
Mitogen-activated protein kinase
- PD-1:
-
Programmed cell death-1
- PD-L1:
-
Programmed cell death-ligand 1
- MSH:
-
Mismatch repair
References
Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17:93–115.
Minton K. Cell cycle inhibitors boost tumour immunogenicity. Nature Reviews Drug Discovery. 2017;16:679.
Helsten T, Kato S, Schwaederle M, et al. Cell-cycle gene alterations in 4,864 tumors analyzed by next-generation sequencing: implications for targeted therapeutics. Mol Cancer Ther. 2016;15:1682–90.
Ingham M, Schwartz GK. Cell-cycle therapeutics come of age. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017;35:2949–59.
Wood DJ, Endicott JA. Structural insights into the functional diversity of the CDK-cyclin family. Open Biol. 2018;8.
Malumbres M. Cyclin-dependent kinases. Genome biology. 2014;15:122–2.
Dalton S. Linking the cell cycle to cell fate decisions. Trends Cell Biol. 2015;25:592–600.
Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–38.
Roberts PJ, Bisi JE, Strum JC, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104:476–87.
Corona SP, Ravelli A, Cretella D, et al. CDK4/6 inhibitors in HER2-positive breast cancer. Crit Rev Oncol Hematol. 2017;112:208–14.
Franco J, Balaji U, Freinkman E, et al. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep. 2016;14:979–90.
Logan JE, Mostofizadeh N, Desai AJ, et al. PD-0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity. Anticancer Res. 2013;33:2997–3004.
Bollard J, Miguela V, Ruiz de Galarreta M, et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut. 2017;66:1286–96.
Saab R, Bills JL, Miceli AP, et al. Pharmacologic inhibition of cyclin-dependent kinase 4/6 activity arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Mol Cancer Ther. 2006;5:1299–308.
Niesvizky R, Badros AZ, Costa LJ, et al. Phase 1/2 study of cyclin-dependent kinase (CDK)4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Leuk Lymphoma. 2015;56:3320–8.
Nemoto A, Saida S, Kato I, et al. Specific antileukemic activity of PD0332991, a CDK4/6 inhibitor, against Philadelphia chromosome-positive lymphoid leukemia. Mol Cancer Ther. 2016;15:94–105.
Yang C, Boyson CA, Di Liberto M, et al. CDK4/6 inhibitor PD 0332991 sensitizes acute myeloid leukemia to cytarabine-mediated cytotoxicity. Cancer Res. 2015;75:1838–45.
Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12.
O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417–30.
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–46.
Brufsky AM. Long-term management of patients with hormone receptor-positive metastatic breast cancer: Concepts for sequential and combination endocrine-based therapies. Cancer Treat Rev. 2017;59:22–32.
Kim ES, Scott LJ. Palbociclib: a review in HR-positive, HER2-negative, advanced or metastatic breast cancer. Target Oncol. 2017;12:373–83.
Wolff AC. CDK4 and CDK6 inhibition in breast cancer - a new standard. N Engl J Med. 2016;375:1993–4.
Clark AS, Karasic TB, DeMichele A, et al. Palbociclib (PD0332991)-a selective and potent cyclin-dependent kinase inhibitor: a review of pharmacodynamics and clinical development. JAMA Oncol. 2016;2:253–60.
Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. The Lancet Oncology. 2015;16:25–35.
Koboldt DC, Fulton RS, McLellan MD, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77.
Eggersmann TK, Degenhardt T, Gluz O, et al. CDK4/6 inhibitors expand the therapeutic options in breast cancer: palbociclib, ribociclib and abemaciclib. BioDrugs. 2019;33:125–35.
Beaver JA, Amiri-Kordestani L, Charlab R, et al. FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res. 2015;21:4760–6.
Turner NC, Slamon DJ, Ro J, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379:1926–36.
Sledge GW Jr, Toi M, Neven P, et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35:2875–84.
Shah A, Bloomquist E, Tang S, et al. FDA approval: ribociclib for the treatment of postmenopausal women with hormone receptor-positive, HER2-negative advanced or metastatic breast cancer. Clin Cancer Res. 2018;24:2999–3004.
Goetz MP, Toi M, Campone M, et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J Clin Oncol. 2017;35:3638–46.
Kim ES. Abemaciclib: first global approval. Drugs. 2017;77:2063–70.
Dickler MN, Tolaney SM, Rugo HS, et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR(+)/HER2(-) metastatic breast cancer. Clin Cancer Res. 2017;23:5218–24.
Finn RS, Martin M, Rugo HS, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375:1925–36.
Masuda N, Nishimura R, Takahashi M, et al. Palbociclib in combination with letrozole as first-line treatment for advanced breast cancer: a Japanese phase II study. Cancer Sci. 2018;109:803–13.
Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. New England Journal of Medicine. 2016;375:1738–48.
Slamon DJ, Neven P, Chia S, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. 2018;36:2465–72.
Tripathy D, Im S-A, Colleoni M, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. The Lancet Oncology. 2018;19:904–15.
Finn RS, Aleshin A, Slamon DJ. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016;18:17.
Maroun JA, Anthony LB, Blais N, et al. Prevention and management of chemotherapy-induced diarrhea in patients with colorectal cancer: a consensus statement by the Canadian Working Group on Chemotherapy-Induced Diarrhea. Curr Oncol. 2007;14:13–20.
Sammons SL, Topping DL, Blackwell KL. HR+, HER2- Advanced breast cancer and CDK4/6 inhibitors: mode of action, clinical activity, and safety profiles. Curr Cancer Drug Targets. 2017;17:637–49.
Patnaik A, Rosen LS, Tolaney SM, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016;6:740–53.
Taylor M, Sosman J, Gonzalez R et al. Phase Ib/II study of LEE011 (CDK4/6 inhibitor) and LGX818 (BRAF inhibitor) in BRAF-mutant melanoma.2014.
Adkins D, Ley J, Neupane P, et al. Palbociclib and cetuximab in platinum-resistant and in cetuximab-resistant human papillomavirus-unrelated head and neck cancer: a multicentre, multigroup, phase 2 trial. Lancet Oncol. 2019.
Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated With CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2:937–40.
Leonard JP, LaCasce AS, Smith MR, et al. Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood. 2012;119:4597–607.
Sosman J, Kittaneh M, Lolkema M et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: Early encouraging clinical activity.2014.
Fujiwara Y, Tamura K, Kondo S, et al. Phase 1 study of abemaciclib, an inhibitor of CDK 4 and 6, as a single agent for Japanese patients with advanced cancer. Cancer Chemother Pharmacol. 2016;78:281–8.
Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the cancer genome Atlas. Cell. 2018;173:321–37 e310.
Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471–5.
Chaikovsky AC, Sage J. Beyond the cell cycle: enhancing the immune surveillance of tumors Via CDK4/6 inhibition. Mol Cancer Res. 2018;16:1454–7.
De Simone M, Arrigoni A, Rossetti G, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016;45:1135–47.
Deng J, Wang ES, Jenkins RW, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018;8:216–33.
Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–96.
Zhang J, Bu X, Wang H, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–5.
Schaer DA, Beckmann RP, Dempsey JA, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Rep. 2018;22:2978–94.
Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273–91.
Cortes J, Im SA, Holgado E, et al. The next era of treatment for hormone receptor-positive, HER2-negative advanced breast cancer: triplet combination-based endocrine therapies. Cancer Treat Rev. 2017;61:53–60.
Teo ZL, Versaci S, Dushyanthen S, et al. Combined CDK4/6 and PI3Kalpha inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Res. 2017;77:6340–52.
Goel S, Wang Q, Watt AC, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29:255–69.
Liu M, Xu S, Wang Y, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, sensitizes lung cancer cells to treatment with epidermal growth factor receptor tyrosine kinase inhibitors. Oncotarget. 2016;7:84951–64.
Zhou J, Wu Z, Wong G, et al. CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in oesophageal squamous cell carcinoma. Nat Commun. 2017;8:13897.
Teh JLF, Cheng PF, Purwin TJ, et al. In vivo E2F reporting reveals efficacious schedules of MEK1/2-CDK4/6 targeting and mTOR-S6 resistance mechanisms. Cancer Discov. 2018;8:568–81.
Chen SH, Gong X, Zhang Y, et al. RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene. 2018;37:821–32.
Small J, Washburn E, Millington K, et al. The addition of abemaciclib to sunitinib induces regression of renal cell carcinoma xenograft tumors. Oncotarget. 2017;8:95116–34.
Pek M, Yatim S, Chen Y, et al. Oncogenic KRAS-associated gene signature defines co-targeting of CDK4/6 and MEK as a viable therapeutic strategy in colorectal cancer. Oncogene. 2017;36:4975–86.
Wood AC, Krytska K, Ryles HT, et al. Dual ALK and CDK4/6 inhibition demonstrates synergy against neuroblastoma. Clin Cancer Res. 2017;23:2856–68.
Cretella D, Ravelli A, Fumarola C, et al. The anti-tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J Exp Clin Cancer Res. 2018;37:72.
Vijayaraghavan S, Karakas C, Doostan I, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916.
Francis AM, Alexander A, Liu Y, et al. CDK4/6 Inhibitors sensitize Rb-positive sarcoma cells to Wee1 kinase inhibition through reversible cell-cycle arrest. Mol Cancer Ther. 2017;16:1751–64.
Yang C, Li Z, Bhatt T, et al. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017;36:2255–64.
Herrera-Abreu MT, Palafox M, Asghar U, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76:2301–13.
Gong X, Litchfield LM, Webster Y et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor abemaciclib. Cancer Cell 2017; 32: 761-776.e766.
Li Z, Razavi P, Li Q et al. Loss of the FAT1 Tumor suppressor promotes resistance to CDK4/6 inhibitors via the Hippo pathway. Cancer Cell 2018; 34: 893-905.e898.
Formisano L, Lu Y, Servetto A, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019;10:1373.
Wander SA, Mayer EL, Burstein HJ. Blocking the cycle: cyclin-dependent kinase 4/6 inhibitors in metastatic, hormone receptor-positive breast cancer. J Clin Oncol. 2017;35:2866–70.
Wang H, Nicolay BN, Chick JM, et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature. 2017;546:426–30.
Cingoz O, Goff SP. Cyclin-dependent kinase activity is required for type I interferon production. Proc Natl Acad Sci U S A. 2018;115:E2950–e2959.
Raspe E, Coulonval K, Pita JM, et al. CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol Med. 2017;9:1052–66.
O'Leary B, Hrebien S, Morden JP, et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun. 2018;9:896.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant NO. 81301912), Beijing Municipal Health System High-level Health Person Foundation Project (Grant NO. 2014-3-005), and Beijing Municipal Science and Technology Commission (Capital Features, Z161100000516083, to Qin Li).
Author information
Authors and Affiliations
Contributions
Qin Li contributed to the design of the review and revised the article, had full access to all of the contents included in this study, and takes responsibility for the integrity of the data and the accuracy of the data analysis. Qi Du and Xiang Guo collected and analyzed the clinical data. Qi Du, Miao Wang, and Yongfu Li wrote the main manuscript text and supplied the figures. Qi Du and Xiaoyi Sun polished and modified the English. All the authors contributed to the review and revision of the manuscript, and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approved the final manuscript for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Du, Q., Guo, X., Wang, M. et al. The application and prospect of CDK4/6 inhibitors in malignant solid tumors. J Hematol Oncol 13, 41 (2020). https://doi.org/10.1186/s13045-020-00880-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00880-8