
Abstract
Inappropriate activation of c-mesenchymal-epithelial transition (MET), the receptor tyrosine kinase (RTK) for hepatocyte growth factor (HGF), has been implicated in tumorigenesis and represented a promising therapeutic target for developing anticancer agents. In contrast to other solid tumors, there are limited data describing the functional role of HGF/c-MET signaling pathway in lymphoma. In the current review, we summarize recent findings about the expression, cellular mechanisms/functions, and therapeutic application of HGF/c-MET in different types of lymphoma, especially B cell lymphoma, T and NK cell lymphoma, and Hodgkin lymphoma. We also discuss the existing problems and future directions about studying the HGF/c-MET pathway in lymphoma cells.
Similar content being viewed by others
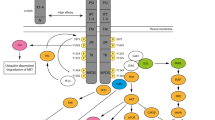

Background
The mesenchymal-epithelial transition (MET) can be utilized as a signaling receptor for hepatocyte growth factor (HGF), and it has been reported that in humans, the MET proto-oncogene encodes for the receptor tyrosine kinase (RTK) of growth factor tyrosine kinase [1, 2]. The structure of MET protein comprises a highly glycosylated 45-kDa extracellular α-subunit and a 145-kDa transmembrane β-subunit, which are linked together by a disulfide bridge (Fig. 1). Upon binding to its ligand, HGF, two MET subunits dimerize leading to auto-phosphorylation of three tyrosine residues (Y1230, Y1234, Y1235) [3, 4]. This initial phosphorylation cascade is followed by the phosphorylation of two other tyrosine residues (Y1349, Y1356), and these residues have been shown as docking sites for downstream signaling molecules that mediate Ras/Raf/MAPK, PI3K/AKT/mTOR, and/or STAT3/5 pathways [5–7]. HGF is known as a paracrine cellular growth and a motility and morphogenic factor. It is secreted by mesenchymal cells and acts as a multi-functional cytokine on cells of mainly epithelial origin after binding to the proto-oncogenic c-MET receptor. In addition, an intricate network of cross-signaling involving the c-MET-epidermal growth factor receptor (EGFR), c-MET-vascular endothelial growth factor receptor (VEGFR), and c-MET-Wnt pathways has also been reported in the past few years [8–10]. Such cross-talk implies/elicits a variety of pleiotropic biological responses leading to increased cell proliferation, survival, migration/invasion, angiogenesis, and metastasis in cancer cells [11]. HGF/c-MET has been extensively studied as a therapeutic target in various cancers for the last two decades, especially in lung cancer therapy. For example, c-MET amplification or activation has been reported as one of the major mechanisms for developing resistance to EGFR tyrosine kinase inhibitor (TKI) treatment in non-small cell lung cancer (NSCLC) patients [8, 12, 13]. However, few studies about the role of HGF/c-MET signaling pathway in lymphoma, a group of lymphocyte-derived cancers, have been documented. Some of them showed the conflicting results with favorable or unfavorable outcome of HGF/c-MET, especially in diffuse large B cell lymphoma (DLBCL). Based on the 2016 World Health Organization (WHO) classification, the major types of lymphoma include mature B cell lymphoma, mature T and NK cell lymphoma, and Hodgkin lymphoma, and each of them has many subtypes [14]. In the USA, lymphoma is the seventh most common cancer with 19.5 and 2.6 of new cases and 6 and 0.4 of death cases per 100,000 persons per year for non-Hodgkin and Hodgkin lymphoma, respectively, from 2009 to 2013. In this review, we will discuss the role of HGF/c-MET pathway in the pathogenesis of lymphoma cells and potential therapies for different types of lymphoma, based on recent published data.

The schematic diagram of HGF/c-MET signal transduction pathway
The expression/activation of HGF/c-MET in different types of lymphoma and its outcome on tumor progression
B cell-derived lymphoma
Weimar et al. reported that in several B cell-derived lymphoma cell lines (BJAB, Raji, Ramos, Daudi, and Jiyoye), two of them (BJAB, Raji) were c-MET positive [15]. Within the same cell lineages, the presence of c-MET could be variant, depending on the stages, specific characteristics of cells in the development of these lineages, and the impact of cell-surrounding environment. For example, c-MET is expressed on immature B cells, e.g., CD19 + CD20− B cells, but not on mature CD19 + CD20+ B cells. In addition, c-MET expression can be upregulated by the activation of mature B cells with CD40 ligand, phorbol 12-myristate 13-acetate (PMA), or Epstein-Barr virus (EBV) infection. The role of HGF has also been implicated in this B lymphoma. HGF induced adhesion of c-MET-positive (but not of c-MET-negative) B lymphoma cells to the extracellular matrix molecules, fibronectin, and collagen [15]. HGF influenced the metastasis of c-MET-positive cells into multiple organs, including the liver, kidney, lymph nodes, lung, gonads, and the central nervous system, in SCID mice but did not affect metastasis of c-MET-negative lymphoma [15]. Since human B lymphoma cells can bind via their α4β1 integrin to murine VCAM-1 molecules [16, 17], HGF induced adhesion of human c-MET-positive B cells to fibronectin probably via the activation of α4β1 integrin [15].
Diffuse large B cell lymphoma
In one of the most common B cell-derived lymphomas, DLBCL, c-MET was overexpressed in 26–73.2% of cases and significantly associated with other signaling molecules such as p-AKT, p-GSK3, and Ki-67 [18, 19]. Interestingly, the overexpression of c-MET in DLBCL was associated with better survival in these studies [18, 19]. This finding was unexpected and conflicting to the previous results with unfavorable outcomes of HGF/c-MET signaling in lymphomas and other cancers. One of the explanation is that c-MET-positive lymphoma cells have retained the physiological growth control by c-MET and possessed high proliferative index, Ki67, resulting in cell cycle progression and possibly chemotherapy sensitivity [20]. However, in vitro c-MET tyrosine kinase activity could activate AKT and its downstream substrates FOXO1 and GSK3 and induced anti-apoptosis [18]. Furthermore, it is reinforced that after being surgically resected, primary intestinal DLBCL with high c-MET copy numbers was associated with an unfavorable prognosis [19]. However, even in one type of cancers, e.g., NSCLC, the impact of c-MET copy number on prognosis is a histological subtype dependent: c-MET amplification with a poorer prognosis in patients with adenocarcinoma but not in those with squamous cell carcinoma [21, 22]. More evidences have been supporting the antiproliferative effect of HGF on melanoma, hepatocellular carcinoma, breast cancer, and leukemia, suggesting an antitumor effect for this growth factor [1, 20, 23–25]. Contradictorily, high levels of serum HGF and overactive HGF/c-MET pathway in DLBCL patients have been also reported to be linked with unfavorable outcome in several studies [26, 27]. HGF increased the adhesion of c-MET-positive B cell lymphoma cells to fibronectin and collagen, mediated via β1-integrin, and furthermore, promoted migration and invasion. These findings might indicate why HGF/c-MET-positive lymphomas have a poorer prognosis [27]. Interestingly, HGF was localized in single and small cell clusters, activated macrophages within the DLBCL tissues [28]. The lack of HGF expression by some DLBCL cell lines indicates a paracrine rather than an autocrine mechanism of c-MET activation in DLBCL [28]. The other DLBCL cell lines expressed HGF activator and are able to process HGF precursor to its active form [28]. HGF-induced activation of c-MET in DLBCL cells resulted in MEK-dependent phosphorylation of the MAP kinases ERK1 and ERK2, the linkages to the regulation of cell proliferation [28]. HGF-induced activation of c-MET induced PI3K-dependent phosphorylation of PKB/AKT and its substrates GSK3 and FOXO3a, contributing to its anti-apoptotic function [28]. Since HGF is an angiogenesis inducer [29] and has been demonstrated to induce expression of vascular endothelial growth factor (VEGF) as well [30, 31], it might also stimulate angiogenesis in DLBCL, thereby promoting tumor growth. It is recently reported that fatty acid synthase (FAS) inhibition triggered caspase-dependent apoptosis and suppressed the expression of c-MET in DLBCL cell lines [32]. Pharmacological FAS blockade might result in rapid changes in the lipid composition of the tumor cell membrane, which could impair a correct cellular localization of c-MET [33]. Moreover, FAS inhibition may cause an imbalance in the membrane phospholipids levels, which may result in decreased c-MET membrane localization and activation [34].
Primary effusion lymphoma
Kaposi’s sarcoma-associated herpesvirus (KSHV) is the etiologic agent of primary effusion lymphoma (PEL), which comprises transformed B cells harboring the viral episome and arises preferentially within the pleural or peritoneal cavities of patients infected with HIV [35]. PEL is a rapidly progressing malignancy with a median survival time of approximately 6 months even under the conventional chemotherapy [36]. Co-expression of HGF and c-MET was found in all KSHV+ PEL cell lines [32, 37]. HGF stimulation of PEL cells rapidly induces c-MET tyrosine phosphorylation [30]. Our recent study found that KSHV de novo infection greatly induced HGF production and the phosphorylation of c-MET from primary endothelial cells [37]. In HIV-infected patients, KSHV-positive group had higher plasma HGF concentration than those from the KSHV-negative group [37]. Blocking of HGF/c-MET pathway can induce caspase-dependent PEL apoptosis through cell cycle arrest, DNA damage, and suppression of downstream MAPK-ERK activity [37]. Interestingly, it has been previously reported that peritoneal fibroblasts produce significant amounts of HGF [38]. Therefore, the activation of HGF/c-MET could be involved in both an autocrine and a paracrine fashion in PEL growth.
Burkitt’s lymphoma
Among Burkitt’s lymphoma cell lines, EB4 and Raji expressed high levels of c-MET, while Namalwa cell line was negative [39]. HGF protected EB4 and Raji lymphoma cells from apoptosis induced by DNA damaging agents via c-MET expression, which is potentially through the anti-apoptotic mitochondrial pore-forming proteins Bcl-XL and Bcl-2 [39]. The HGF/c-MET-mediated function or pathogenesis can be reinforced in Namalwa cell line transfected with c-MET [40]. CD44v3 splice variant promotes HGF-induced phosphorylation of c-MET, phosphorylation of several downstream proteins, and activation of the MAPK-ERK1 [40]. This might be instrumental in promoting tumor growth and metastasis, since ERK1/2 are intermediates in signaling pathways linking extracellular signals to gene transcription in the nucleus and have been implicated in a wide variety of biological responses including cell proliferation [40].
Mucosa-associated lymphoid tissue lymphoma
c-MET was found in the lymphocytes composing mucosa-associated lymphoid tissue (MALT) lymphoma, and HGF was recognized mostly in the endothelial cells and macrophages in the MALT lymphoma [41]. HGF activator was localized on the mesenchymal cells other than the lymphocytes. c-MET inhibition suppressed the hepatic and pulmonary MALT lymphoma, while the gastric MALT lymphoma showed only a tendency to decrease in size [41].
Hodgkin lymphoma
Hodgkin lymphoma is a B cell neoplasm characterized by a minority of Reed-Sternberg (RS) cells in the infiltrating reactive cells. In classical Hodgkin lymphoma (cHL) [30], the expression of c-MET was detected in the tumor cells from 38 to 52% of the patients, and the expression of HGF was found in 8% of the patients [42, 43]. The c-MET expression is positive in L428, L1236, and U-HO1 but negative in KMH2 cell lines. Expression of c-MET in tumor cells from cHL patients strongly correlated with a favorable prognosis with a higher 5-year survival rate compared with c-MET-negative tumor patients. It was explained that the percentage of cancer cells in total cells of cHL is small, typically less than 1%, whereas the percentage of cancer cells in DLBCL and solid malignancies is generally between 40 and 80% [42]. In the infiltrating cells, several known effects of c-MET, such as reducing organ fibrosis and upregulating pro-inflammatory cytokines [44, 45], might explain the favorable effect of c-MET in cHL. In addition, c-MET could reduce the TGF-β production, an immuno-suppressant, and enhance the antitumor immunity [42, 43]. However, it has confusedly been shown that high expression of c-MET was associated with lower complete remission of cHL in another study [46]. This conflicting could be due to c-MET positivity limited to RS cells in the former study, whereas c-MET in the latter one was inferred in the whole cHL tissues. Another explanation is that it was probably due to cross-talk between the malignant cells and the reactive cells in the cHL tissue [46]. Interestingly, RS cells expressed c-MET but not HGF and several HGF-positive dendritic-reticulum cells were found scattered around c-MET-positive RS cells [47]. This indicated that RS cells and the infiltrated reactive cells in cHL were cross-talked via HGF/c-MET signaling pathway.
T and NK cell-derived lymphoma
In anaplastic large cell lymphoma (ALCL), a type of T cell lymphoma, c-MET is expressed in Karpas299 cells and some other ALCL cell lines [48]. However, nucleophosmin-anaplastic lymphoma kinase (ALK) but not c-MET has been implicated in the pathogenesis of ALCL [48]. In addition, HGF/c-MET were also expressed in natural killer/T cell lymphoma (NKTCL) cell lines. NKTCL cells were found to produce HGF and activate the HGF/c-MET signaling pathway for the tumor cell proliferation in an autocrine manner [49]. The novel c-MET T helper (Th) cell epitopes, c-MET278–292, 817–831, and 1244–1258, which were restricted by HLA-DR molecules, HLA-DR9, HLA-DR12, and HLA-DR53, respectively, have been identified [49]. The c-MET-specific T helper (Th) cells could also recognize dendritic cells (DCs) pulsed with c-MET expressed tumor cell lysates. In addition, it has been observed that c-MET inhibition augmented Th cell recognition by decreasing the TGF-β production by tumor cells. Anti-autophagy is one of the protective mechanisms of NKTCL because chaperon-dependent autophagy inhibitor, 17-DMAG, have been reported to suppress the cell growth of NKTCL directly [50, 51]. Moreover, 17-DMAG significantly reduced NKTCL T cell recognition [49].
In addition, the expression of c-MET was found in PBMCs from adult T cell leukemia/lymphoma (ATLL) patients and HTLV-1-infected T cell lines. HGF secreted in paracrine rather than in autocrine mechanism seems to be involved in the pathogenesis of ATLL [52]. ATLL cells became more aggressive by the acquisition of c-MET expression on its surface [52]. The proliferation of primary cells in acute but not chronic ATLL cases was induced by HGF [53]. Infiltrated ATLL cells and adjacent stromal cells in the liver of acute ATLL patients have been shown to be positive for HGF/c-MET, suggesting that the activation of HGF/c-MET was involved in the aggression of ATLL cells [53]. Moreover, the activity of HGF/c-MET was enhanced by surrounding cytokine environment. It was shown that ATLL cells as well as HTLV-1-infected cells secreted not only IL-6 but other HGF inducers such as IL-1ß and TNF-α, through which the production of HGF in stromal cells could be upregulated [54–56]. The functional role and underlying mechanisms of HGF/c-MET pathway in different types of lymphomas were listed in Table 1.
Targeting c-MET in lymphoma
Because of its pleiotropic role in oncogenesis and cancer progression, HGF/c-MET is considered to be an important target in anticancer therapy. However, currently, there are no HGF inhibitors that have been tried in lymphoma treatment, so here, we will focus on c-MET inhibitors, their application in lymphoma treatment (Table 2). c-MET inhibitors are a class of small molecules that inhibit the enzymatic activity of the c-MET tyrosine kinase. PF-2341066, an ATP-competitive small-molecule inhibitor of the catalytic activity of c-MET and the ALK protein, was administered by oral gavage in mice-bearing ALCL. PF-2341066 potently inhibited cell proliferation, which was associated with G1 cell cycle arrest and induction of apoptosis in ALK-positive ALCL cells [48]. PF-2341066 was also administered intraperitoneally in non-obese diabetic/severe-combined immunodeficiency (NOD/SCID) mice with KSHV-positive PEL tumors. PF-2341066 treatment dramatically suppressed PEL tumor progression including reducing ascites formation and spleen enlargement over this time frame and reduced expression of phosphor-c-MET and phosphor-ERK within the spleen tissues [37]. As mentioned above, PF-2341066 has been shown as the inhibitor to several other RTKs such as ALK and ROS1 in different types of cancer cells [48, 57]. Moreover, ALK inhibitors especially second- and third-generation inhibitors have displayed promising effects on ALK+ NSCLC patients even with mutations [58–61]. However, PEL cell lines do not express the p-ALK (Tyr1604), t-ALK, p-ROS1 (Tyr2274), or t-ROS1 reported in these previous studies [37, 48, 57], implying that these virus-associated lymphoma cell lines may have unique expressional pattern of RTKs.
SU11274, another ATP-competitive c-MET selective inhibitor, blocked the constitutive phosphorylation of c-MET and downstream kinases and induced G2/M cell cycle arrest and significantly suppressed cell growth in a dose-dependent manner [42]. Treatment with PHA665752, a second generation of the highly selective c-MET inhibitor, causes inactivation of c-MET, leading to the inactivation of AKT and downstream signaling of molecules FOXO1 and GSK3 in DLBCL cell lines. PHA665752 treatment induced the activation of Caspase-9, Caspase-3, and downstream PARP cleavage, the hallmarks of cells undergoing apoptosis [18]. PHA-665752 was also injected intraperitoneally in mice with MALT lymphoma, which induced the significant decrease in the hepatic and pulmonary MALT lymphoma size and the marked activation of Caspase 3 in fundic, hepatic, and pulmonary MALT lymphoma [41]. NKTCL cell lines treated with ARQ197, an ATP-non-competitive selective c-MET inhibitor which is currently in phase 3 clinical trials in NSCLC patients, reduced the proliferative response by approximately 50% in two of three cell lines, indicating that the HGF/c-MET signaling is partly responsible for NKTCL proliferation [49].
Conclusions
There are only limited data studying the role of HGF/c-MET signaling pathway in lymphomas, when compared to those from other solid tumors. For example, MET gene mutation and amplification represent one of the major mechanisms causing constitutive active HGF/c-MET signaling in cancer cells. However, no amplification of the MET gene was found in B cell lymphoma cell lines and patient samples [28]. Currently, only two germ line missense mutations of c-MET have been found within B cell lymphoma, especially DLBCL (R1166Q in the tyrosine kinase domain in one patient and R988C in the juxtamembrane domain in four patients) [28]. Therefore, future work highlighting more potential mechanisms of activating HGF/c-MET signaling from lymphoma cells may uncover innovative therapeutic strategies for these malignancies. To date, only a few c-MET inhibitors have entered clinical trials, among which crizotinib and cabozantinib were the first to be approved by the US FDA for the treatment of certain solid tumor [62, 63]. However, none of them have been used in clinical trials for any type of lymphoma. We should accelerate clinical trial process by choosing those promising c-MET inhibitors which have been shown to be dramatically effective in lymphoma animal models.
Abbreviations
- ALCL:
-
Anaplastic large cell lymphoma
- ALK:
-
Anaplastic lymphoma kinase
- ATLL:
-
Adult T cell leukemia/lymphoma
- cHL:
-
Classical Hodgkin lymphoma
- DCs:
-
Dendritic cells
- DLBCL:
-
Diffuse large B cell lymphoma
- EBV:
-
Epstein-Barr virus
- EGFR:
-
Epidermal growth factor receptor
- FASN:
-
Fatty acid synthase
- HGF:
-
Hepatocyte growth factor
- KSHV:
-
Kaposi’s sarcoma-associated herpesvirus
- MALT:
-
Mucosa-associated lymphoid tissue
- MET:
-
Mesenchymal-epithelial transition
- NKTCL:
-
Natural killer/T cell lymphoma
- NOD/SCID:
-
Non-obese diabetic/severe-combined immunodeficiency
- NSCLC:
-
Non-small cell lung cancer
- PEL:
-
Primary effusion lymphoma
- PMA:
-
Phorbol 12-myristate 13-acetate
- RS cells:
-
Reed-Sternberg cells
- RTK:
-
Receptor tyrosine kinase
- Th cell:
-
T helper cell
- TKI:
-
Tyrosine kinase inhibitor
- VEGFR:
-
Vascular endothelial growth factor receptor
- WHO:
-
World Health Organization
References
Shiota G, Rhoads DB, Wang TC, Nakamura T, Schmidt EV. Hepatocyte growth factor inhibits growth of hepatocellular carcinoma cells. Proc Natl Acad Sci U S A. 1992;89(1):373–7.
Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci U S A. 1987;84(18):6379–83.
Ferracini R, Longati P, Naldini L, Vigna E, Comoglio PM. Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J Biol Chem. 1991;266(29):19558–64.
Zhen Z, Giordano S, Longati P, Medico E, Campiglio M, Comoglio PM. Structural and functional domains critical for constitutive activation of the HGF-receptor (Met). Oncogene. 1994;9(6):1691–7.
Ponzetto C, Bardelli A, Maina F, Longati P, Panayotou G, Dhand R, Waterfield MD, Comoglio PM. A novel recognition motif for phosphatidylinositol 3-kinase binding mediates its association with the hepatocyte growth factor/scatter factor receptor. Mol Cell Biol. 1993;13(8):4600–8.
Weidner KM, Di Cesare S, Sachs M, Brinkmann V, Behrens J, Birchmeier W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature. 1996;384(6605):173–6.
Zhu H, Naujokas MA, Fixman ED, Torossian K, Park M. Tyrosine 1356 in the carboxyl-terminal tail of the HGF/SF receptor is essential for the transduction of signals for cell motility and morphogenesis. J Biol Chem. 1994;269(47):29943–8.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–43.
Sulpice E, Ding S, Muscatelli-Groux B, Berge M, Han ZC, Plouet J, Tobelem G, Merkulova-Rainon T. Cross-talk between the VEGF-A and HGF signalling pathways in endothelial cells. Biol Cell. 2009;101(9):525–39.
Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12(5):468–76.
Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103.
Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–5.
Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17(1):77–88.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–90.
Weimar IS, Weijer K, van den Berk PC, Muller EJ, Miranda N, Bakker AQ, Heemskerk MH, Hekman A, de Gast GC, Gerritsen WR. HGF/SF and its receptor c-MET play a minor role in the dissemination of human B-lymphoma cells in SCID mice. Br J Cancer. 1999;81(1):43–53.
Tsuzuki S, Toyama-Sorimachi N, Kitamura F, Tobita Y, Miyasaka M. FK506 (tacrolimus) inhibits extravasation of lymphoid cells by abrogating VLA-4/VCAM-1 mediated transendothelial migration. FEBS Lett. 1998;430(3):414–8.
Renz ME, Chiu HH, Jones S, Fox J, Kim KJ, Presta LG, Fong S. Structural requirements for adhesion of soluble recombinant murine vascular cell adhesion molecule-1 to alpha 4 beta 1. J Cell Biol. 1994;125(6):1395–406.
Uddin S, Hussain AR, Ahmed M, Al-Dayel F, Bu R, Bavi P, Al-Kuraya KS. Inhibition of c-MET is a potential therapeutic strategy for treatment of diffuse large B-cell lymphoma. Lab Invest. 2010;90(9):1346–56.
Huang WT, Chuang SS. High MET gene copy number predicted poor prognosis in primary intestinal diffuse large B-cell lymphoma. Diagn Pathol. 2013;8:16.
Ronen D, Altstock RT, Firon M, Mittelman L, Sobe T, Resau JH, Vande Woude GF, Tsarfaty I. Met-HGF/SF mediates growth arrest and differentiation in T47D breast cancer cells. Cell Growth Differ. 1999;10(2):131–40.
Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, Legrain M, Mennecier B, Wihlm JM, Massard G, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thorac Oncol. 2008;3(4):331–9.
Shibata T, Uryu S, Kokubu A, Hosoda F, Ohki M, Sakiyama T, Matsuno Y, Tsuchiya R, Kanai Y, Kondo T, et al. Genetic classification of lung adenocarcinoma based on array-based comparative genomic hybridization analysis: its association with clinicopathologic features. Clin Cancer Res. 2005;11(17):6177–85.
Koh YW, Hwang HS, Jung SJ, Park C, Yoon DH, Suh C, Huh J. Receptor tyrosine kinases MET and RON as prognostic factors in diffuse large B-cell lymphoma patients receiving R-CHOP. Cancer Sci. 2013;104(9):1245–51.
Tajima H, Matsumoto K, Nakamura T. Hepatocyte growth factor has potent anti-proliferative activity in various tumor cell lines. FEBS Lett. 1991;291(2):229–32.
Accordi B, Pillozzi S, Dell’Orto MC, Cazzaniga G, Arcangeli A, Kronnie GT, Basso G. Hepatocyte growth factor receptor c-MET is associated with FAS and when activated enhances drug-induced apoptosis in pediatric B acute lymphoblastic leukemia with TEL-AML1 translocation. J Biol Chem. 2007;282(40):29384–93.
Hsiao LT, Lin JT, Yu IT, Chiou TJ, Liu JH, Yen CC, Wang WS, Chen PM. High serum hepatocyte growth factor level in patients with non-Hodgkin’s lymphoma. Eur J Haematol. 2003;70(5):282–9.
Kawano R, Ohshima K, Karube K, Yamaguchi T, Kohno S, Suzumiya J, Kikuchi M, Tamura K. Prognostic significance of hepatocyte growth factor and c-MET expression in patients with diffuse large B-cell lymphoma. Br J Haematol. 2004;127(3):305–7.
Tjin EP, Groen RW, Vogelzang I, Derksen PW, Klok MD, Meijer HP, van Eeden S, Pals ST, Spaargaren M. Functional analysis of HGF/MET signaling and aberrant HGF-activator expression in diffuse large B-cell lymphoma. Blood. 2006;107(2):760–8.
Jiang W, Hiscox S, Matsumoto K, Nakamura T. Hepatocyte growth factor/scatter factor, its molecular, cellular and clinical implications in cancer. Crit Rev Oncol Hematol. 1999;29(3):209–48.
Wojta J, Kaun C, Breuss JM, Koshelnick Y, Beckmann R, Hattey E, Mildner M, Weninger W, Nakamura T, Tschachler E, et al. Hepatocyte growth factor increases expression of vascular endothelial growth factor and plasminogen activator inhibitor-1 in human keratinocytes and the vascular endothelial growth factor receptor flk-1 in human endothelial cells. Lab Invest. 1999;79(4):427–38.
Xin X, Yang S, Ingle G, Zlot C, Rangell L, Kowalski J, Schwall R, Ferrara N, Gerritsen ME. Hepatocyte growth factor enhances vascular endothelial growth factor-induced angiogenesis in vitro and in vivo. Am J Pathol. 2001;158(3):1111–20.
Capello D, Gaidano G, Gallicchio M, Gloghini A, Medico E, Vivenza D, Buonaiuto D, Fassone L, Avanzi GC, Saglio G, et al. The tyrosine kinase receptor met and its ligand HGF are co-expressed and functionally active in HHV-8 positive primary effusion lymphoma. Leukemia. 2000;14(2):285–91.
Uddin S, Hussain AR, Ahmed M, Bu R, Ahmed SO, Ajarim D, Al-Dayel F, Bavi P, Al-Kuraya KS. Inhibition of fatty acid synthase suppresses c-Met receptor kinase and induces apoptosis in diffuse large B-cell lymphoma. Mol Cancer Ther. 2010;9(5):1244–55.
Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, Lupu R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci U S A. 2004;101(29):10715–20.
Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332(18):1186–91.
Chen YB, Rahemtullah A, Hochberg E. Primary effusion lymphoma. Oncologist. 2007;12(5):569–76.
Dai L, Trillo-Tinoco J, Cao Y, Bonstaff K, Doyle L, Del Valle L, Whitby D, Parsons C, Reiss K, Zabaleta J, et al. Targeting HGF/c-MET induces cell cycle arrest, DNA damage, and apoptosis for primary effusion lymphoma. Blood. 2015;126(26):2821–31.
Yashiro M, Chung YS, Inoue T, Nishimura S, Matsuoka T, Fujihara T, Sowa M. Hepatocyte growth factor (HGF) produced by peritoneal fibroblasts may affect mesothelial cell morphology and promote peritoneal dissemination. Int J Cancer. 1996;67(2):289–93.
Skibinski G, Skibinska A, James K. Hepatocyte growth factor (HGF) protects c-met-expressing Burkitt’s lymphoma cell lines from apoptotic death induced by DNA damaging agents. Eur J Cancer. 2001;37(12):1562–9.
van der Voort R, Taher TE, Wielenga VJ, Spaargaren M, Prevo R, Smit L, David G, Hartmann G, Gherardi E, Pals ST. Heparan sulfate-modified CD44 promotes hepatocyte growth factor/scatter factor-induced signal transduction through the receptor tyrosine kinase c-Met. J Biol Chem. 1999;274(10):6499–506.
Nakamura M, Takahashi T, Matsui H, Baniwa Y, Takahashi S, Murayama SY, Serizawa H, Suzuki H, Hibi T. Alteration of angiogenesis in Helicobacter heilmannii-induced mucosa-associated lymphoid tissue lymphoma: interaction with c-Met and hepatocyte growth factor. J Gastroenterol Hepatol. 2014;29 Suppl 4:70–6.
Xu C, Plattel W, van den Berg A, Ruther N, Huang X, Wang M, de Jong D, Vos H, van Imhoff G, Viardot A, et al. Expression of the c-Met oncogene by tumor cells predicts a favorable outcome in classical Hodgkin’s lymphoma. Haematologica. 2012;97(4):572–8.
Wha Koh Y, Park C, Hyun Yoon D, Suh C, Huh J. MET and MST1R as prognostic factors for classical Hodgkin’s lymphoma. Mod Pathol. 2013;26(9):1172–82.
Ueki T, Kaneda Y, Tsutsui H, Nakanishi K, Sawa Y, Morishita R, Matsumoto K, Nakamura T, Takahashi H, Okamoto E, et al. Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nat Med. 1999;5(2):226–30.
Beilmann M, Vande Woude GF, Dienes HP, Schirmacher P. Hepatocyte growth factor-stimulated invasiveness of monocytes. Blood. 2000;95(12):3964–9.
Bedewy M, El-Maghraby S, Bedewy A. CD163 and c-Met expression in the lymph node and the correlations between elevated levels of serum free light chain and the different clinicopathological parameters of advanced classical Hodgkin’s lymphoma. Blood Res. 2013;48(2):121–7.
Teofili L, Di Febo AL, Pierconti F, Maggiano N, Bendandi M, Rutella S, Cingolani A, Di Renzo N, Musto P, Pileri S, et al. Expression of the c-met proto-oncogene and its ligand, hepatocyte growth factor, in Hodgkin disease. Blood. 2001;97(4):1063–9.
Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, Yamazaki S, Alton GR, Mroczkowski B, Los G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6(12 Pt 1):3314–22.
Kumai T, Matsuda Y, Ohkuri T, Oikawa K, Ishibashi K, Aoki N, Kimura S, Harabuchi Y, Celis E, Kobayashi H. c-Met is a novel tumor associated antigen for T-cell based immunotherapy against NK/T cell lymphoma. Oncoimmunology. 2015;4(2):e976077.
Iwata S, Saito T, Ito Y, Kamakura M, Gotoh K, Kawada J, Nishiyama Y, Kimura H. Antitumor activities of valproic acid on Epstein-Barr virus-associated T and natural killer lymphoma cells. Cancer Sci. 2012;103(2):375–81.
Murata T, Iwata S, Siddiquey MN, Kanazawa T, Goshima F, Kawashima D, Kimura H, Tsurumi T. Heat shock protein 90 inhibitors repress latent membrane protein 1 (LMP1) expression and proliferation of Epstein-Barr virus-positive natural killer cell lymphoma. PLoS One. 2013;8(5):e63566.
Imaizumi Y, Murota H, Kanda S, Hishikawa Y, Koji T, Taguchi T, Tanaka Y, Yamada Y, Ikeda S, Kohno T, et al. Expression of the c-Met proto-oncogene and its possible involvement in liver invasion in adult T-cell leukemia. Clin Cancer Res. 2003;9(1):181–7.
Onimaru Y, Tsukasaki K, Murata K, Imaizumi Y, Choi YL, Hasegawa H, Sugahara K, Yamada Y, Hayashi T, Nakashima M, et al. Autocrine and/or paracrine growth of aggressive ATLL cells caused by HGF and c-Met. Int J Oncol. 2008;33(4):697–703.
Yamada Y, Ohmoto Y, Hata T, Yamamura M, Murata K, Tsukasaki K, Kohno T, Chen Y, Kamihira S, Tomonaga M. Features of the cytokines secreted by adult T cell leukemia (ATL) cells. Leuk Lymphoma. 1996;21(5–6):443–7.
Tendler CL, Greenberg SJ, Burton JD, Danielpour D, Kim SJ, Blattner WA, Manns A, Waldmann TA. Cytokine induction in HTLV-I associated myelopathy and adult T-cell leukemia: alternate molecular mechanisms underlying retroviral pathogenesis. J Cell Biochem. 1991;46(4):302–11.
Yamano Y, Machigashira K, Ijichi S, Usuku K, Kawabata M, Arimura K, Osame M. Alteration of cytokine levels by fosfomycin and prednisolone in spontaneous proliferation of cultured lymphocytes from patients with HTLV-I-associated myelopathy (HAM/TSP). J Neurol Sci. 1997;151(2):163–7.
Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, McTigue M, Deng YL, Liu W, Brooun A, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci U S A. 2015;112(11):3493–8.
Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Mittal V, Cang S, Liu D. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015;8:17.
Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34.
Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol. 2016;9(1):59.
Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. 2016;9:19.
Kazandjian D, Blumenthal GM, Chen HY, He K, Patel M, Justice R, Keegan P, Pazdur R. FDA approval summary: crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist. 2014;19(10):e5–11.
Ernani V, Kumar M, Chen AY, Owonikoko TK. Systemic treatment and management approaches for medullary thyroid cancer. Cancer Treat Rev. 2016;50:89–98.
Jucker M, Gunther A, Gradl G, Fonatsch C, Krueger G, Diehl V, Tesch H. The Met/hepatocyte growth factor receptor (HGFR) gene is overexpressed in some cases of human leukemia and lymphoma. Leuk Res. 1994;18(1):7–16.
Accornero P, Lattanzio G, Mangano T, Chiarle R, Taulli R, Bersani F, Forni PE, Miretti S, Scuoppo C, Dastru W, et al. An in vivo model of Met-driven lymphoma as a tool to explore the therapeutic potential of Met inhibitors. Clin Cancer Res. 2008;14(7):2220–6.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from a DOD Career Development Award to Z.Q. (CA140437), the SOM Research Enhancement Funding (2015–2016) to Z.Q., the Leukemia Research Foundation (2016–2017) to Z.Q., and awards from the National Natural Science Foundation of China (81272191, 81472547, 81672924 to Z.Q. and 81400164 to L.D.). Funding sources had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Authors’ contributions
LBQ and ZQ drafted the manuscript. LD discussed and revised the manuscript. All authors read and approved final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
This is not applicable for this review.
Ethics approval and consent to participate
This is not applicable for this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
