
Abstract
Hematopoietic stem cell (HSC) aging, which is accompanied by reduced self-renewal ability, impaired homing, myeloid-biased differentiation, and other defects in hematopoietic reconstitution function, is a hot topic in stem cell research. Although the number of HSCs increases with age in both mice and humans, the increase cannot compensate for the defects of aged HSCs. Many studies have been performed from various perspectives to illustrate the potential mechanisms of HSC aging; however, the detailed molecular mechanisms remain unclear, blocking further exploration of aged HSC rejuvenation. To determine how aged HSC defects occur, we provide an overview of differences in the hallmarks, signaling pathways, and epigenetics of young and aged HSCs as well as of the bone marrow niche wherein HSCs reside. Notably, we summarize the very recent studies which dissect HSC aging at the single-cell level. Furthermore, we review the promising strategies for rejuvenating aged HSC functions. Considering that the incidence of many hematological malignancies is strongly associated with age, our HSC aging review delineates the association between functional changes and molecular mechanisms and may have significant clinical relevance.
Similar content being viewed by others

Background
A key step in hematopoietic stem cell (HSC) aging research was achieved in 1996, revealing that HSCs from old mice were only one-quarter as efficient as those from young mice at homing to and engrafting the bone marrow (BM) of irradiated recipients [1]. This landmark discovery established that the HSC aging process is accompanied by functional decline. Since then, differences between young and aged HSCs have been elucidated from multiple aspects, and the mechanisms of HSC aging have been gradually illustrated. Furthermore, in the clinic, donor age is carefully considered in HSC transplantation, and young donors result in better survival after HSC transplantation [2,3,4].
Aged HSCs are inferior to young HSCs and show incomplete reconstitution potential. For example, in primary transplantation experiments, compared with young HSCs, aged HSCs showed an overall reduction in long-term repopulating potential [1] and differentiation bias [5]. In the second transplantation experiments, BM cells from old animals were less able to engraft later passage recipients than those from young animals [6]. These findings demonstrated that HSC functions are partially dysregulated during aging and that approaches to rejuvenate aged HSCs should be further elucidated.
Different studies have explored the mechanisms by which aged HSC dysfunction occurs. Altered expression levels of multiple genes and mutation of some specific genes were shown to lead to HSC aging [7]. In addition, inhibition of some signaling pathways, such as the mammalian target of rapamycin (mTOR) and p38 mitogen-activated protein kinase (MAPK) pathways, was closely related to HSC aging [8, 9]. Furthermore, epigenetic perturbations also drove both cellular functional attenuation and other aging manifestations [10]. Finally, some factors in the HSC niche, such as cytokines and enzymes, are also crucial during the aging process [11].
In the present review, we compare the differences in the hallmarks, signaling pathways, and epigenetics of young and aged HSCs, and provide an overview of the BM niche wherein HSCs reside. Notably, we summarize the very recent studies which dissect HSC aging at the single-cell level. In addition, we review the promising strategies for rejuvenating aged HSC functions. Considering that the incidence of many hematological malignancies is strongly associated with age, our HSC aging review delineates the association between functional changes and molecular mechanisms and may have significant clinical relevance.
Changes in the HSC hallmarks during aging
The functions of HSCs, one of the most important blood cell types, decline in both mice [12] and humans [13] during the aging process. Here, we summarize the changes in the hallmarks of HSC aging with regard to self-renewal, differentiation bias, homing, and engraftment.
Self-renewal
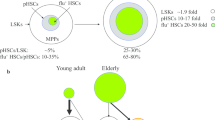
HSCs are characterized by their capacity for long-term self-renewal and the ability to generate all functional blood cells. Although different studies demonstrated a dramatic increase in the number of mouse HSCs with age [14, 15], the ability of HSCs to self-renew did not increase accordingly. To further compare young and old HSC self-renewal activity in vivo, Dykstra et al. performed secondary transplantations and found that old HSCs showed less self-renewal activity and generated smaller daughter clones in extended serial transplants than their young counterparts [6]. These phenomena are consistent with the results that most HSCs are actively cycling during fetal life and old age, while HSCs in adulthood are often associated with quiescence [1, 16, 17]. Studies on HSCs in aged mice show an overall decrease in cell cycle activity, with old HSCs undergoing fewer cell divisions than young HSCs [18, 19]. For example, the transition from active cell cycling in fetal HSCs to quiescence in adult HSCs was associated with changes in gene expression programs, including a marked reduction in the expression of Sox17, a transcription factor required for the maintenance of fetal but not adult hematopoiesis [20, 21]. The expression levels of other genes associated with the cell cycle, such as Xrcc5, Cdadc1, Cct5, and Polr2h, are also changed during HSC aging [22].
Differentiation bias
Compared with young HSCs, aged HSCs have more myeloid differentiation potential and less B cell and T cell output after transplantation into young irradiated recipients. For example, Rossi et al. [23] found a significant reduction in the ability of old long-term HSCs (LT-HSCs) to give rise to peripheral B lymphocytes and a corresponding trend of old LT-HSCs toward increased myelopoiesis. In 2016, Nilsson et al. [24] further found that the levels of common lymphoid progenitors decreased and the frequencies of megakaryocytes and erythrocyte progenitors increased with age. Another important feature in the differentiation of aged HSCs is platelet bias. Grover et al. observed that a very high proportion of aged HSCs almost exclusively produced platelets and that HSC aging was accompanied by a coordinated upregulation of platelet lineage gene expression [25]. The lineage bias during aging was accompanied by the systemic downregulation of genes mediating lymphoid specification and function (e.g., Bcl11b, Blnk, Cd160, Cd86, Csk) and upregulation of genes involved in specifying myeloid fate and function (e.g., Amp3, Anxa7, Ap3b1, Arhgef12, Cbfa2t1h) [7, 23, 26]. In humans, the expression levels of some specific genes have been found to exhibit the same change tendencies as those in mice [13, 27]. For example, upregulated genes in aged HSCs, such as Selp, specify myeloerythroid fate, while downregulated genes, such as Flt3 and Sox4, are usually associated with lymphopoiesis [13].
Homing and engraftment
HSC transplantation is a normal and effective way to assess the functions and potential of HSCs. To test stem cell implantation ability, Liang et al. [28] injected young or old BM cells into congenic mice, and they found that the homing efficiency of old mouse was approximately three-fold lower than that of young mouse. Some specific genes have been demonstrated to be crucial in regulating HSC repopulation, such as Cdc42, Ccr9, Gnrh2, and Lep [29, 30]. CD44 is critical in the maintenance and migration of HSCs [31], and the absence of CD44 in neonatal BM was shown to enhance the long-term engraftment potential of HSCs. Additionally, p16Ink4a, a cyclin-dependent kinase inhibitor, has been shown to play an important role in stem cell regulation and HSC aging [18]. p16Ink4a-positive cells accumulate during adulthood, and this accumulation negatively influences lifespan and promotes age-dependent changes in the kidney and heart [32, 33]. Janzen et al. found that p16Ink4a expression in HSCs increased with age and that the absence of p16Ink4a could mitigate the repopulating defects and apoptosis in HSCs [18]. Moreover, Klf5 is associated with BM homing, and its enrichment is also found in LT-HSCs during aging [34,35,36].
Changes in the intrinsic signaling pathways during HSC aging
The functional decline in aged HSCs is also associated with some important signaling pathways. Here, we review the current understanding of the signaling pathways that are differentially activated or repressed during HSC aging, including the DNA damaging, Janus kinase and signal transducer and activator of transcription (JAK/STAT), nuclear factor (NF)-κB, mTOR, transforming growth factor (TGF)-β, Wnt, reactive oxygen species (ROS), and mitochondrial unfolded protein response (UPRmt) pathways.
DNA damaging pathways
DNA damage is caused by physical, chemical, and biological factors [37] and can block genome replication and transcription. The accumulation of DNA damage during aging has been observed in many studies. Rübe et al. [38] observed an increase in endogenous γH2AX-foci (a sensitive parameter for detecting DNA double-strand breaks) levels in HSCs from elderly donors. Beerman et al. [39] found that age-associated DNA damage accrual was greatest within the HSC compartment among diverse hematopoietic progenitor cells. Genome-wide analysis of young and old HSCs also identified some genes involved in DNA repair that are downregulated with age, such as Xab2, Rad52, and Xrcc1 [7]. A specific type of DNA damage is caused by the erosion of telomeres [40], and telomere shortening also occurs during aging [41, 42].
DNA damage leads to a cascade of cellular events known as the DNA damage response (DDR). The DDR is associated with age and is regulated by some important pathways, such as the nucleotide excision repair (NER) and nonhomologous end-joining (NHEJ) pathways. NER plays an important role in maintaining the functional capacity of LT-HSCs during aging by preserving the reconstitution ability, self-renewal potential, and proliferative capacity and by preventing programmed cell death under conditions of stress [43]. The NER pathway-associated gene Xab2 was shown to be downregulated in aged HSCs [7], suggesting that the NER pathway acts to restore HSC function but is weakened during aging. Another DNA repair pathway is the NHEJ pathway. Nijnik et al. [44] reported that hypomorphic mutations of murine ligase IV (Lig4y288c), a protein implicated in the NHEJ pathway, led to an age-dependent defect in hematopoiesis during aging. In addition, mice deficient in KU70 (a key component of the NHEJ pathway) exhibited severe defects in self-renewal, competitive repopulation, and BM hematopoietic niche occupancy [45]. Consistently, KU70 expression in HSCs was negatively correlated with donor age [46]. Taken together, these observations suggest that the NHEJ pathway may act to preserve HSC functions, and its downregulation during aging may contribute to HSC functional loss.
The JAK/STAT, NF-κB, and mTOR pathways
The JAK/STAT signaling pathway is a conserved metazoan signaling system that plays an important role in the immune response, homeostasis, and regenerative processes [47]. Recently, a study using single-cell transcriptomics revealed JAK/STAT signaling functions in stem cell exhaustion during aging [48]. Kirschner et al. showed that approximately 25% of p53-activated old HSCs coexpressed cell cycle inhibitory and proliferative transcripts from JAK/STAT signaling, partially explaining the prolonged cell proliferation, myeloid skewing, and stem cell exhaustion [48].
NF-κB is also known to be an important regulator of HSC aging, and its activity varies at different developmental stages [49]. Stein et al. showed that loss of the NF-κB subunit RelA/p65 severely impaired HSC functions, which occurred in conjunction with increased HSPC cycling, extramedullary hematopoiesis, and differentiation defects [50]. Chambers et al. demonstrated that 71% of 22-month-old HSCs showed enhanced nuclear localization of the p65 protein (an NF-κB subunit), in contrast to only 3% in 2-month-old HSCs, suggesting improved NF-κB activity in aged HSCs [7]. In addition, aged HSCs failed to downregulate Rad21/cohesion, a critical mediator of NF-κB signaling [51]. These results suggested that aged HSCs exhibit increased NF-κB activity.
The mTOR pathway regulates cell growth, memory, and aging by receiving signals from mitogenic growth factors, nutrients, and cellular energy levels [52,53,54]. Chen et al. observed that the levels of phosphorylated (p-)mTOR and mTOR activity were significantly higher in HSCs from aged mice than in those from young mice [8].
The TGF-β signaling pathway
The TGF-β pathway plays important roles in regulating HSC behaviors, such as quiescence, self-renewal, and differentiation [55]. Challen et al. showed that exposure to TGF-β1 exerted a stimulatory effect on myeloid-biased HSC proliferation and inhibited the turnover of lymphoid-biased HSCs in young mice [56]. In contrast, a striking reduction in myeloid cell production was found in old mice treated with TGF-β1, and aged HSCs were shown to be more sensitive to TGF-β1 than young HSCs [57]. A genome-wide transcriptome analysis also directly showed that the expression levels of regulatory genes in the TGF-β pathway (such as Smad4, Endoglin, Spectrin b2, Nr4a1, Cepba, Jun, and Junb) were reduced during HSC aging, demonstrating that TGF-β signaling in HSCs declines with age [7, 58].
The Wnt pathway
Polarity is associated with specialized functions in HSC, such as migration or division, while the loss of polarity has been correlated with reduced self-renewal and altered differentiation of HSC [59, 60]. The small RhoGTPase Cdc42 showed elevated activity in aged HSCs [61] and a correlation with polarity loss in aged HSCs [59]. Further study of the Cdc42 polarity pathway revealed that a shift from canonical to noncanonical Wnt signaling caused HSC aging [62]. Wnt5a treatment of young HSCs activated Cdc42 and induced aging-associated stem cell polarity, a reduction in regenerative capacity, and aging-like myeloid-lymphoid differentiation skewing.
The ROS and UPRmt pathway
HSCs reside in a low-oxygen BM niche and maintain low ROS levels [63]. The hypoxic niches are essential for quiescence and protect HSC from apoptosis and loss of self-renewal potential. However, with the accumulation of ROS levels, the self-renewal capacity and repopulation ability of HSC decline during aging process [64]. The thioredoxin-interacting protein (TXNIP) is a regulator of p53 and plays a pivotal role in the maintenance of the hematopoietic cells by regulating intracellular ROS during oxidative stress [65]. TXNIP-p38 axis also acts as a regulatory mechanism in HSC aging by causing lineage skewing, a decrease in engraftment, and an increase in ROS [66]. In addition, Forkhead O (FoxO) proteins play essential roles in the response to physiologic oxidative stress and thereby mediate quiescence and enhance survival in the HSC compartment [67].
Mitochondrial stress generates a reactive oxygen species (ROS)-dependent retrograde signal that modulates neutral stem cell proliferation and differentiation [68]. The UPRmt is an emerging adaptive stress response pathway that ensures optimal quality and function of the mitochondrial proteome [69]. The expression of a UPRmt regulator, Sirt7, was found to be reduced in aged HSCs, and its inactivation compromised the regenerative capacity of HSCs; furthermore, Sirt7 upregulation improved the regenerative capacity of aged HSCs [70]. Moreover, treatment with the UPRmt stimulator nicotinamide riboside induced the synthesis of prohibition proteins and rejuvenated muscle stem cells in aged mice [71].
In summary, these studies demonstrate that signaling pathways can form a network of metabolic processes and provide a systematic way to explain alterations in HSCs during aging (Fig. 1). Future studies should focus on the crosstalk among different signaling pathways and how pathways act synergistically and antagonistically with each other during HSC aging.

Functional alterations and HSC aging mechanisms. Aging negatively affects HSC functions, including decreasing self-renewal ability and myeloid/platelet-biased differentiation and impairing implantation ability. The intrinsic mechanisms are illustrated at the gene level, signaling pathway level, and epigenetic level. HSC aging is accompanied by some cell surface markers being upregulated with age (such as CD28, CD38, CD41, CD47, CD62, CD 69, CD74, and CD81) and some being downregulated with age (such as CD27, CD34, CD37, CD44, CD48, CD52, CD63, CD79b, CD86, CD97, CD97b, and CD160). Furthermore, aged HSCs show different expression levels of specific genes. For example, Amp3, Anxa7, Ap3b1, SELP, Egr1, Arhgef12, and Cbfa2t1h are upregulated, and Flt3, Xab2, Rad52, Xrcc1, Sox17, Bcl11b, and Blnk are downregulated. In addition, some signaling pathways are activated/repressed during HSC aging, including the JAK/STAT-NF-κB-mTOR pathway, TGF-β pathway, Wnt pathway, and ROS and UPRmt pathway. Age-related epigenetic regulation includes DNMT1, DNMT3A, DNMT3B, H3K4me3, and H3K27me3. Extrinsic mechanisms include HSC-surrounding cells (including MSCs, neutrophils, megakaryocytes, and macrophages), cytokines (including IL-6 and IL-1B), enzymes (including caspase-1), and β-adrenergic nerve signaling (including increased β2-AR signaling and decreased β3-AR signaling). The red box indicates that the molecule is upregulated with age, and the green box indicates that the molecule is downregulated with age
Changes at the intrinsic epigenetic level during HSC aging
Epigenetics refers to changes in gene expression but does not involve changes in the DNA sequence of organisms. Loss of epigenetic regulation at the chromatin level may drive both cellular functional attenuation and other manifestations during aging [72]. In this section, we compare the differences in the DNA methylation, histone modification, and noncoding RNAs in young and old HSCs. In addition, we summarize the research on epigenetic modification by using single-cell epigenetic technologies.
DNA methylation
DNA methylation has been shown to increase during the HSC aging process [58]. Beerman et al. [10] showed that DNA methylation changes during HSC aging occur in regions associated with HSC proliferation and lineage-biased differentiation. Furthermore, genome-wide epigenetic and transcriptome profiling identified some important age-associated regulators of methylation, such as DNA methyltransferase 1 (DNMT1), DNMT3A/B, and TET1/2 [15, 73].
DNMT1 has been shown to be essential for HSC self-renewal [74], and loss of DNMT1 causes myeloid skewing [75, 76]. Challen et al. demonstrated that conditional knockout of DNMT3A in HSCs led to increased self-renewal at the expense of differentiation [77]. Another DNMT family member, DNMT3B, was also found to be essential for HSC differentiation [77]. TET2 has also been reported to regulate HSC differentiation and increase myeloid output [78, 79]. In addition, 5-methylcytosine (5-mC) and 5-hydroxymethylcytosine (5-hmC) are two important epigenetic modifications during HSC aging [80]. The loss of the mean 5-mC content in aged leukocytes is 2% of that in young adult leukocytes [81]. Sun et al. showed a decrease in the 5-hmC level in aged mouse HSCs [58], and Busque et al. found the same phenomenon in human peripheral blood cells [82]. These alterations in aged HSC epigenetics are summarized in Table 1.
Histone modifications
Histone modifications include acetylation, methylation, phosphorylation, sumoylation, and ubiquitination and can impact gene expression by altering chromatin structure and affecting the accessibility of the DNA. There is abundant evidence that histone modifications regulate HSC functions, such as self-renewal and differentiation [86,87,88,89,90,91]. By comparing young and old mouse HSCs, Sun et al. demonstrated that H3K4me3 expression increased with age and showed a strong relationship with age-associated changes in gene expression [58]. Furthermore, broadening of the coverage and intensity of the H3K27me3 signal was observed in aged HSCs. H4K16ac is another age-associated epigenetic marker and can be pharmacologically regulated by the Cdc42 activity-specific inhibitor (CASIN) [83]. Aged LT-HSCs showed an overall lower level of H4K16ac than young LT-HSCs [59]. A recent study also demonstrated that aging was associated with significant reductions in H3K4me1, H3K27ac, and H3K4me3 [15]. Remarkably, some novel age-associated chromatin markers in hematopoietic progenitors, including H3K23ac, H2BS14ph, and H3K9me2, were observed at the single-cell level by highly multiplexed mass cytometry [85, 92]. These alterations in histone modification during HSC aging are summarized in Table 1. The regions in which histone modifications occur are not random but are associated with specific functions. For example, sites with decreased H3K4me1 were linked to genes involved in myeloid and erythroid differentiation and functions, while loss of H3K27ac was linked to genes associated with leukocyte activation, apoptotic signaling, and histone modifications [15].
Noncoding RNAs
Noncoding RNAs are RNAs that are not translated into proteins, including transfer RNA, ribosomal RNA, piwi-interacting RNA, microRNA, and long noncoding RNA (lncRNA). Many noncoding RNAs act as regulatory molecules that control gene expression and impact the epigenetic state [93, 94]. Recently, some noncoding RNAs have been demonstrated to play important roles in HSC functions, and their expression levels change with age. Djeghloul et al. observed that the expression of microRNA miR-125b increased with age in human HSCs [95]. These authors also found that inhibition of miR-125 improved the capacity of HSCs from elderly individuals to generate B cells. Another microRNA cluster, the microRNA-132/212 cluster, was upregulated and enriched during HSC aging [96]. Both overexpression and deletion of the above microRNAs led to inappropriate hematopoiesis with age [28]. In addition to miRNAs, some lncRNAs also show differential expression during aging [97].
In general, DNA methylation, histone modifications, and noncoding RNAs play important roles in regulating HSC functions during aging; however, different layers of epigenetic modifications are not independent. Age-associated histone modification changes are accompanied by alterations in DNA methylation. In HSCs, the inability to remove H3K4me1/2 methylation may prevent DNA methylation from repressing the self-renewal program [87]. Furthermore, regions in which H3K36me3 expression decreased in aged HSCs also displayed DNA hypomethylation [58, 98]. However, few studies have examined the relationship between noncoding RNAs and other epigenetic modifications. Therefore, the synergistic and antagonistic effects among different epigenetic modifications should be further elucidated.
Changes in the extrinsic HSC niche during aging
In addition to intrinsic factors, some studies also demonstrated that extrinsic factors affected HSC aging [99, 100]. The BM microenvironment niche is a crucial factor for HSC functions [101]. In the HSC niche, megakaryocytes promote the quiescence of neighboring HSCs [102]. VCAM-1+ macrophages guide the homing of HSPCs to a vascular niche [103]. On the other hand, BM CD169+ macrophages [104] and F4/80+Ly-6G+CD11b+ phagocytic macrophages [105] maintain HSC niches, but their depletion mobilizes HSCs. Furthermore, a recent study found that CD150 high BM Tregs controlled HSC quiescence and engraftment [106]. In addition to the abovementioned hematopoietic cells, nonhematopoietic cells (including mesenchymal stromal cells (MSCs), arteriolar and sinusoidal endothelial cells, and perivascular cells) also play crucial roles in the HSC niche [100, 107, 108]. Moreover, the sympathetic nervous system (SNS) regulates HSC mobilization and orchestrates the release of adrenergic neurotransmitters into the microenvironment in a circadian manner [109,110,111,112]. In this section, the effects of HSCs surrounding cells, the SNS, and other factors on HSC aging are summarized (Fig. 2).

Cell-extrinsic mechanisms of HSC aging involved in HSC-surrounding cells (including megakaryocytes, MSCs, macrophages, and neutrophils), cytokines and enzymes (including IL-6, IL-1B, and caspase-1). Disrupted β-adrenergic nerve signaling (increased β2-AR-IL6-mediated megakaryocyte differentiation and reduced β3-AR-Nos1 activity) is an important determinant of niche alterations during aging, resulting in impaired lymphoid differentiation and myeloid expansion. Dysfunction of aged marrow macrophages directs HSC platelet bias; aged mice have markedly more senescent neutrophils and higher levels of cytokines IL-1B and caspase-1 in their BM niche than young mice. The number of MSCs increases significantly during aging and is associated with replicative senescence and HSC homing
In 2018, Maryanovich found that HSC aging critically depended on BM innervation by the SNS around arteriolar niches, as loss of SNS nerves or adrenoreceptor β3 (ADRβ3) signaling in the young mouse BM microenvironment led to premature HSC aging [109]. Furthermore, the distance between HSCs and megakaryocytes regulates HSC proliferation and increases in β3-adrenergic receptor (AR) mice and in natural aging. In 2019, Ho et al. also identified disrupted β-adrenergic nerve signaling (increased β2-AR-interleukin (IL)-6 levels and decreased β3-AR-Nos1 activity) as an important determinant of niche alterations during aging, resulting in impaired lymphoid differentiation and myeloid expansion [113]. In addition, Frisch et al. showed that dysfunction of aged marrow macrophages directed HSC platelet bias and that aged mice exhibited markedly more senescent neutrophils than young mice [11]. Finally, the aged mouse BM niche also expressed elevated levels of cytokines IL-1B and caspase-1. These works highlighted the instructive role of megakaryocytes and macrophages in the age-associated lineage skewing of HSCs.
For nonhematopoietic cells, the number of MSC subsets (such as PDGFRa+SCA1+ and PDGFRa+CD51+ cells) in aged mouse BM was found to be significantly higher than that in young mouse BM [11]. Furthermore, in 2018, Hennrich et al. presented proteome-wide atlases of age-associated alterations in HSPCs, lymphocytes and precursors, monocytes/macrophages and precursors, granulocytes, erythroid precursors, and MSCs [114]. In aged MSCs, prominent alterations included differential regulation of proteins that were associated with cellular responses to stress, replicative senescence, and HSC homing.
Single cell technologies for HSC aging
Single-cell RNA sequencing
Single-cell RNA sequencing has become a powerful tool to characterize distinct functional states at single-cell resolution [115,116,117]. Several studies using single-cell RNA sequencing have been performed to reveal cell-intrinsic differences during HSC aging [22, 25].
Upon comparing young and old mouse HSCs, Kowalczyk et al. found that cell cycle-related genes dominated the transcriptomic variability and observed fewer cells in the G1 phase among old HSCs [22]. Moreover, old short-term HSCs (ST-HSCs) transcriptionally resembled young LT-HSCs, suggesting that ST-HSCs remained in a less differentiated state. Grover et al. showed that depletion of HSC platelet programming through loss of the Fog-1 transcription factor was accompanied by increased lymphoid output [25]. Mann et al. found that LT-HSCs from young and aged mice had differential responses to inflammatory challenge and that age-dependent inflammatory myeloid bias was intrinsic to LT-HSCs [34].
In addition to obtaining single-cell transcriptional results from mice, Oetjen et al. also performed a comprehensive assessment of human BM cells using single-cell RNA sequencing [118]. They found consistent increases in chromatin markers in a broad array of cell subtypes from hematopoietic progenitors to terminally differentiated immune cells. Hennrich et al. revealed that the mRNA levels of age-increased glycolytic enzymes were higher in myeloid-primed hematopoietic stem/progenitor cells (HSPCs) than in lymphoid-primed HSPCs, whereas the transcript levels of age-unaffected enzymes were similar in both subsets [114]. These results suggested that the lineage skewing of HSPCs toward myeloid differentiation upon aging was associated with the glycolytic pathway. Adelman et al. observed a decrease in cycling-HSCs and lymphoid-primed multipotent progenitors during aging and identified 364 genes that are differentially expressed with age, such as Egr1, Klf6, and Jun [15]. Overall, these single-cell analyses, summarized in Table 2, helped to demonstrate the intrinsic molecular changes during HSC aging.
Single-cell transcriptomics data provide abundant information about the differential gene expression of young and aged HSCs. The expression of surface molecules is a research hotspot, as these molecules can be used to track the aging process of HSCs and assess HSC functions. Using normalized transcription expression matrices, different studies [7, 11, 13, 22, 24, 25, 120, 121] found that CD28, CD38, CD41, CD61, CD47, CD62, CD69, CD74, and CD81 were upregulated with age and that CD27, CD34, CD37, CD44, CD48, CD52, CD63, CD79b, CD86, CD97, CD97b, and CD160 were downregulated with age. Furthermore, some molecules (such as CD9 and CD151) showed contrasting expression tendencies with age in different studies [7, 10, 24]. In addition, a histone 2B-green fluorescent protein label in HSCs (an HSPC-specific GFP label-retaining system) was used to label a reserve stem cell population [122, 123]. These molecules show an exact tendency during aging and could be used to identify aged HSCs and assess HSC functions (Table 3).
Epigenetics at the single-cell level
The development of single-cell epigenomic technologies has allowed the identification of DNA methylation, histone modifications, chromatin accessibility, and chromosome conformation at the single-cell level [117, 124]. For example, single-cell chromatin immunoprecipitation sequencing and single-cell assays for transposase-accessible chromatin using sequencing (scATAC-seq) have been applied to investigate histone modifications and to map accessible chromatin regions.
Florian et al. observed that young HSCs divided mainly asymmetrically, while aged HSCs divided primarily symmetrically [119]. Moreover, the potential of daughter cells was linked to the amount of the epigenetic marker H4K16ac and to the amount of open chromatin. Cheung et al. developed epigenetic landscape profiling using cytometry by time-of-flight (EpiTOF) to measure epigenetic modifications and profile the global levels of a broad array of chromatin modifications in primary human immune cells at the single-cell level [85]. Consistent increases in chromatin markers were found in a broad array of cell subtypes from hematopoietic progenitors to terminally differentiated immune cells, suggesting that systemic changes may result from the reprogrammed chromatin state in hematopoietic progenitors or further upstream in HSCs. These single-cell epigenetic analyses contributed to our understanding of the distinct types of epigenetic alterations occurring during aging at single-cell resolution (Table 2).
Aged HSC rejuvenation strategies
Currently, there is no doubt that HSCs show declining function during aging, but whether this dysfunction is reversible remains unclear. In 2014, Villeda et al. reported that exposure of an aged animal to young blood can counteract and reverse pre-existing effects of brain aging at the molecular, structural, functional, and cognitive levels, suggesting that some aging-related phenotypes can be improved [125]. In this section, we review the approaches to achieve at least partial rejuvenation of aged HSC functions, including prolonged fasting, genetic modulators, pharmacological intervention, and changing the BM niche (Table 4).
Prolonged fasting
Prolonged fasting reduces progrowth signaling and activates pathways that enhance cellular resistance to toxins in mice and humans [131,132,133]. Prolonged fasting can protect mice from chemotoxicity by reducing circulating insulin-like growth factor-1 (IGF-1) expression [134, 135]. Notably, Cheng et al. showed that prolonged fasting can rejuvenate HSCs. Prolonged fasting reduces circulating IGF-1 levels and protein kinase A (PKA) activity in various cell populations and promotes stress resistance, self-renewal, and lineage-balanced regeneration [126].
Genetic modulators (Satb1, Sirt3, and Sirt7)
HSC aging is accompanied by alterations in gene expression. Therefore, overexpressing some downregulated genes or knocking down the expression of some upregulated genes might be strategies to prevent HSC dysfunction. Reduced Satb1 expression was found in aged HSCs and associated with compromised lymphopoietic potential, and forced Satb1 overexpression partially restored that potential [127]. In addition, the expression of Sirt3, which regulates the global acetylation landscape of mitochondrial proteins, was suppressed by aging, and Sirt3 upregulation in aged HSCs improved the HSC regenerative capacity [128]. A similar phenomenon was also found for Sirt7 [70]. Although overexpression of some regulators has been found to rejuvenate HSCs, few studies have revealed a relationship between gene knockdown and HSC rejuvenation. In the future, gene knockdown might be another credible way to restore HSC functions and identify the functions of some genes in rejuvenation.
Pharmacological intervention
Rapamycin (mTOR inhibitor)
Aged mice exhibit increased mTOR signaling in HSCs, and rapamycin can enhance the regenerative capacity of HSCs from aged mice, improve their immune response, and extend their life span [8].
CASIN (Cdc42 inhibitor)
Cdc42 regulates diverse cellular functions, including cellular transformation, cell division, migration, enzyme activity, and cell polarity [136]. Aged HSCs show elevated Cdc42 activity, and Cdc42 inhibition has been demonstrated to rejuvenate HSC functions. Treated with CASIN in vitro, aged HSCs showed an increase in the percentage of polarized cells, and their H4K16ac level and spatial distribution were rejuvenated to a status similar to that in young HSCs [59]. In addition, CASIN treatment increased the contribution to the B cell compartment in peripheral blood and reduced the contribution to the myeloid lineage. Recently, another study also found that aged HSCs treated with CASIN reestablished an immune system similar to that of young animals [137].
TN13 and SB203580 (p38 MAPK inhibitor)
Inhibition of p38 MAPK reduces ROS levels and contributes to HSC rejuvenation. TN13, a cell-penetrating peptide-conjugated peptide, inhibited p38 activity and rejuvenated aged HSCs by reducing ROS [66]. Another inhibitor of p38 MAPK, SB203580, was also found to rescue ROS-induced defects in the repopulating capacity of HSCs and the maintenance of HSC quiescence [9]. These results are consistent with the phenomenon that HSCs reside in a low-oxygen BM niche and that the ROS level of HSCs increases during aging [138, 139].
ABT263 (clearance of senescent cells)
One strategy to delay aging is to restore cell functions, while another is to clear senescent cells. Senescent cells accumulate with age and contribute to the development of aging-related diseases [140,141,142]. Depletion of senescent cells mitigated irradiation-induced premature aging of the hematopoietic system and rejuvenated aged HSCs in normally aged mice. Chang et al. found that ABT263, a specific inhibitor of the antiapoptotic proteins BCL-2 and BCL-xL, selectively killed senescent cells [129]. Oral administration of ABT263 to sublethally irradiated mice and normally aged mice effectively depleted senescent cells, including senescent BM HSCs.
Changing the BM niche
Kuribayashi et al. showed that engrafting aged HSCs into young niches restored the age-related transcriptional profiles of HSCs. They transplanted 20-month-old aged HSCs into 10-week-old young mice and later collected aged HSCs engrafted in young mice (aged/Y). The gene expression profiles of aged/Y HSCs were reprogrammed to a large extent similar to those of young HSCs [130]. Another study showed that BM innervation by the SNS influenced the function of HSCs and that supplementation with a sympathomimetic (β3-AR agonist, BRL37344) significantly rejuvenated the in vivo functions of aged HSCs in old mice [109]. Furthermore, chronic treatment of progeroid mice with BRL37344 decreased premature myeloid and HSC expansion and restored the proximal association of HSCs to megakaryocytes [113].
Conclusions
In the present review, we summarize the hallmarks of HSC aging in self-renewal, differentiation bias, and implantation ability. On the one hand, HSC aging is driven by multiple cell-intrinsic factors, including gene expression alterations, signaling pathway activation/repression, and epigenetic regulation. We review recent HSC aging studies with high-throughput single-cell sequencing at both the transcriptomic and epigenomic levels and summarize the single-cell sequencing data on age-associated surface molecules. On the other hand, some cell-extrinsic factors, including HSC-surrounding cells (such as megakaryocytes, MSCs, macrophages, and neutrophils), β-adrenergic nerve signals, cytokines, and enzymes (including IL-6, IL-1B, and caspase-1), also affect HSC aging. Finally, we review some strategies that have been employed to rejuvenate aged HSCs based on the above intrinsic and extrinsic mechanisms, including prolonged fasting, gene expression regulation, pharmacological intervention, and changing the BM niche.
Classical knowledge about hematopoiesis, which is built on a system defined with cell surface markers, is rather restricted and has been challenged. Compared with bulk sequencing, single-cell RNA sequencing technologies allow the dissection of gene expression at single-cell resolution, which provide unprecedented insight into cellular heterogeneity and HSC aging mechanisms. With evolvement of single cell technologies, researchers can profile multiple epigenetic marks within the same single cell and do so in combination with transcriptional information. However, considering the limitations of transcript coverage, low capture efficiency, high costs, and restricted cell throughput facing current single-cell sequencing methods, future single-cell strategies should be designed to conduct full-length sequencing and achieve a balance between high-throughput analysis and sufficient sequencing depth. In the future, single cell technologies and other emerging new technologies will pave the way for manipulation of the transcriptome and epigenome to rejuvenate aged HSC.
Although the latest single-cell studies delineated the association between functional decline and molecular mechanism, these studies did not conclusively identify driving factors of HSC aging. Whether these alterations drive HSC aging or whether these alterations are only accompanied by HSC aging remains unknown. Therefore, gene editing experiments should be performed to determine the specific functions of each gene. In addition, while many studies have directly studied aged HSCs, few studies have examined the HSC niche or revealed the specific functions of each cell subset in the HSC niche during aging. Furthermore, most strategies to rejuvenate aged HSCs directly act on HSCs, and few studies have tried to affect the HSC niche. Therefore, future work should emphasize the mechanisms of the HSC niche during aging. Moreover, expanding long-term HSCs in vitro is still a challenge, and the findings of HSC aging could be applied to this challenge [143,144,145,146]. For example, some rejuvenation molecules might be added to expand HSCs. Finally, the declining immune ability of the elderly population might be associated with aged hematopoietic cell dysfunction [147, 148]. These problems may be effectively overcome once HSC aging mechanisms are fully revealed and rejuvenation strategies are optimized.
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Abbreviations
- HSCs:
-
Hematopoietic stem cells
- ROS:
-
Reactive oxygen species
- mTOR:
-
Mammalian target of rapamycin
- MAPK:
-
Mitogen-activated protein kinase
- LT-HSCs:
-
Long-term HSCs
- BM:
-
Bone marrow
- ST-HSCs:
-
Short-term HSCs
- MPP:
-
Multipotent progenitors
- HSPCs:
-
Hematopoietic stem and progenitor cells
- DDR:
-
DNA damage response
- NER:
-
Nucleotide excision repair
- NHEJ:
-
Non-homologous end-joining
- JAK/STAT:
-
Janus kinase and signal transducer and activator of transcription
- NF-κB:
-
Nuclear factor-κB
- TGF-β:
-
Transforming growth factor-β
- UPRmt :
-
Mitochondrial unfolded protein response
- TXNIP:
-
Thioredoxin-interacting protein
- FoxO:
-
Forkhead O
- 5-mC:
-
5-Methylcytosine
- 5-hmC:
-
5-Hydroxymethylcytosine
- scATAC-seq:
-
Single-cell assay for transposase-accessible chromatin using sequencing
- EpiTOF:
-
Epigenetic landscape profiling using cytometry by time-of-flight
- SNS:
-
Sympathetic nervous system
- ADRβ3:
-
Adrenoreceptor β3
- IL-6:
-
Interleukin-6
- IL-1B:
-
Interleukin-1B
- MSCs:
-
Mesenchymal stromal cell
- IGF-1:
-
Insulin-like growth factor-1
References
Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2(9):1011–6.
Xu L, Chen H, Chen J, Han M, Huang H, Lai Y, Liu D, Liu Q, Liu T, Jiang M, et al. The consensus on indications, conditioning regimen, and donor selection of allogeneic hematopoietic cell transplantation for hematological diseases in China-recommendations from the Chinese Society of Hematology. J Hematol Oncol. 2018;11(1):33.
Xu LP, Wang SQ, Ma YR, Gao SJ, Cheng YF, Zhang YY, Mo WJ, Mo XD, Zhang YP, Yan CH, et al. Who is the best haploidentical donor for acquired severe aplastic anemia? Experience from a multicenter study. J Hematol Oncol. 2019;12(1):87.
Wang Y, Chang YJ, Xu LP, Liu KY, Liu DH, Zhang XH, Chen H, Han W, Chen YH, Wang FR, et al. Who is the best donor for a related HLA haplotype-mismatched transplant? Blood. 2014;124(6):843–50.
Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, Rossi DJ. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A. 2010;107(12):5465–70.
Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. 2011;208(13):2691–703.
Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5(8):e201.
Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009;2(98):ra75.
Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446–51.
Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, Rossi DJ. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013;12(4):413–25.
Frisch BJ, Hoffman CM, Latchney SE, LaMere MW, Myers J, Ashton J, Li AJ, Saunders J 2nd, Palis J, Perkins AS, et al. Aged marrow macrophages expand platelet-biased hematopoietic stem cells via Interleukin1B. JCI Insight. 2019;5(10):e124213.
de Haan G, Nijhof W, Van Zant G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 1997;89(5):1543–50.
Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, Weissman IL. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A. 2011;108(50):20012–7.
Dykstra B, de Haan G. Hematopoietic stem cell aging and self-renewal. Cell Tissue Res. 2008;331(1):91–101.
Adelman ER, Huang HT, Roisman A, Olsson A, Colaprico A, Qin T, Lindsley RC, Bejar R, Salomonis N, Grimes HL, et al. Aging human hematopoietic stem cells manifest profound epigenetic reprogramming of enhancers that may predispose to leukemia. Cancer Discov. 2019;9(8):1080–101.
Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest. 2006;116(10):2808–16.
Nygren JM, Bryder D, Jacobsen SE. Prolonged cell cycle transit is a defining and developmentally conserved hemopoietic stem cell property. J Immunol. 2006;177(1):201–8.
Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443(7110):421–6.
Nygren JM, Bryder D. A novel assay to trace proliferation history in vivo reveals that enhanced divisional kinetics accompany loss of hematopoietic stem cell self-renewal. PLoS One. 2008;3(11):e3710.
Kim I, Saunders TL, Morrison SJ. Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell. 2007;130(3):470–83.
Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195(5):709–20.
Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, Regev A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015;25(12):1860–72.
Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102(26):9194–9.
Rundberg Nilsson A, Soneji S, Adolfsson S, Bryder D, Pronk CJ. Human and murine hematopoietic stem cell aging is associated with functional impairments and intrinsic megakaryocytic/erythroid bias. PLoS One. 2016;11(7):e0158369.
Grover A, Sanjuan-Pla A, Thongjuea S, Carrelha J, Giustacchini A, Gambardella A, Macaulay I, Mancini E, Luis TC, Mead A, et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat Commun. 2016;7:11075.
Moehrle BM, Geiger H. Aging of hematopoietic stem cells: DNA damage and mutations? Exp Hematol. 2016;44(10):895–901.
Chambers SM, Boles NC, Lin KY, Tierney MP, Bowman TV, Bradfute SB, Chen AJ, Merchant AA, Sirin O, Weksberg DC, et al. Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell. 2007;1(5):578–91.
Liang Y, Van Zant G, Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106(4):1479–87.
Gur-Cohen S, Kollet O, Graf C, Esmon CT, Ruf W, Lapidot T. Regulation of long-term repopulating hematopoietic stem cells by EPCR/PAR1 signaling. Ann N Y Acad Sci. 2016;1370(1):65–81.
Kode J, Khattry N, Bakshi A, Amrutkar V, Bagal B, Karandikar R, Rane P, Fujii N, Chiplunkar S. Study of stem cell homing & self-renewal marker gene profile of ex vivo expanded human CD34(+) cells manipulated with a mixture of cytokines & stromal cell-derived factor 1. Indian J Med Res. 2017;146(1):56–70.
Cao H, Heazlewood SY, Williams B, Cardozo D, Nigro J, Oteiza A, Nilsson SK. The role of CD44 in fetal and adult hematopoietic stem cell regulation. Haematologica. 2016;101(1):26–37.
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530(7589):184–9.
Liu Y, Johnson SM, Fedoriw Y, Rogers AB, Yuan H, Krishnamurthy J, Sharpless NE. Expression of p16(INK4a) prevents cancer and promotes aging in lymphocytes. Blood. 2011;117(12):3257–67.
Mann M, Mehta A, de Boer CG, Kowalczyk MS, Lee K, Haldeman P, Rogel N, Knecht AR, Farouq D, Regev A, et al. Heterogeneous responses of hematopoietic stem cells to inflammatory stimuli are altered with age. Cell Rep. 2018;25(11):2992–3005.e2995.
Shahrin NH, Diakiw S, Dent LA, Brown AL, D'Andrea RJ. Conditional knockout mice demonstrate function of Klf5 as a myeloid transcription factor. Blood. 2016;128(1):55–9.
Taniguchi Ishikawa E, Chang KH, Nayak R, Olsson HA, Ficker AM, Dunn SK, Madhu MN, Sengupta A, Whitsett JA, Grimes HL, et al. Klf5 controls bone marrow homing of stem cells and progenitors through Rab5-mediated beta1/beta2-integrin trafficking. Nat Commun. 2013;4:1660.
Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA. Environmental and chemical carcinogenesis. Semin Cancer Biol. 2004;14(6):473–86.
Rube CE, Fricke A, Widmann TA, Furst T, Madry H, Pfreundschuh M, Rube C. Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS One. 2011;6(3):e17487.
Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell. 2014;15(1):37–50.
Wang L, Xiao H, Zhang X, Wang C, Huang H. The role of telomeres and telomerase in hematologic malignancies and hematopoietic stem cell transplantation. J Hematol Oncol. 2014;7:61.
Zimmermann S, Martens UM. Telomeres, senescence, and hematopoietic stem cells. Cell Tissue Res. 2008;331(1):79–90.
Vaziri H, Dragowska W, Allsopp RC, Thomas TE, Harley CB, Lansdorp PM. Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc Natl Acad Sci U S A. 1994;91(21):9857–60.
Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–9.
Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, Rodrigues NP, Crockford TL, Cabuy E, Vindigni A, et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447(7145):686–90.
Qing Y, Wang Z, Bunting KD, Gerson SL. Bcl2 overexpression rescues the hematopoietic stem cell defects in Ku70-deficient mice by restoration of quiescence. Blood. 2014;123(7):1002–11.
Prall WC, Czibere A, Jager M, Spentzos D, Libermann TA, Gattermann N, Haas R, Aivado M. Age-related transcription levels of KU70, MGST1 and BIK in CD34+ hematopoietic stem and progenitor cells. Mech Ageing Dev. 2007;128(9):503–10.
Schubert C, Allhoff M, Tillmann S, Maie T, Costa IG, Lipka DB, Schemionek M, Feldberg K, Baumeister J, Brummendorf TH, et al. Differential roles of STAT1 and STAT2 in the sensitivity of JAK2V617F- vs. BCR-ABL-positive cells to interferon alpha. J Hematol Oncol. 2019;12(1):36.
Kirschner K, Chandra T, Kiselev V, Flores-Santa Cruz D, Macaulay IC, Park HJ, Li J, Kent DG, Kumar R, Pask DC, et al. Proliferation drives aging-related functional decline in a subpopulation of the hematopoietic stem cell compartment. Cell Rep. 2017;19(8):1503–11.
Bottero V, Withoff S, Verma IM. NF-kappaB and the regulation of hematopoiesis. Cell Death Differ. 2006;13(5):785–97.
Stein SJ, Baldwin AS. Deletion of the NF-kappaB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood. 2013;121(25):5015–24.
Chen Z, Amro EM, Becker F, Holzer M, Rasa SMM, Njeru SN, Han B, Di Sanzo S, Chen Y, Tang D, et al. Cohesin-mediated NF-kappaB signaling limits hematopoietic stem cell self-renewal in aging and inflammation. J Exp Med. 2019;216(1):152–75.
Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23(18):3151–71.
Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–84.
Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–45.
Blank U, Karlsson S. TGF-beta signaling in the control of hematopoietic stem cells. Blood. 2015;125(23):3542–50.
Challen GA, Boles NC, Chambers SM, Goodell MA. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell. 2010;6(3):265–78.
Quere R, Saint-Paul L, Carmignac V, Martin RZ, Chretien ML, Largeot A, Hammann A, Pais de Barros JP, Bastie JN, Delva L. Tif1gamma regulates the TGF-beta1 receptor and promotes physiological aging of hematopoietic stem cells. Proc Natl Acad Sci U S A. 2014;111(29):10592–7.
Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14(5):673–88.
Florian MC, Dorr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, Filippi MD, Hasenberg A, Gunzer M, Scharffetter-Kochanek K, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10(5):520–30.
Singh AK, Althoff MJ, Cancelas JA. Signaling pathways regulating hematopoietic stem cell and progenitor aging. Curr Stem Cell Rep. 2018;4(2):166–81.
Etienne-Manneville S. Cdc42--the centre of polarity. J Cell Sci. 2004;117(Pt 8):1291–300.
Florian MC, Nattamai KJ, Dorr K, Marka G, Uberle B, Vas V, Eckl C, Andra I, Schiemann M, Oostendorp RA, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503(7476):392–6.
Lee J, Cho YS, Jung H, Choi I. Pharmacological regulation of oxidative stress in stem cells. Oxid Med Cell Longev. 2018;2018:4081890.
Jang Y-Y, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110(8):3056–63.
Jung H, Kim MJ, Kim DO, Kim WS, Yoon S-J, Park Y-J, Yoon SR, Kim T-D, Suh H-W, Yun S, et al. TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab. 2013;18(1):75–85.
Jung H, Kim DO, Byun J-E, Kim WS, Kim MJ, Song HY, Kim YK, Kang D-K, Park Y-J, Kim T-D, et al. Thioredoxin-interacting protein regulates haematopoietic stem cell ageing and rejuvenation by inhibiting p38 kinase activity. Nat Commun. 2016;7(1):13674.
Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–39.
Xavier JM, Morgado AL, Rodrigues CM, Sola S. Tauroursodeoxycholic acid increases neural stem cell pool and neuronal conversion by regulating mitochondria-cell cycle retrograde signaling. Cell Cycle. 2014;13(22):3576–89.
Jovaisaite V, Auwerx J. The mitochondrial unfolded protein response-synchronizing genomes. Curr Opin Cell Biol. 2015;33:74–81.
Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, Haynes CM, Chen D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347(6228):1374–7.
Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D'Amico D, Ropelle ER, Lutolf MP, Aebersold R, et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352(6292):1436–43.
Choudry FA, Frontini M. Epigenetic control of haematopoietic stem cell aging and its clinical implications. Stem Cells Int. 2016;2016:5797521.
Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, Yu Y, Bhattacharyya S, Shaknovich R, Geng H, Lobry C, et al. TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol. 2015;16(6):653–62.
Liu X, Jia X, Yuan H, Ma K, Chen Y, Jin Y, Deng M, Pan W, Chen S, Chen Z, et al. DNA methyltransferase 1 functions through C/ebpa to maintain hematopoietic stem and progenitor cells in zebrafish. J Hematol Oncol. 2015;8:15.
Broske AM, Vockentanz L, Kharazi S, Huska MR, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009;41(11):1207–15.
Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009;5(4):442–9.
Challen GA, Sun D, Mayle A, Jeong M, Luo M, Rodriguez B, Mallaney C, Celik H, Yang L, Xia Z, et al. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell. 2014;15(3):350–64.
Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci U S A. 2011;108(35):14566–71.
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24.
Oshima M, Iwama A. Epigenetics of hematopoietic stem cell aging and disease. Int J Hematol. 2014;100(4):326–34.
Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, Miura K, Miyazaki T, Ogura C, Okazaki Y, Jinno Y. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Ann Hum Genet. 2004;68(Pt 3):196–204.
Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179–81.
Grigoryan A, Guidi N, Senger K, Liehr T, Soller K, Marka G, Vollmer A, Markaki Y, Leonhardt H, Buske C, et al. LaminA/C regulates epigenetic and chromatin architecture changes upon aging of hematopoietic stem cells. Genome Biol. 2018;19(1):189.
Towbin BD, González-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 2012;150(5):934–47.
Cheung P, Vallania F, Warsinske HC, Donato M, Schaffert S, Chang SE, Dvorak M, Dekker CL, Davis MM, Utz PJ, et al. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell. 2018;173(6):1385–97 e1314.
Stewart MH, Albert M, Sroczynska P, Cruickshank VA, Guo Y, Rossi DJ, Helin K, Enver T. The histone demethylase Jarid1b is required for hematopoietic stem cell self-renewal in mice. Blood. 2015;125(13):2075–8.
Kerenyi MA, Shao Z, Hsu YJ, Guo G, Luc S, O'Brien K, Fujiwara Y, Peng C, Nguyen M, Orkin SH. Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. Elife. 2013;2:e00633.
Katsumoto T, Yoshida N, Kitabayashi I. Roles of the histone acetyltransferase monocytic leukemia zinc finger protein in normal and malignant hematopoiesis. Cancer Sci. 2008;99(8):1523–7.
Cellot S, Hope KJ, Chagraoui J, Sauvageau M, Deneault E, MacRae T, Mayotte N, Wilhelm BT, Landry JR, Ting SB, et al. RNAi screen identifies Jarid1b as a major regulator of mouse HSC activity. Blood. 2013;122(9):1545–55.
Attema JL, Papathanasiou P, Forsberg EC, Xu J, Smale ST, Weissman IL. Epigenetic characterization of hematopoietic stem cell differentiation using miniChIP and bisulfite sequencing analysis. Proc Natl Acad Sci U S A. 2007;104(30):12371–6.
Maes J, Maleszewska M, Guillemin C, Pflumio F, Six E, Andre-Schmutz I, Cavazzana-Calvo M, Charron D, Francastel C, Goodhardt M. Lymphoid-affiliated genes are associated with active histone modifications in human hematopoietic stem cells. Blood. 2008;112(7):2722–9.
Towbin BD, Gonzalez-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 2012;150(5):934–47.
Liu Y, Cheng Z, Pang Y, Cui L, Qian T, Quan L, Zhao H, Shi J, Ke X, Fu L. Role of microRNAs, circRNAs and long noncoding RNAs in acute myeloid leukemia. J Hematol Oncol. 2019;12(1):51.
James AR, Schroeder MP, Neumann M, Bastian L, Eckert C, Gokbuget N, Tanchez JO, Schlee C, Isaakidis K, Schwartz S, et al. Long non-coding RNAs defining major subtypes of B cell precursor acute lymphoblastic leukemia. J Hematol Oncol. 2019;12(1):8.
Djeghloul D, Kuranda K, Kuzniak I, Barbieri D, Naguibneva I, Choisy C, Bories JC, Dosquet C, Pla M, Vanneaux V, et al. Age-associated decrease of the histone methyltransferase SUV39H1 in HSC perturbs heterochromatin and B lymphoid differentiation. Stem Cell Reports. 2016;6(6):970–84.
Mehta A, Zhao JL, Sinha N, Marinov GK, Mann M, Kowalczyk MS, Galimidi RP, Du X, Erikci E, Regev A, et al. The microRNA-132 and microRNA-212 cluster regulates hematopoietic stem cell maintenance and survival with age by buffering FOXO3 expression. Immunity. 2015;42(6):1021–32.
Luo M, Jeong M, Sun D, Park HJ, Rodriguez BA, Xia Z, Yang L, Zhang X, Sheng K, Darlington GJ, et al. Long non-coding RNAs control hematopoietic stem cell function. Cell Stem Cell. 2015;16(4):426–38.
Beerman I, Rossi DJ. Epigenetic control of stem cell potential during homeostasis, aging, and disease. Cell Stem Cell. 2015;16(6):613–25.
Ergen AV, Boles NC, Goodell MA. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood. 2012;119(11):2500–9.
Kusumbe AP, Ramasamy SK, Itkin T, Mae MA, Langen UH, Betsholtz C, Lapidot T, Adams RH. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. 2016;532(7599):380–4.
Ventura Ferreira MS, Bergmann C, Bodensiek I, Peukert K, Abert J, Kramann R, Kachel P, Rath B, Rutten S, Knuchel R, et al. An engineered multicomponent bone marrow niche for the recapitulation of hematopoiesis at ectopic transplantation sites. J Hematol Oncol. 2016;9:4.
Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20(11):1315–20.
Li D, Xue W, Li M, Dong M, Wang J, Wang X, Li X, Chen K, Zhang W, Wu S, et al. VCAM-1+ macrophages guide the homing of HSPCs to a vascular niche. Nature. 2018;564(7734):119–24.
Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, Battista M, Leboeuf M, Prophete C, van Rooijen N, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011;208(2):261–71.
Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116(23):4815–28.
Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, Robson SC, Frenette PS, Fujisaki J. CD150(high) bone marrow tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell. 2018;22(3):445–53 e445.
Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457–62.
Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, Jaiyeola C, Zhao Z, Luby-Phelps K, Morrison SJ. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526(7571):126–30.
Maryanovich M, Zahalka AH, Pierce H, Pinho S, Nakahara F, Asada N, Wei Q, Wang X, Ciero P, Xu J, et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat Med. 2018;24(6):782–91.
Katayama Y, Battista M, Kao W-M, Hidalgo A, Peired AJ, Thomas SA, Frenette PS. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124(2):407–21.
Méndez-Ferrer S, Battista M, Frenette PS. Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann N Y Acad Sci. 2010;1192:139–44.
Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452(7186):442–7.
Ho YH, Del Toro R, Rivera-Torres J, Rak J, Korn C, Garcia-Garcia A, Macias D, Gonzalez-Gomez C, Del Monte A, Wittner M, et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell. 2019;25(3):407–18 e406.
Hennrich ML, Romanov N, Horn P, Jaeger S, Eckstein V, Steeples V, Ye F, Ding X, Poisa-Beiro L, Lai MC, et al. Cell-specific proteome analyses of human bone marrow reveal molecular features of age-dependent functional decline. Nat Commun. 2018;9(1):4004.
Tang F, Barbacioru C, Nordman E, Li B, Xu N, Bashkirov VI, Lao K, Surani MA. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc. 2010;5(3):516–35.
Tang F, Barbacioru C, Bao S, Lee C, Nordman E, Wang X, Lao K, Surani MA. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell. 2010;6(5):468–78.
Ye F, Huang W, Guo G. Studying hematopoiesis using single-cell technologies. J Hematol Oncol. 2017;10(1):27.
Oetjen KA, Lindblad KE, Goswami M, Gui G, Dagur PK, Lai C, Dillon LW, McCoy JP, Hourigan CS. Human bone marrow assessment by single-cell RNA sequencing, mass cytometry, and flow cytometry. JCI Insight. 2018;3(23).
Florian MC, Klose M, Sacma M, Jablanovic J, Knudson L, Nattamai KJ, Marka G, Vollmer A, Soller K, Sakk V, et al. Aging alters the epigenetic asymmetry of HSC division. PLoS Biol. 2018;16(9):e2003389.
Gekas C, Graf T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood. 2013;121(22):4463–72.
Shimazu T, Iida R, Zhang Q, Welner RS, Medina KL, Alberola-Lla J, Kincade PW. CD86 is expressed on murine hematopoietic stem cells and denotes lymphopoietic potential. Blood. 2012;119(21):4889–97.
Qiu J, Papatsenko D, Niu X, Schaniel C, Moore K. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Rep. 2014;2(4):473–90.
Bernitz JM, Kim HS, MacArthur B, Sieburg H, Moore K. Hematopoietic stem cells count and remember self-renewal divisions. Cell. 2016;167(5):1296–309 e1210.
Clark SJ, Lee HJ, Smallwood SA, Kelsey G, Reik W. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016;17:72.
Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med. 2014;20:659.
Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, Da Sacco S, Mirisola M, Quinn DI, Dorff TB, et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell. 2014;14(6):810–23.
Satoh Y, Yokota T, Sudo T, Kondo M, Lai A, Kincade Paul W, Kouro T, Iida R, Kokame K, Miyata T, et al. The Satb1 protein directs hematopoietic stem cell differentiation toward lymphoid lineages. Immunity. 2013;38(6):1105–15.
Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, Zhang D, Scadden DT, Chen D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013;3(2):319–27.
Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22(1):78–83.
Kuribayashi W, Iwama A, Oshima M. Incomplete rejuvenation of aged hscs in young bone marrow niche. Exp Hematol. 2019;76:S72.
Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421(6919):182–7.
Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010;328(5976):321–6.
Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW, Hwang D, Martin-Montalvo A, Saavedra J, Ingles S, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3(70):70ra13.
Lee C, Safdie FM, Raffaghello L, Wei M, Madia F, Parrella E, Hwang D, Cohen P, Bianchi G, Longo VD. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 2010;70(4):1564–72.
Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, Longo VD. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A. 2008;105(24):8215–20.
Qadir MI, Parveen A, Ali M. Cdc42: role in cancer management. Chem Biol Drug Des. 2015;86(4):432–9.
Leins H, Mulaw M, Eiwen K, Sakk V, Liang Y, Denkinger M, Geiger H, Schirmbeck R. Aged murine hematopoietic stem cells drive aging-associated immune remodeling. Blood. 2018;132(6):565–76.
Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A. 2001;98(18):10469–74.
Akunuru S, Geiger H. Aging, clonality, and rejuvenation of hematopoietic stem cells. Trends Mol Med. 2016;22(8):701–12.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424–35.
Aguayo-Mazzucato C, Andle J, Lee TB Jr, Midha A, Talemal L, Chipashvili V, Hollister-Lock J, van Deursen J, Weir G, Bonner-Weir S. Acceleration of beta cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. 2019;30(1):129–42 e124.
Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Misra Sen J, Gorospe M, et al. Senolytic therapy alleviates Abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22(5):719–28.
Xu Y, Shan W, Li X, Wang B, Liu S, Wang Y, Long Y, Tie R, Wang L, Cai S, et al. A synthetic three-dimensional niche system facilitates generation of functional hematopoietic cells from human-induced pluripotent stem cells. J Hematol Oncol. 2016;9(1):102.
Jiang M, Chen H, Lai S, Wang R, Qiu Y, Ye F, Fei L, Sun H, Xu Y, Jiang X, et al. Maintenance of human haematopoietic stem and progenitor cells in vitro using a chemical cocktail. Cell Discov. 2018;4:59.
Guo B, Huang X, Lee MR, Lee SA, Broxmeyer HE. Antagonism of PPAR-gamma signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat Med. 2018;24(3):360–7.
Bai T, Li J, Sinclair A, Imren S, Merriam F, Sun F, O'Kelly MB, Nourigat C, Jain P, Delrow JJ, et al. Expansion of primitive human hematopoietic stem cells by culture in a zwitterionic hydrogel. Nat Med. 2019.
Kasakovski D, Xu L, Li Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J Hematol Oncol. 2018;11(1):91.
Nikolich-Zugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol. 2018;19(1):10–9.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81900176, 81770130, 81730008, 81870080, and 91949115), National Key R&D Program of China, Stem Cell and Translation Research (2018YFA0109300), and Zhejiang Province Science Foundation for Distinguished Young Scholars (LR19H080001).
Author information
Authors and Affiliations
Contributions
XL and XZ wrote the manuscript. YX, BW, YZ, XL, PQ, and HH provided suggestions and revisions. All authors contributed to the writing of the review. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This is not applicable for this review.
Consent for publication
This is not applicable for this review.
Competing interests
The authors have no competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, X., Zeng, X., Xu, Y. et al. Mechanisms and rejuvenation strategies for aged hematopoietic stem cells. J Hematol Oncol 13, 31 (2020). https://doi.org/10.1186/s13045-020-00864-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00864-8