
Abstract
Due to their post-mitotic state, metabolic demands and often large polarised morphology, the function and survival of neurons is dependent on an efficient cellular waste clearance system both for generation of materials for metabolic processes and removal of toxic components. It is not surprising therefore that deficits in protein clearance can tip the balance between neuronal health and death. Here we discuss how autophagy and lysosome-mediated degradation pathways are disrupted in several neurological disorders. Both genetic and cell biological evidence show the diversity and complexity of vesicular clearance dysregulation in cells, and together may ultimately suggest a unified mechanism for neuronal demise in degenerative conditions. Causative and risk-associated mutations in Alzheimer’s disease, Frontotemporal Dementia, Amyotrophic Lateral Sclerosis, Parkinson’s disease, Huntington’s disease and others have given the field a unique mechanistic insight into protein clearance processes in neurons. Through their broad implication in neurodegenerative diseases, molecules involved in these genetic pathways, in particular those involved in autophagy, are emerging as appealing therapeutic targets for intervention in neurodegeneration.
Similar content being viewed by others


Introduction
Neurodegenerative diseases are defined by the progressive and irreversible destruction of neurons, with age-associated cell death occurring through heterogeneous, only partially defined mechanisms. A varied range of behavioural, cognitive and physiological symptoms are associated with neurodegenerative diseases, dependent on the affected neuronal populations. The most common neurodegenerative diseases broadly cause two primary symptoms, cognitive decline such as the profound dementia presented in Alzheimer’s disease (AD), and motor system dysfunction such as the slowing of movement and eventual paralysis seen in Parkinson’s disease (PD). Almost without exception, effective preventative therapeutics are unavailable for neurodegenerative diseases, with only palliative treatments currently in use. As with most neurological disorders [1] neurodegenerative diseases are distributed globally with an increasing incidence correlating with the ageing populations, and associated with a growing health and socioeconomic burden.
A paucity of effective treatments for neurodegenerative diseases has led to an urgent search for candidate cellular mechanisms for therapeutic intervention. Protein turnover has long been implicated in many of the most common neurodegenerative diseases, through the discovery that several proteins genetically linked to familial forms of disorders form stable aggregates within cells. Well-described examples include AD associated amyloid plaques and hyperphosphorylated Tau containing neurofibrillary tangles, PD associated Lewy bodies and neurites, and cytosolic inclusions of Amyotrophic Lateral Sclerosis (ALS). The accumulation of these mono- and oligomeric peptides suggests ineffective cellular clearance of macromolecules, in particular via the endo-lysosomal and autophagic machinery. Emerging genetic and molecular biological evidence now suggests that both systems may be dysfunctional across a broad spectrum of neurodegenerative disorders, their contribution expanding beyond just the turnover of aggregation prone proteins in neurons (Table 1). Here we summarily review the evidence for a role of endo-lysosomal and autophagy dysfunction in progressive neurodegenerative disorders, using specific examples of their contribution from common disorders to illustrate key concepts.
Autophagy
Autophagy is a process of ‘self-eating’ through which unwanted or toxic macromolecules and organelles are sequestered and delivered to the lysosome to generate raw materials including proteins, lipids, carbohydrates and nucleic acids for use in metabolic processes. In most cell types, autophagy functions primarily in response to starvation [54] and some forms of apoptosis [55]. However, in post-mitotic neurons, where the programmed death and replacement of unhealthy cells is not a viable option, autophagy takes on a more crucial role in maintaining normal cellular homeostasis, in particular the critical turnover of misfolded proteins and damaged organelles. This is demonstrated by observations of increased autophagy in response to acute brain damage such as strokes and traumatic brain injuries, however there is still controversy as to whether this response is homeostatic or pathological (reviewed by [56]). There are three mechanistically distinct forms of autophagy that function within neurons; macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy, each of which have been implicated in maintaining normal neuronal function or in neurodegeneration.
Macroautophagy signalling cascade
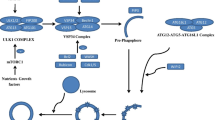
During macroautophagy, macromolecules and organelles such as mitochondria and peroxisomes are sequestered within specialised vesicles and digested for removal or generation of raw material (Fig. 1). Macroautophagy is a complex sequential process composed of multiple steps which are generally considered consistent between cell types, predominantly facilitated by a cascade of Autophagy Related Genes (ATG). Sensing is the crucial first step in autophagy induction, where the cell makes a choice to induce degradation of toxic or superfluous cellular components. In normal healthy physiological conditions, the serine-threonine kinase mammalian target of Rapamycin (mTOR), the master sensor for autophagy, forms the mTOR complex (MTORC1) to promote cell growth [57]. Depleted levels of cellular cyclic adenosine monophosphate (cAMP) activate 5′-adenosine monophosphate activated protein kinase (AMPK), which in turn phosphorylates unc-51 like autophagy activating kinase 1 (ULK1), promoting it to form a complex with Focal Adhesion Kinase Family Kinase-Interacting protein Of 200 KDa (FIP200), ATG13 and ATG101 [58, 59]. Initiation/nucleation triggers formation of the ‘phagophore’, a lipid double membrane produced to encapsulate the target cargo, restricting it to a smaller cytoplasmic region for further processing. To enable phagophore formation, ULK1 phosphorylates and activates the vacuolar protein sorting 34 (VPS34) complex, consisting of the class III phosphatidylinositol-3-kinase VPS34, Beclin1, VPS14 and VPS15 [60]. The activated VPS34 complex enriches the isolation membrane with phosphatidylinositol 3-phosphate (PI(3)P), recruiting additional autophagy machinery. The phagophore next undergoes elongation, facilitated by two processes. Firstly, phosphatidylethanolamine is covalently bound to cytosolic Microtubule Associated Protein 1 Light Chain 3 and GABARAP family proteins (herein LC3-I), producing an autophagosome-associated LC3-II [61]. Secondly a complex of ATG5-ATG12-ATG16 associates with the isolation membrane, allowing it to entirely enclose the whole target organelle [62]. Selection of cargos occurs in parallel to sensing, initiation and elongation, marking substrates for autophagy. Proteins are targeted for autophagy by ubiquitination and labelling primarily with p62/Sequestosome-1 (p62), which through an ATG8 interaction motif/LC3 interacting region (AIM/LIR) [63, 64] recruits LC3-II to the isolation membrane [65]. Other cargo recognition proteins including Neighbour Of BRCA1 Gene 1 (NBR1), Nuclear Domain 10 Protein 52/ Calcium Binding And Coiled-Coil Domain 2 (NDP52), and Optineurin (OPTN), also contribute to specific targets for autophagy [66]. Once target cargos are bound by LC3-II, further initiation machinery is recruited. Closure of the membrane leads to formation of a double membraned vesicle called an ‘autophagosome’, containing the target cargos. Since their formation can occur in synapses and neurites significant distances from the neuronal soma [67], transport of autophagosomes is often necessary for their delivery to appropriate cellular compartments for degradation. Autophagosomes finally undergo fusion with late-endosomes or lysosomes to deliver substrates for hydrolytic enzymatic degradation.

Autophagy and endo-lysosomal mechanisms and related genes associated with neurodegenerative diseases. Macroautophagy begins with formation of an isolation membrane to engulf cargos selected for degradation. Elongation of the isolation membrane results in formation of the double membrane autophagosome marking the final step before lysosomal fusion and degradation. In parallel the endosomal system sorts molecules for either recycling or targeting to the lysosome, with chaperone-mediated autophagy (CMA) and microautophagy also delivering cargos to the lysosomes. Hydrolytic enzymes within the acidic lysosomal lumen digest the target and the constituents resulted from this are released into the cellular cytoplasm. Neurodegenerative disease causing or associated genes affecting various stages of autophagy are listed alongside the process in which they are involved. For additional information relating to disease association of listed genes, refer to Table 1
Macroautophagy
Macroautophagy is a highly conserved process [68, 69] and unsurprisingly several key molecules and mechanisms are associated with neuronal dysfunction and degenerative conditions. The critical importance of autophagy in neuronal health is best documented in model organisms deficient for genes required for the initial steps of autophagy. The ULK1 homologue, serine/threonine-protein kinase unc-51 was first identified in a C. elegans screen for genes associated with ‘uncoordinated’ phenotypes, with its dysfunction resulting in incomplete developmental axon outgrowth and elongation [70], clearly demonstrating the importance of autophagy in normal neurodevelopment. Current research however, also suggests that aberrant autophagy plays a fundamental role in ageing and neurodegeneration. Conditional deletion of essential autophagy genes in mice has demonstrated the critical requirement for neuronal autophagy in adult animals. Mice lacking neuronal expression of the autophagosome membrane elongation genes ATG5 [71] or ATG7 [72] are viable into adulthood, however they show significantly reduced autophagy, associated with progressive motor dysfunction and neurodegeneration. A notable observation from both ATG5 and ATG7 deletion models was the formation of intraneuronal inclusion bodies composed of ubiquitinated protein. Large protein aggregates are considered a hallmark histopathological feature of many neurodegenerative disorders, though their precise contribution (or even protection) is poorly defined.
Genetic evidence from several neurodegenerative diseases supports a contribution for protein accumulation in pathogenesis, with hereditary mutations in many aggregation prone proteins, and also dysfunction of cargo selection genes required for targeting inclusions to the autophagy system. Natively unfolded alpha-synuclein [29] forms Lewy bodies and neurites in PD and associated Parkinsonisms [73], with hereditary point mutations increasing aggregation propensity [74]. Hyperphosphorylation of Microtubule Associated Protein Tau (Tau) accumulates in neurofibrillary tangles [75,76,77] in several neurodegenerative diseases including AD and familial forms of Frontotemporal Dementia (FTD) [78]. Extracellular amyloid beta (Aβ) plaques are a hallmark of AD, composed of fragments of amyloid precursor protein (APP) [79, 80] generated by presenilin secretases (PSEN1/2) [81], with APP [82, 83] and PSEN genes [84, 85] mutated in familial AD. Both familial and sporadic Amyotrophic Lateral Sclerosis (ALS) is associated with cytosolic aggregation of proteins, most frequently TDP-43 [86, 87], with FUS [88] and SOD1 [89,90,91] seen in some familial cases. Mutations resulting in repeat expansion are also associated with protein aggregation such as the ALS/FTD linked gene C9ORF72 [92,93,94], or polyglutamine (polyQ) expansions seen in several disorders, most prominently Huntingtin (HTT) in Huntington’s Disease (HD) [39, 95]. Typically intracellular inclusion bodies are ubiquitinated and labelled with autophagy receptor proteins, most commonly p62 [96], suggesting autophagy plays an active role in their clearance. Mutations in p62 [44, 45] and OPTN [43] have been identified in ALS and FTD, directly implicating effective protein targeting for clearance in disease pathogenesis.
Mutations in core autophagy genes have not been identified as directly causative in any common neurodegenerative disorders, though some rare conditions have been reported [97]. There is however a wealth of data implicating the mis-regulation of autophagy sensing and initiation/nucleation in neurodegenerative disorders, particularly HD. Experiments in cellular [98], Drosophila and mouse models of polyQ expanded HTT [99] have demonstrated that promoting autophagy through pharmacological inhibition of mTOR is sufficient to rescue phenotypes associated with HD toxicity. Contrasting evidence suggests that expression of the mTOR activators RAS homolog enriched in brain (Rheb) or RAS homolog enriched in the striatum (Rhes) can also alleviate symptoms in HD mouse models [100], while only transient protection is seen in R6/2 mouse treated with mTOR inhibitors [101]. The contribution of autophagy to HD is complicated by the fact that mTOR is found within poly-Q rich protein aggregates [99] and that polyQ-expanded HTT enhances mTORC1 activity [102], suggesting a direct interaction of mTOR with HTT. PolyQ-expanded HTT can disrupt nucleation of the isolation membrane, by impairing the phosphorylation of Beclin-1 associated ATG14 and VPS34 complex activity [103]. Pro-nucleation has also been implicated in polyQ-repeat associated Machado-Joseph disease/spinocerebellar ataxia type 3, with increased expression of the VPS34 complex component Beclin-1 showing protective activity in mouse models [104, 105]. Beclin-1 mediated nucleation may also contribute to the pathology of AD. In transgenic mouse AD models expressing human APP, reduction of Beclin-1 expression leads to increased intraneuronal Aβ accumulation, extracellular Aβ deposition and neurodegeneration [3]. Intriguingly, peptide fragments produced through caspase cleavage of Beclin-1 have been detected in the brains of AD patients and murine models, which exacerbated neurodegenerative phenotypes when overexpressed [4]. Conversely, caspase-resistant Beclin-1 was found to be neuroprotective, suggesting some post-translationally processed species of Beclin-1 may themselves be toxic [4].
A direct function of ALS/FTD gene C9ORF72 in the initiation of autophagy has also been suggested recently. ALS/FTD can manifest through a 5′ hexanucleotide repeat expansion in the C9ORF72 gene, which can contain several thousand repeats generating both RNAi and protein products that accumulate over time [92, 94]. In addition to clear gain-of-function pathologies, mutant C9ORF72 alleles may also reduce expression of some isoforms of the gene, suggesting partial loss-of-function may partially contribute to disease [92]. C9ORF72 has been found to interact with RAB1A, a RAB GTPase effector molecule required for the recruitment of ULK1 complex to the phagophore [106]. Decreased levels of autophagy have been reported in neurons derived from C9ALS/FTD patients, and reduction of C9ORF72 expression in cultured neurons was found to attenuate autophagy and accumulation of intracellular p62 puncta, indicative of protein accumulation [106, 107].
Chaperone-mediated autophagy
Chaperone-mediated autophagy (CMA) is a selective form of autophagy, whereby peptides carrying a KFERQ-like motif are recognised by cytoplasmic chaperone proteins, which then deliver the target directly to lysosomes for degradation [108]. Target peptides are bound by cytoplasmic chaperones including Heat shock protein 90 (HSP90), delivered to the lysosome-associated membrane protein 2A (LAMP2A) receptor on the lysosomal membrane and transported into the lysosome lumen for hydrolytic degradation. Unlike micro- and macroautophagy, CMA is not evolutionarily conserved and has only been observed in mammalian cells [109, 110].
CMA contributes to the clearance of proteins associated with several neurodegenerative disorders [111, 112], with compelling evidence to suggest a role in the dopaminergic neuron loss seen in PD. Several genes genetically associated with familial forms of PD appear to disrupt CMA. The natively unfolded alpha-synuclein peptide is a substrate for CMA [113], however both stabilised dopamine-bound peptides, [114] and PD-associated mutant species [113] are ineffectively degraded through this process. Ubiquitin carboxyl-terminal esterase L1 (UCHL-1), has been shown to interact with heat shock protein 70 (HSC70), HSP90 and LAMP2A, with disease associated mutations further increasing binding and impeding CMA of alpha-synuclein [115]. PD-associated Leucine Rich Repeat Kinase 2 (LRRK2) also appears to be degraded through CMA, with the PD associated mutations rendering the protein a poor substrate but also impeding the CMA translocation complex [116]. Most recently, PD-associated deglycase DJ-1, which functions in neuronal response to oxidative stress and mitochondrial turnover, has also been found to undergo CMA-mediated degradation, with a preference for non-functional oxidised forms [117]. Reduced CMA and turnover of non-functional DJ-1 was associated with increased mitochondrial dysfunction and cell death in repose to toxin induced oxidative stress [117]. CMA and PD have also been associated through the degradation of myocyte enhancer factor 2D (MEF2D), a transcription factor that contributes to neuronal survival under stress [118]. Inhibition of CMA through knockdown of HSC70 or LAMP2A results in accumulation of cytoplasmic non-functional MEF2D in neuronal cultures, with increased cytoplasmic MEF2D also reported in alpha-synuclein transgenic mice and PD patient tissues [119]. Taken together, these findings suggest processing of PD-associated peptides through CMA may be a contributing factor in disease pathogenesis and progression and that this process may be critical for the maintenance of dopamine neurons in particular.
Microautophagy
Microautophagy is the least well characterised of the three forms of autophagy, with its role in neurodegeneration mostly unexplored. In this process, proteins entering the endo-lysosomal system through invagination are engulfed by the late endosome and lysosomal membrane [120]. The synapse appears to be a particularly vulnerable neuronal compartment in many neurodegenerative disorders, in part due to the constant turnover of SNARE proteins required for neurotransmitter release, which can form dysfunctional neurotoxic species [121]. Experiments in Drosophila have demonstrated that an endosomal form of microautophagy can be perturbed through knockdown of the synapse enriched chaperone HSC70–4, required for recognition of the peptide degradation motif, resulting in significantly perturbed neurotransmitter release [122]. As microautophagy appears to support normal neuronal function, particularly at sensitive synaptic terminals, further investigation should be conducted in the context of neurodegenerative disorders to define its contribution.
Selective autophagy
Autophagy mechanisms can also be subclassified into those involving selective degradation of specific organelles, such as peroxisomes (pexophagy), nuclei (nucleophagy) and endoplasmic reticulum (ER-phagy), as well as those involving degradation of molecular materials such as lipids (lipophagy), stress granules (granulophagy) and myelin (myelinophagy) (reviewed by [123]). Autophagy receptors for selective targeting of organelles which, under specific conditions, link these organelles with the cellular autophagy machinery leading to their destruction, are being continuously discovered. Receptors important for pexophagy include NBR1 [124], Atg30 [125] and Atg36 [126], whereas FAM134B [127] and Atg40 [128] are required for ER-phagy. Once bound to these adaptors, cargos enter the autophagy cascade for lysosomal degradation.
Whilst the contribution of most cargo selective forms of autophagy to neuronal health is largely unexplored, mitophagy, perhaps the most thoroughly characterised, has been strongly implicated in neurodegenerative disease. Mitophagy is the process by which dysfunctional mitochondria are selectively targeted by autophagosomes and degraded via autophagosome-lysosome fusion, facilitating a quality-control mechanism which maintains a healthy mitochondrial network (Fig. 2). Due to their high metabolic demand and post-mitotic state, neurons are particularly sensitive to mitochondrial dysfunction and thus mitophagy is vitally important in this cell type. Like other forms of selective autophagy, the targeting of mitochondria for mitophagy occurs though a mechanism which parallels that of general macroautophagy cargo targeting, but with specific adapters that allow for the selective targeting of damaged organelles. The canonical mitophagy model is that mitochondrial insult results in the dissipation of mitochondrial membrane potential (ΔΨm), followed by a block of PTEN-induced kinase 1 (PINK1) import into the intermembrane space, where it is usually cleaved by Presenilin Associated Rhomboid-Like (PARL) [129]. PINK1 accumulates on the mitochondrial outer membrane (MOM) and phosphorylates ubiquitin at Ser65 (pS65-Ub), leading to the recruitment of Parkin E3 Ubiquitin Protein Ligase (PRKN) from the cytosol [130]. PINK1 also phosphorylates PRKN at Ser65 of its ubiquitin-like domain, stimulating PRKN E3 ubiquitin ligase activity [131]. This triggers a positive-feedback mechanism during which subsequent PRKN recruitment and ubiquitination of MOM proteins [132, 133] results in the recruitment of AIM/LIR autophagy adapters including p62, OPTN and TAX1 Binding Protein 1 (TAX1BP1). The kinase domain of PINK1 has been shown to recruit OPTN and NDP52 independent of PRKN and recruitment of these two adapters is essential for mitophagy [134]. Though responsible for recruiting LC3-II to the poly-ubiquitinated MOM [135], p62 is dispensable [134, 136] but can improve the efficiency of mitochondrial incorporation into autophagosomes at a later stage in the process. The ULK1 complex transiently assembles at depolarised mitochondria [137], in a PRKN-dependant, LC3-II-independent fashion. ATG9A vesicles are also recruited / formed de novo at depolarised mitochondria, independently of ULK1 recruitment. ULK1 and ATG9A foci only partially co-localise at mitochondria and neither are required for the recruitment of LC3-II, though both are required for mitophagy to occur [137].

Mitophagy and related genes associated with neurodegenerative diseases. Dysfunctional mitochondria are targeted for autophagic clearance by a number of specific adapters which are associated with neurodegenerative disorders. Upon depolarisation, PTEN-induced kinase 1 (PINK1) accumulates on the mitochondrial outer membrane (MOM), where it phosphorylates Ser65 of ubiquitin and the ubiquitin-like domain of PRKN. pS65-Ub acts as a positive-feedback mechanism for the further recruitment of PRKN to the MOM and activation of its E3 ubiquitin-ligase activity. PRKN ubiquitinates a number of targets on the MOM, including mitochondrial fusion proteins such as Mitofusin1 (MFN1), decorating the damaged organelle in poly-ubiquitin chains. F-Box Only Protein 7 (FBXO7) also participates in MFN1 ubiquitination. PINK1, PRKN and pS65-Ub chains on the MOM facilitates the recruitment of autophagy adapters Phosphotyrosine-Independent Ligand For The Lck SH2 Domain Of 62 KDa (p62), Nuclear Domain 10 Protein 52 (NDP52) and Optinuerin (OPTN). Parkinson’s disease-associated mutations in β-glucocerebrosidase (GBA) and Leucine Rich Repeat Kinase 2 (LRRK2) are considered to impair PRKN-mediated mitophagy. Phosphorylation of ALS-associated TBK1 in response to mitochondrial damage is dependent on NDP52 and OPTN recruitment, but subsequently increases the affinity of OPTN for poly-ubiquitin on the MOM. TBK1 also phosphorylates RAB7A, which in turn facilitates the recruitment / formation of ATG9 vesicles. The ULK1 complex and ATG9 vesicles are recruited / form de novo at damaged mitochondria and initiate autophagic engulfment. This is enhanced by the recruitment of LC3-II by p62. Neurodegenerative disease causing or associated genes affecting various stages of mitophagy are listed. For additional information relating to disease association of listed genes, refer to Table 1
The most well-established association between defective mitophagy and neurodegeneration is with PD since PRKN was discovered as the causation of autosomal-recessive juvenile parkinsonism (ARJP) in a Japanese population [26, 27] and PINK1 was subsequently identified as a second ARJP associated gene [24, 25]. Initial functional characterisation of both genes was performed in Drosophila, demonstrating loss of function mutations in the Drosophila PRKN homologue parkin cause aberrant mitochondrial morphology in energy demanding cell types, such as sperm, flight muscle and, more relevant to PD, dopaminergic neurons [138, 139]. Similar phenotypes were observed in Pink1 mutant Drosophila and genetic epistasis experiments showed that overexpression of parkin rescued Pink1 mutant phenotypes but not vice versa, placing PRKN downstream of PINK1 in a common pathway of mitochondrial quality control [140,141,142]. PINK1 and PRKN patients feature the loss of DA neurons of the substantia nigra pars compacta and mitochondria are enlarged in induced pluripotent stem cells (iPSC)-derived DA neurons from these patients [143]. Taken together, PINK1 and PRKN genetic and experimental evidence strongly associate loss of normal mitophagy with ARJP.
Histopathological post-mortem analysis of PD patient brains also suggests disrupted turnover of mitochondria. Mitochondrial complex I defects in the post-mortem substantia nigra are a hallmark of PD pathology [144], indicating that deficient mitochondrial quality control is a common feature across familial and sporadic cases of PD (reviewed [145]). A signature of damaged mitochondria, polymeric pS65-Ub, accumulates in cytoplasmic granules, beaded neurites and granulovacuolar degeneration bodies with age in healthy individuals [146, 147]. In sporadic PD and Dementia with Lewy Bodies patients, these structures have been identified in the proximity of Lewy bodies. Their abundance positively correlates with both age and Braak stage, demonstrating age and disease-associated increases in mitochondrial quality control [147]. Expectedly, given that PINK1 and PRKN are responsible for generating pS65-Ub chains on the MOM, pS65-Ub positive structures are markedly reduced in PINK1 and PRKN post-mortem brains, inferring that defects in mitophagy are observed in these patients [146, 147].
Mitophagy-related roles can further be attributed to several other PD-associated genes. Mutations in another E3 ubiquitin ligase, F-Box Only Protein 7 (FBXO7), were identified as the cause of parkinsonian pyramidal syndrome, a rare form of ARJP which presents with pyramidal tract dysfunction [17, 18]. FBXO7 enhances PRKN recruitment to depolarised mitochondria and also participates in the ubiquitination of Mitofusin 1 (MFN1), facilitating the segregation of damaged mitochondria from the healthy mitochondrial network [148]. The most frequent cause of autosomal dominant PD is the G2019S hypermorphic variant of LRRK2. Cold-shock induced mitophagy is impaired in fibroblasts derived from patients with either PRKN mutations or the G2019S LRRK2 variant. This effect is reversed by treatment with LRRK2 inhibitor LRRK2-in-1 in G2019S LRRK2 but not PRKN loss-of-function fibroblasts [149]. LRRK2-in-1 may also protect against oxidative stress by restoring basal mitophagy levels in a subset of sporadic PD derived fibroblasts [150].
Mitophagy has more recently been implicated in other neurodegenerative diseases. PINK1 has been identified in GWAS of genetic modifiers of HD progression, along with a number of regulators of mitochondrial fission/fusion dynamics [151]. In a Drosophila model of HD, Pink1 overexpression rescues mitochondrial morphology, conveys neuroprotection and extends lifespan, indicating that defects in mitophagy may also contribute to aspects of HD pathogenesis [152]. ALS-associated mutations in OPTN or TANK-binding protein 1 (TBK1) block efficient clearance of depolarised mitochondria in PRKN-expressing HeLa cells, indicating that both of these proteins are important, though not essential, in mitophagy [153]. TBK1 is rapidly phosphorylated and activated upon mitochondrial damage and this is dependent on the mitochondrial recruitment of NDP52 and the ubiquitin binding domain of OPTN. In turn, p-TBK1 phosphorylates OPTN, enhancing its affinity for polyubiquitin and thus its retention at depolarised mitochondria [154]. p-TBK1 also phosphorylates RAB7A [155] required for ATG9-vesicle formation and efficient mitophagy in PRKN-expressing HeLa cells [155,156,157]. Some ALS-associated missense mutations in TBK1 abolish its phosphorylation, activation and ability to phosphorylate OPTN [158], thus defects in mitophagy could play a role in the pathogenesis of patients with these mutations.
Changes in mitophagy may also occur in diseases where genetic evidence does not clearly suggest a mitochondrial contribution to pathology. In AD, post-mortem hippocampal tissues exhibit strikingly lower levels of mitophagy, assessed by mitochondria-lysosome co-localisation and visualisation of mitophagy events by transmission electron microscopy. These deficits correlate with decreased PINK1, p-TBK1 and p-ULK1 in the same samples and also in iPSCs derived from apolipoprotein E4 (APOE4) and APP-mutation carrying patients [159]. Furthermore, there is evidence that upregulation of mitophagy may protect against AD phenotypes. Urolithin A (UA), a metabolite produced in the gut from the ellagitannin class of polyphenols found in pomegranate, raspberries and walnuts, upregulated mitophagy in nematodes and rodents in a manner dependant on PINK1/PRKN and independent of general macroautophagy [159, 160]. UA treatment ameliorated learning and memory defects in both Aß and hyperphosphorylated Tau in a C. elegans models, and improved cognition in mouse models of AD [159]. Intriguingly this study found microglial activation and neuroinflammation were reduced upon UA treatment in APP-PSEN1 mice, suggesting mitophagy deficits are tied to chronic inflammation in the brain, a hallmark of many neurodegenerative diseases. The anti-inflammatory cytokine Interleukin 10 was increased in hippocampal microglia of these mice upon UA treatment, in a PINK1-dependent fashion, indicating that this anti-inflammatory response is also likely dependent on mitophagy. Interleukin 10 has previously been shown to promote mitophagy through inhibition of the mTOR pathway in lipopolysaccharide-activated macrophages, maintaining a healthy mitochondrial network and a metabolic profile based on oxidative phosphorylation as opposed to glycolysis [161]. Microglial activation is associated with a respiratory switch from oxidative phosphorylation to glycolysis, facilitated by the glucose transporter GLUT1. The GLUT1-specific inhibitor STF31 supresses neuroinflammation and neurodegeneration in a mouse model of light-induced retinal degeneration [162]. These studies thus identify a promising strategy for combatting both mitochondrial dysfunction and chronic neuroinflammation in neurodegenerative disease, through upregulation of mitophagy and rebalancing of metabolic state.
Therapeutic targeting of autophagy in neurodegeneration
Through their implication in a broad range of neurodegenerative disorders, endo-lysosomal and autophagy mechanisms have become appealing targets for therapeutic intervention [163]. Autophagy targeting compounds fall in two broad categories, acting through mTOR-dependent or -independent mechanisms. Modulation of mTOR-dependent autophagy via inhibition of mTORC1 with rapamycin has been widely explored across a spectrum of human diseases, including various forms of cancers, auto-immune and neurodegenerative disorders [164]. Rapamycin possesses strong immunosuppressant and anti-proliferation properties which, though beneficial for treatment of cancer and autoimmune disorders, are undesirable for chronic treatment of neurodegenerative disorders. As rapamycin has been found beneficial in treatment of neurodegeneration in preclinical models [165], attempts to circumvent its immunosuppressant activity have been made through “Rapalog” derivative molecules, several of which have been demonstrated to improve phenotypes in models of neurodegenerative disorders including HD [99], spinocerebellar ataxia type-3 [166] and FTD-associated tauopathy [167].
Several mTOR-independent modifiers of autophagy are gaining interest as therapeutics, with AMPK activating molecules such as trehalose and metformin proving effective in reducing neurodegenerative phenotypes in models of AD [168, 169], ALS [170,171,172], HD [173, 174] and tauopathies [175]. Cellular targets not directly associated with the core autophagy machinery have also been found to modify neurodegeneration, including Estrogen Related Receptor α [176] and cAMP [177, 178]. Interestingly, the widely used AD-therapeutic memantine has emerged from a screen of clinically approved molecules which enhance autophagy [179], suggesting a potential mode of action for the drug which may be repurposed in other neurodegenerative disorders.
Endosomes
Endosomes capture surface molecules through internalisation of the plasma membrane, or acquire cargo intracellularly following trans-golgi trafficking. Multiple checkpoints along the endosomal pathway either designate cargos for degradation at the lysosome or recycle them back to the plasma membrane or golgi via the retromer complex [180, 181]. Endosomes exist in three specific states: early (also called sorting), recycling, or late depending on their post internalisation stage and association with distinct Rab guanosine triphophatases (RAB GTPases) [182].
Early endosome
The early endosome (EE) serves as the primary sorting compartment of the endocytic pathway, receiving extracellular material, lipid membranes and membrane-bound proteins from small endocytic vesicles, formed from specialised clathrin-coated invaginations of the plasma membrane. Upon their delivery to the EE, cargos are separated within minutes and assigned for either degradation or recycling. Proteins destined for recycling back to the plasma membrane first cluster within tubular EE extension membranes, whereas the larger and rounder EE compartment houses proteins targeted for degradation. Retrograde transport of cargos from the EE to the trans-golgi network is facilitated by the Retromer complex, which consists of VPS26-VPS29-VPS36 cargo recognition and sorting nexin (SNX) membrane recognition components [183]. Endosomal cargo separation is regulated primarily by RAB4 [184] and RAB5 [185], in addition to some other less well characterised GTPases including RAB10 [186], RAB14 [187], RAB21 [188] and RAB22 [189]. These RAB proteins facilitate either the recruitment of additional RABs to enable vesicle maturation or provide a platform for other proteins and protein complexes to dissociate and re-associate with the vesicle membrane [190] for trafficking or sorting purposes. The PI(3)P rich EE membrane itself is also generated through recruitment of PI 3-kinase VPS34 by RAB5 [191].
Initial endocytosis is disturbed in several age-dependent neurological disorders, notably PD where mutations have been identified in several EE genes. The synaptic enriched inositol-phosphatase Synaptojanin 1 (SYNJ1) binds clathrin and associated proteins, likely contributing to the uncoating of clathrin coated vesicles. Loss of SYNJ1 is associated with dysfunctional endocytosis [192, 193], through disruption of the earliest stages of EE formation [30, 31]. Indeed, enlarged EEs and altered trafficking have been seen in fibroblasts derived from early onset PD patients carrying SYNJ mutations [194]. Endocytosis in PD may further be perturbed by disruption of DnaJ/Heat Shock Protein Family (HSP40) co-chaperone (DNAJC) proteins [195], notably DNAJC6/Auxilin-1 and DNAJC13/RME-8. Neuron-specific DNAJC6/Auxilin-1 interacts with HSC70, facilitating the uncoating of clathrin vesicles [15, 196], whilst DNAJC13/RME-8 decreases retromer-mediated cargo transport sorting through interaction with SNX1 thereby preventing the formation of the necessary tubular structure of the EE membrane [197, 198]. Disruption of retromer activity has also been directly implicated in PD through mutations in the retromer complex gene VPS35 [34, 35]. PD-associated mutations in LRRK2 have also recently been found to alter expression of essential endocytic proteins and also impair endocytosis of clathrin-associated synaptic vesicles in patient derived dopaminergic neuron cultures [199]. Lipophilic and aggregation prone alpha-synuclein may itself inhibit retromer recycling of some membrane proteins through blocking VPS17 and SNX3 from EE association [200].
Beyond PD, several other neurodegenerative disorders have been linked to the EE system. RAB5 interacts with Early Endosome Antigen 1 (EEA1), a soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors (SNARE) complex interacting protein, to enable vesicle fusion [191] and recruitment of HTT via HTT associated protein 40 (HAP40) to enable endosome motility [201]. The poly-Q repeat expansion found in disease associated alleles of the HTT gene has been found to upregulate HAP40, facilitating a shift of EEs from microtubules to actin thereby decreasing trafficking speeds [39, 40]. EE dysfunction may also contribute to juvenile-onset ALS through mutations to Alsin Rho Guanine Nucleotide Exchange Factor (ALS2) [202, 203]. ALS2 contains several domains required for guanine-nucleotide exchange required for RAB activation. Loss-of-function mutations in the ALS2 gene have been found to interfere with GDP/GTP exchange required by RAB5 [204], resulting in EE accumulation and trafficking abnormalities [205]. EE function has long been of interest in AD pathogenesis, initially due to enlargement of RAB5 positive vesicles being one of the earliest pathological events seen in patient tissue [206]. This is not surprising given that the EE pathway is compromised at many levels from the endocytosis of secretases residing on the plasma membrane, intracellular trafficking of key enzymes through the internalisation of extracellular Aβ. More recently, emerging evidence suggests that genes associated with an increased risk for developing late-onset sporadic AD may converge on microglia [207], with several endocytic genes potentially contributing to pathology. Proteins encoded by AD risk genes, including Bridging Integrator 1 (BIN1) [6], CD2AP, EPHA1 [10, 11], PICALM [8], Sortilin Related Receptor 1 (SORL1) [14], amongst others [7, 208, 209] may all interfere with EE function. SNPs in BIN1 represent one of the most common AD risk associated mutations after APOE. RAS and RAB interactor 1 (RIN1), a BIN1 interacting protein, functions as guanine nucleotide exchange factor (GEF) for the RAB5 GTPase family. This interaction was found to promote epidermal growth factor receptor (EGFR) downregulation [210]. SNPs associated with BIN1 are also likely to affect other critical RAB5 dependent processes, which require further investigation.
Retromer-mediated sorting from the EE further controls intracellular shuttling proteins relevant to AD including APP and Beta-Secretase 1 (BACE1), which are required to generate Aβ [211]. Knockout of retromer associated Vps35 in a mouse model of AD enhanced levels of amyloidogenic Aβ species [212], suggesting that retromer signalling from the EE is a negative regulator of Aβ production. This theory is bolstered by genetic evidence linking SNPs and gene expression of Sortilin Related VPS10 Domain Containing Receptor 1 (SORCS1) [213], a membrane homologue of SORL1, to impaired retromer-associated sorting which may lead to APP processing deficits [214]. SNPs in APOE [215] and Clusterin may also accelerate extracellular Aβ-uptake and clearance [216, 217] decreasing endocytosis capacity later in disease. Given the role of EE dysfunction in a diverse range of neurological disorders, this pathway may represent a common mechanism of either disease manifestation or disease progression.
Recycling endosome
Portions of neuronal plasma membrane and residing surface receptors that have been internalised and lost through EE formation are replenished by recycling endosomes (REs). At the ultrastructural level, REs have a tubular formation and form a non-continuous network [218], identified in tissues through association with RAB11 [219]. Lipids to be recycled are sorted away from those ubiquitylated receptors and ligands that are destined for degradation due to the REs acidic environment (pH ∼ 6.0) [220]. Endosomal recycling can be rapid, occurring within 2–3 min or can take around 10 mins from initial endocytosis. Different RAB subtypes appear to be required for either fast or slow kinetics, with RAB35 associated with fast moving vesicles and RAB11 slow [221,222,223], although why these different mechanisms exist and under what cellular conditions they occur is not well understood.
Although the RE compartment is relatively understudied compared to EE, several links to neurodegenerative diseases have been made. The activity of recycling endosome associated RAB11 is at least in part controlled through interaction with HTT. The removal of GDP from RAB11 is compromised by mutant polyQ expanded HTT in human cells, leading to deficits in RE size and receptor recycling [224], impacting dendritic spine complexity in rodent models and patients [225] and electrophysiology, lifespan and locomotion in Drosophila models of HD [225, 226]. RAB11 and its role in regulating recycling endosome activity has been implicated in disease beyond HD. Charcot-Marie-Tooth peripheral neuropathy type 4C associated SH3 domain and tetratricopeptide repeats 2 (SH3TC2) [49] is considered a RAB11 effector protein, localising to GTP-bound species [227]. In CMT4C, mislocalisation of SH3TC2 and lack of RE trafficking is considered a causative feature of disease progression [227]. Increasingly targeting RAB11 activity is now considered a keen therapeutic target for HD, with potential benefits for other neurodegenerative diseases.
Another important molecule bound to the EE/RE following secretion from the golgi is the gamma-secretase component PSEN1. Although enhanced amyloidogenic Aβ production is likely to play a role in disease manifestation, a recent report also suggests that it is the accumulation of β C-terminal fragments which cause RE dysfunction [228]. In this model, mutant forms of PSEN1 and APP decrease RAB11 dependent trafficking from the cell body to the axon [228]. Hence neurons have a decreased capacity to deliver lipoproteins, receptors and transporters back to the plasma membrane in vulnerable sub-compartments. Lysosome restricted PSEN2, which also cleaves APP, may play a more important role in normal cellular Aβ production, with more toxic species generated by the mislocalisation of mutant PSEN1 from the EE/RE to the lysosome [229].
Genetic evidence indicates that recycling of specific proteins confers neurodegenerative disease specificity. However, general RE disruption may contribute to neuronal demise indirectly. Several substrates of the RE pathway suggest why its disruption is so clearly detrimental to neuronal function. RAB11 vesicles have been found to carry important neurotrophic factors, such as BDNF [230] and critical synaptic receptors, such as AMPA [231]. Although loss of recycling capacity may not initially drive cell death, it may be key to understanding why synapses are preferentially lost early in disease.
Late endosome
Late endosomes (LEs) / multi-vesicular bodies (MVBs) are generated through the maturation of EEs. Endosomal Sorting Complex Required for Transport (ESCRT) complexes 0-III and several VPS proteins are also recruited to ubiquitinated surface molecules on the cytosol facing endosomal membrane. ESCRT complexes facilitate the invagination of endosome membrane proteins and lipids, producing a MVB, an endosome containing smaller intraluminal vesicles [232]. During this maturation process RAB5 and RAB4 dissociate from the endocytic membrane and RAB7 and RAB9 are recruited. Genetic and cell biological evidence suggest that adequate RAB7 function to initiate clearance through LE-lysosomes fusions may be a critical factor in maintaining normal neuronal function [233]. RAB7 can also assist the recruitment of the retromer to late endosomes through interaction with VPS35 [234]. LEs/MVBs acidify to pH levels of 6.0–4.9 [235] in the final step of the endocytic pathway before intraluminal cargos are delivered to the lysosome for degradation (discussed below).
Dysfunction of LE activity in neurodegenerative appears mostly restricted to PD, spearheaded by genetic association. GWAS approaches have identified LRRK2 mutations as common risk factors for the development of sporadic PD [21, 22], in addition to mutations in Cyclin G Associated Kinase (GAK) and the LE associate RAB7L1 [19, 236]. Highlighting the importance of the LE pathway in the maintenance of the large highly arborised dopamine neurons, a protein complex of LRRK2, RAB7 and GAK was previous uncovered by an unbiased protein-protein interaction experiment [237]. Overexpression of these molecules promotes protein clearance from the trans-Golgi network, suggesting that trafficking from golgi to LE is compromised in PD. LRRK2 was also found to impede cargo trafficking by prohibiting budding of the LE membrane to form smaller vesicles, via decreased RAB7 activity [238], a process exacerbated by PD associated mutations. LRRK2 kinase inhibition via small molecules increases lysosome formation [150], suggesting that LE dysregulation directly impacts on clearance. Loss of LRRK2 or RAB7 also downregulates VSP35 [239] which may further perpetuate the dysregulation of the endosomal pathway upstream of the LE. The ESCRT-III complex has also been implicated in neurodegeneration through its function to concentrate endosomal cargos into LE intralumenal vesicles. ALS/FTD associated charged multi-vesicular body protein 2B (CHMP2B) mutations [41, 42] were found to cause severe lysosome pathologies [240] and metabolic disturbances in neurons [241]. This evidence shows that LE perturbations may therefore lead to downstream lysosome-mediated clearance complications or initiate dysfunction in earlier in the endocytic pathway.
Lysosomes
Lysosomes are the terminal compartment through which macromolecules are degraded and recycled to generate nutrients. Lysosomes are generated through the maturation of LEs, achieved via delivery of hydrolytic enzymes from the golgi and also active acidification of the lumen via the lysosomal vATPase hydrogen pump. Once acidified to approximately pH 5, over 50 hydrolytic enzymes, including a broad range of glycosidases and proteases degrade the contents. The matured lysosome is able to fuse with and degrade the contents of other vesicular compartments including endosomes, autophagosomes, amphisomes (fused endosome-autophagosome) and phagosomes (phagocytosed material). A wide range of cellular macromolecules can be processed through the lysosome, including nucleic acids, proteins, carbohydrates and lipids. Several aspects of lysosomal biology including enzymatic dysfunction and positioning have been implicated in neurodegenerative disorders.
Lysosome function
As lysosomal degradation is one of the primary mechanisms of cellular waste removal, it is unsurprising that genes facilitating this essential process have been linked to a broad range of diseases. Lysosomal Storage Disorders (LSD) are a family of hereditary conditions in which substrates of lysosomal degradation accumulate within the lumen, caused by mutations in a range of lysosome specific hydrolases, enzymatic regulators, membrane proteins and transporters. Many of the > 50 genes linked to LSDs cause juvenile neurodegenerative disorders, though pathologies of the liver, spleen and bones are also common (reviewed [242]). Several LSDs which feature neurodegeneration are associated with mutations in hydrolytic enzymes responsible for the processing of specific lipids, resulting in their build up within the lysosomal lumen. Examples include the Neuronal ceroid lipofuscinoses (NCLs), a family of 14 genetically distinct, autosomal recessive LSDs that present juvenile onset vision loss, seizures, cognitive decline and motor dysfunction, unified by the accumulation within neuronal lysosomes of auto-fluorescent lipofuscin, a heterogenous mixture of oxidised lipids, proteins and carbohydrates [243]. Neuroinflammation and neuronal death are seen in juvenile onset Sandhoff disease and heterogenous onset Tay-Sach’s disease, both GM2-gangliosidosis disorders caused by accumulation GM2-gangliosides within the lysosomal lumen [244].
The sphingolipidosis Gaucher’s disease is of particular interest due to implications in the pathogenesis of PD. Autosomal recessive Gaucher’s disease is characterised by the accumulation of glucosylceramide (GluCer) due to mutation of the β-glucocerebrosidase (GBA) gene [245], with progressive neurological dysfunction is seen in the severe early-onset type II and milder late-onset type III forms. In addition to accumulation of lysosomal GluCer, misfolded mutant GBA accumulates in the ER [246, 247], with mutant GBA associated with activated unfolded protein response in model systems [248,249,250]. There is a wealth of emerging data to suggest a strong association between impediment of lysosomal enzymatic function and synucleinopathies, in particular PD [251]. Genetic studies of PD patients have identified a strong association with heterozygosity for GBA loss-of-function mutations and increased risk of developing PD [252]. Both wild type alpha-synuclein and PD associated variants interact with lipids [253]. Dysfunction of GBA can disrupt alpha-synuclein function [254] and exacerbate its aggregation [255], with GluCer stabilising the peptide in oligomeric species and promoting its aggregation [256]. Alpha-synuclein is degraded through the lysosome via chaperone-mediate autophagy (see above) [257, 258]. Further still, accumulation of alpha-synuclein itself is able to inhibit lysosomal GBA function, suggesting a feed-forward loop of alpha-synuclein aggregation promoting lysosomal dysfunction and further accumulation of aggregated protein [256]. Intriguingly, PD-associated GBA variants GBAL444P and GBAN370S can also impede normal PRKN ubiquitination of mitochondrial substrates [259, 260], and heterozygous GBAL444P mutations decrease the delivery of the mitochondria to lysosomes [261]. As mitochondrial dysfunction is not observed in heterozygous GBA knockout neurons [262] PD-associated GBA variants may convey specific gain-of-function effects in neurons, aside from lysosomal function. Due to the implications of GBA-associated lysosomal dysfunction and PD-associated pathologies, GluCer synthesis and metabolism have become promising targets for therapeutic intervention [254, 263,264,265], as have molecules such as ubiquitin ligase NEDD4, which target alpha-synuclein for lysosomal destruction [266].
Lysosomal dysfunction can also contribute to neurodegeneration through mutations that do not directly affect hydrolytic enzymes. Niemann-Pick disease type C (NPC) is a juvenile onset neurodegenerative condition with death occurring in young adulthood, primarily effecting the cerebellum, associated with accumulation of a range of lipids within the lysosomal lumen including cholesterol, sphingomyelin and sphingosine [267,268,269,270,271]. The disorder has two genetically distinct forms; NPC1 is a sterol-sensing transmembrane protein acidic compartments [50, 51, 272] and rarer mutations in NPC2 that disrupt a lysosomal soluble peptide with a cholesterol binding domain [52, 53, 273]. As NPC1 is expressed in most cell types, why neurons are particularly vulnerable to its dysfunction is unclear. Experimental data has suggested defective regulation of lysosomal calcium may contribute to NPC associated phenotypes, with increased storage of lysosomal sphingosine causing a reduction in luminal calcium levels, subsequent accumulation of further lipids and defects in endocytic trafficking [274]. Since NPC shares formation of the hyperphosphorylated Tau neurofibrillary tangles typically seen in AD and PD [275], understanding the mechanistic role of NPC1/2 in lysosome function may have broader implications for other neurodegenerative diseases and their treatment.
Lysosome positioning
The positioning of lysosomes within a cell is intertwined with the function of these vesicles, particularly with regard to acidification of the lumen. In non-polarised cells, lysosomes are distributed into two groups; a relatively stationary perinuclear “cloud” [276] where early endosomes mature through to lysosomes, and a highly motile population in the periphery [277]. Lysosome transportation to the periphery generally occurs along microtubule networks, with anterograde transport to the periphery mediated by kinesin motor proteins [278], and returning retrograde transport by the dynactin motor complex [279]. Lysosome distribution differs somewhat in highly polarised neurons, where the vast length and volume of many axons requires effective delivery of acidified lysosomes. Though lysosomes can be detected throughout the soma, axon, dendrites and synapses of neurons, their positioning appears to define their function. Mature, acidified lysosomes are enriched in the soma, with a decreasing gradient of acidity along the distal-proximal length of the axon, suggesting degradation within lysosomes occurs in the cell body [280, 281]. Directionality of lysosome transport within the axon has not been fully resolved, in part due to differences in the assays used for their detection [282]. Further research into the basic neurobiology of lysosome maintenance and trafficking is long overdue and would enable us to better understand neurodegenerative disease.
Abnormal transport and positioning of lysosomes may contribute towards the pathogenesis of AD, particularly as disruption of the endo-lysosomal system is one of the earliest detectable histopathological features [206]. Swollen, dystrophic neurites are a common histopathological feature of AD [283, 284], with lysosomes and related vesicles found to accumulate within these axonal swellings. Curiously, such lysosome enriched swellings are often in regions proximal to amyloid plaques in patient brains [285,286,287] and rodent models of the disease [288]. Whether accumulation of lysosomes cause amyloid pathology, or a secondary event downstream of plaque formation, remains unanswered. It is conceivable that plaques and the neuroinflammation may alter local intra-axonal processes such as transportation, however emerging experimental data suggests that dysregulation of lysosome axonal transport may actively drive amyloid accumulation. Proteins in the c-Jun N-terminal kinase-interacting proteins (JIP) family of conserved mitogen activated protein kinases (MAPKs) regulate microtubule mediated transport of cargos along axons [289, 290]. Mutation of JIP3 causes accumulation of lysosomal vesicles, amyloid processing enzymes and increased production of toxic species of Aβ in an AD mouse model [291]. Further links between AD pathology and lysosome function can be found in the function of PSEN proteins. Early onset AD patients with PSEN1 or 2 mutations present elevated lysosomal pathology levels or lysosome associated pathology [292]. Experimental disruption of PSEN1 in cell culture models results in reduced assembly of the vacuolar-type H+ ATPase (vATPase) complex at the lysosomal membrane and subsequent failure of the lumen to reach correct acidic pH [293]. Consequentially, lysosomal acid-sensitive hydrolytic enzymes have reduced function and increased efflux of luminal Ca2+. Taken together, evidence of accumulation of axonal lysosomes, increasing amyloid plaque burden and lysosomal dysfunction associated hereditary AD genes are suggestive that impaired lysosomal positioning may be a contributing factor in AD that warrants further investigation. Furthermore, as mislocalisation of lysosomes has also been reported in cellular models of HD [294] and ALS associated mutant dynactin-p150glued [295], disruption of their trafficking may be a common pathogenic event in neurodegeneration.
Conclusions and future perspectives
Changes in autophagy, mitophagy and endo-lysosomal processes have been implicated in most neurodegenerative diseases, however their contribution is still only partially defined, with several outstanding questions. Though the regulatory processes underpinning autophagy are well understood with regard to starvation, its regulation in neurons in health and disease is poorly defined. The initiating signals for upregulation of autophagy in times of neurotoxic stress are not well understood, particularly how the right balance between homeostatic autophagy and the clearance of toxic material is achieved. More so, it is not entirely clear if autophagy is indeed protective, or is instead contributing to neuronal stress and destruction. This is in part due to gaps in our basic understanding of the cellular mechanism driving autophagy. Though a peripheral origin and retrograde transport of autophagosomes has been demonstrated in tissue culture experiments [67], the source of the phagophore isolation membrane in neurons is not clear and requires further description in vivo. Experiments in Drosophila have also suggested an important role for microautophagy in the maintenance of synapses [122], a highly vulnerable neuronal compartment, and this process warrants further investigation in the context of neurodegenerative disease.
Despite extensive exploration of the function of PINK1 and PRKN in Parkinsonisms, robust evidence for defective mitophagy as a direct cause of pathology in patient brains is lacking. It is clear that PINK1 and PRKN have independent roles outside of mitophagy [296, 297], and furthermore, there is evidence to suggest that basal mitophagy can occur independently of PINK1 or PRKN [298, 299]. The importance of PINK1/PRKN mediated mitophagy to the viability of dopaminergic neurons in the substantia nigra thus needs to be clarified. The link between mitophagy and neuroinflammation is not well characterised, but new findings indicate that the metabolic state of microglia influence their activation [162] and this can be regulated by mitochondrial turnover [159]. The relationship between mitochondrial quality control in glial cells and neurodegenerative disorders may reflect the convergence of two key processes in neurodegenerative disorders and therefore requires further investigation.
It is curious why specific mechanisms within the endosomal pathway appear to be dysregulated in different neurological conditions, suggesting divergent pathological roles in neurodegeneration. While EE dysfunction appears to be a characteristic feature of multiple neurodegenerative diseases, RE is primarily implicated in HD and AD, whereas LE pathway dysfunction is largely restricted to PD. Disruption of synaptic receptor recycling, observed when the RE is compromised [231], may be of a significant importance to medium spiny neurons and cortical/hippocampal neurons that underpin learning and memory through spine remodelling. The LE pathway may on the other hand play a more prominent feature in dopaminergic neurons, where the disposal of mitochondria and alpha-synuclein is prioritised [156, 300]. It is currently unclear why EE dysfunction appears to be a pathological feature of many different diseases. Does EE dysfunction always lead to lysosomal problems downstream or is the LE system adaptable enough to correct itself despite endocytosis and sorting issues? Cumulative evidence supports the latter, suggesting that lack of LE-lysosome fusion can to some degree be compensated for by autophagic clearance. The endosomal pathway is a dynamic continuum and a shift in its balance may result in neuronal demise, as evidenced by both causative and enhanced disease risk associated mutations.
Finally, abundant data suggests that defects in autophagy and the endo-lysosomal system contribute to disease, supporting the concept that their stimulation is a feasible target for therapeutic intervention in neurodegeneration. Several pharmacological modifiers of autophagy with blood brain barrier permeable properties exist, with some experimental evidence to support their use [98, 99, 301, 302]. These are generally not considered appropriate for long-term use due to global alterations of essential cellular processes [303]. Identifying potent, neuro-specific modulators of autophagy and endo-lysosomal function will be essential to determine if these pathways are truly viable targets for therapeutics, in order to ultimately treat devastating neurodegenerative disorders.
Availability of data and materials
All work cited is in the public domain.
Abbreviations
- AD:
-
Alzheimer’s disease
- AIM/LIR:
-
ATG8 Interaction motif/LC3 interacting region
- ALS:
-
Amyotrophic Lateral Sclerosis
- ALS2:
-
Alsin Rho Guanine Nucleotide Exchange Factor
- AMPK:
-
5′- Adenosine Monophosphate-activated Protein Kinase
- APOE4:
-
Apolipoprotein E4
- APP:
-
Amyloid Precursor Protein
- ARJP:
-
Autosomal-Recessive Juvenile Parkinsonism
- ATG:
-
Autophagy Related Gene
- Aβ:
-
Amyloid Beta
- BACE:
-
Beta-Secretase 1
- BECN1:
-
Beclin1
- BIN1:
-
Bridging Integrator 1
- C9ORF72:
-
Chromosome 9 Open Reading Frame 72
- cAMP:
-
Cyclic Adenosine Monophosphate
- CHMP2B:
-
Charged Multi-Vesicular Body Protein 2B
- CMA:
-
Chaperone-Mediated Autophagy
- DJ-1:
-
Parkinsonism Associated Deglycase
- DNAJC:
-
DnaJ/Heat Shock Protein Family (HSP40) co-chaperone
- EE:
-
Early Endosome
- EEA1:
-
Early Endosome Antigen 1
- EGFR:
-
Epidermal Growth Factor Receptor
- ER:
-
Endoplasmic Reticulum
- ESCRT:
-
Endosomal Sorting Complex Required for Transport
- FAM134B:
-
Family with sequence similarity 134, Member B
- FBXO7:
-
F-Box Only Protein 7
- FIP200:
-
Focal Adhesion Kinase Family Kinase-Interacting Protein Of 200KDa
- FTD:
-
Frontotemporal Dementia
- GAK:
-
Cyclin G Associated Kinase
- GBA:
-
β-glucocerebrosidase
- GDP:
-
Guanosine Diphosphate
- GEF:
-
Guanine Nucleotide Exchange Factor
- GluCer:
-
Glucosylceramide
- GTP:
-
Guanosine Triphosphate
- GWAS:
-
Genome Wide Association Study
- HAP40:
-
HTT associated protein 40
- HD:
-
Huntington’s disease
- HSC70:
-
Heat Shock Protein Family A (Hsp70) Member 8
- HSP90:
-
Heat Shock Protein 90
- HTT:
-
Huntingtin
- iPSC:
-
induced Pluripotent Stem Cell
- JIP:
-
c-Jun N-terminal kinase-interacting proteins
- LAMP2A:
-
Lysosome-Associated Membrane Protein 2A
- LC3-I:
-
Microtubule Associated Protein 1 Light Chain 3 – non-lipidated
- LC3-II:
-
Microtubule Associated Protein 1 Light Chain 3 - phosphatidylethanolamine lipidated
- LE:
-
Late Endosome
- LRRK2:
-
Leucine Rich Repeat Kinase 2
- MAPK:
-
Mitogen Activated Protein Kinases
- MEF2D:
-
Myocyte Enhancer Factor 2D
- MFN1:
-
Mitofusin 1
- MOM:
-
Mitochondrial Outer Membrane
- mTOR:
-
mammalian Target Of Rapamycin
- mTORC1:
-
mammalian Target Of Rapamycin Complex 1
- MVB:
-
Multi-Vesicular Body
- NADH:
-
Nicotinamide adenine dinucleotide
- NBR1:
-
Neighbour Of BRCA1 Gene 1 Autophagy Cargo Receptor
- NCL:
-
Neuronal ceroid lipofuscinosis
- NDP52:
-
Nuclear Domain 10 Protein 52/ Calcium Binding And Coiled-Coil Domain 2
- NEDD4:
-
Neural Precursor Cell Expressed, Developmentally Down-Regulated 4
- NPC:
-
Niemann-Pick disease type C
- OPTN:
-
Optineurin
- p62:
-
Phosphotyrosine-Independent Ligand For The Lck SH2 Domain Of 62 KDa / Sequestosome-1
- PARL:
-
Presenilin Associated Rhomboid-Like
- PD:
-
Parkinson’s disease
- PI(3)P:
-
Phosphatidylinositol 3-phosphate
- PINK1:
-
PTEN-Induced Kinase 1
- poly-Q:
-
poly-Glutamine
- PRKN:
-
Parkin RBR E3 Ubiquitin Protein Ligase
- PSEN:
-
Presenlin
- RAB:
-
GTPase RAB guanosine triphophatase
- RE:
-
Recycling Endosome
- Rheb:
-
RAS homolog enriched in the brain
- Rhes:
-
RAS homolog enriched in the striatum
- RIN:
-
RAS and RAB interactor 1
- SH3TC2:
-
SH3 domain and tetratricopeptide repeats 2
- SNARE:
-
Soluble N-ethylmaleimide-sensitive fusion protein attachment protein Receptor
- SNX:
-
Sorting Nexin
- SORCS1:
-
Sortilin Related VPS10 Domain Containing Receptor 1
- SORL1:
-
Sortilin Related Receptor 1
- SYNJ1:
-
Synaptojanin 1
- TAX1BP1:
-
TAX1 Binding Protein 1
- TBK1:
-
TANK Binding Kinase 1
- UA:
-
Urolithin A
- UCHL-1:
-
Ubiquitin carboxyl-terminal esterase L1
- ULK1:
-
Unc-51-like Autophagy Activating Kinase 1
- vATPase:
-
vacuolar-Type H+ ATPase
- VPS:
-
Vacuolar Protein Sorting
References
Feigin VL, Abajobir AA, Abate KH, Abd-Allah F, Abdulle AM, Abera SF, et al. Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017;16(11):877–97.
Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–72.
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118(6):2190–9.
Bieri G, Lucin KM, O’Brien CE, Zhang H, Villeda SA, Wyss-Coray T. Proteolytic cleavage of Beclin 1 exacerbates neurodegeneration. Mol Neurodegener. 2018;13(1) Dec [cited 2019 Mar 7]. Available from: https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-018-0302-4.
Rohn TT, Wirawan E, Brown RJ, Harris JR, Masliah E, Vandenabeele P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol Dis. 2011;43(1):68–78.
Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303(18):1832–40.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1088–93.
Lambert J-C, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–9.
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–35.
Naj AC, Jun G, Beecham GW, Wang L-S, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–41.
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376(6543):775–8.
Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry. 2010;68(10):885–93.
Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39(2):168–77.
Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim Y-I, Zenvirt S, et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PloS One. 2012;7(5):e36458.
Olgiati S, Quadri M, Fang M, Rood JPMA, Saute JA, Chien HF, et al. DNAJC6 Mutations Associated With Early-Onset Parkinson’s Disease. Ann Neurol. 2016;79(2):244–56.
Di Fonzo A, Dekker MCJ, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, et al. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology. 2009;72(3):240–5.
Shojaee S, Sina F, Banihosseini SS, Kazemi MH, Kalhor R, Shahidi G-A, et al. Genome-wide Linkage Analysis of a Parkinsonian-Pyramidal Syndrome Pedigree by 500 K SNP Arrays. Am J Hum Genet. 2008;82(6):1375–84.
Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124(6):593–605.
Tayebi N, Walker J, Stubblefield B, Orvisky E, LaMarca ME, Wong K, et al. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab. 2003;79(2):104–9.
Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7.
Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. 2002;51(3):296–301.
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–60.
Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. 2001;68(4):895–900.
Matsumine H, Saito M, Shimoda-Matsubayashi S, Tanaka H, Ishikawa A, Nakagawa-Hattori Y, et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2–27. Am J Hum Genet. 1997;60(3):588–96.
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–8.
Tucci A, Nalls MA, Houlden H, Revesz T, Singleton AB, Wood NW, et al. Genetic variability at the PARK16 locus. Eur J Hum Genet. 2010;18(12):1356–9.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–7.
Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat. 2013;34(9):1208–15.
Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 2013;34(9):1200–7.
Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395(6701):451–2.
Lowe J, McDermott H, Landon M, Mayer RJ, Wilkinson KD. Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J Pathol. 1990;161(2):153–60.
Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89(1):162–7.
Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89(1):168–75.
Metzger S, Walter C, Riess O, Roos RAC, Nielsen JE, Craufurd D, et al. The V471A polymorphism in autophagy-related gene ATG7 modifies age at onset specifically in Italian Huntington disease patients. PloS One. 2013;8(7):e68951.
Metzger S, Saukko M, Van Che H, Tong L, Puder Y, Riess O, et al. Age at onset in Huntington’s disease is modified by the autophagy pathway: implication of the V471A polymorphism in Atg7. Hum Genet. 2010;128(4):453–9.
Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, et al. Regulation of Intracellular Accumulation of Mutant Huntingtin by Beclin 1. J Biol Chem. 2006;281(20):14474–85.
The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–83 The Huntington’s Disease Collaborative Research Group.
Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature. 1983;306(5940):234–8.
Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 2006;67(6):1074–7.
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–8.
Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–6.
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–6.
Rubino E, Rainero I, Chiò A, Rogaeva E, Galimberti D, Fenoglio P, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79(15):1556–62.
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436–41.
Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–6.
Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet. 2003;72(3):722–7.
Senderek J, Bergmann C, Stendel C, Kirfel J, Verpoorten N, De Jonghe P, et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am J Hum Genet. 2003;73(5):1106–19.
Carstea ED, Polymeropoulos MH, Parker CC, Detera-Wadleigh SD, O’Neill RR, Patterson MC, et al. Linkage of Niemann-Pick disease type C to human chromosome 18. Proc Natl Acad Sci U S A. 1993;90(5):2002–4.
Higgins ME, Davies JP, Chen FW, Ioannou YA. Niemann-Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol Genet Metab. 1999;68(1):1–13.
Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290(5500):2298–301.
Vanier MT, Duthel S, Rodriguez-Lafrasse C, Pentchev P, Carstea ED. Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. Am J Hum Genet. 1996;58(1):118–25.
He L, Zhang J, Zhao J, Ma N, Kim SW, Qiao S, et al. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv Nutr Bethesda Md. 2018;9(4):493–504.
Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. 2018;20(10):1110–7.
Wolf MS, Bayır H, Kochanek PM, Clark RSB. The role of autophagy in acute brain injury: A state of flux? Neurobiol Dis. 2019;122:9–15.
Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery. Cell. 2002;110(2):163–75.
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–91.
Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009;5(5):649–62.
Russell RC, Tian Y, Yuan H, Park HW, Chang Y-Y, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–50.
Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117(Pt 13):2805–12.
Romanov J, Walczak M, Ibiricu I, Schüchner S, Ogris E, Kraft C, et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012;31(22):4304–17.
Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, et al. ATG8 Family Proteins Act as Scaffolds for Assembly of the ULK Complex: SEQUENCE REQUIREMENTS FOR LC3-INTERACTING REGION (LIR) MOTIFS. J Biol Chem. 2012;287(47):39275–90.
Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53(2):167–78.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–45.
Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233–42.
Fu MM, Nirschl JJ, Holzbaur ELF. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev Cell. 2014;29(5):577–90.
Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007;581(11):2156–61.
Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333(1–2):169–74.
Ogura K, Wicky C, Magnenat L, Tobler H, Mori I, Müller F, et al. Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev. 1994;8(20):2389–400.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–9.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–4.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–40.
Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4(11):1318–20.
Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85(11):4051–5.
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83(13):4913–7.
Ihara Y, Nukina N, Miura R, Ogawara M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J Biochem (Tokyo). 1986;99(6):1807–10.
Goedert M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018;592(14):2383–91.
Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–90.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82(12):4245–9.
Alzheimer’s Disease Collaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet. 1995;11(2):219–22.
St George-Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Nee L, Watkins PC, et al. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science. 1987;235(4791):885–90.
Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, et al. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235(4791):880–4.
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–90.
Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–7.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–72.
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–11.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62.
Shaw CE, Enayat ZE, Powell JF, Anderson VE, Radunovic A, al Sarraj S, et al. Familial amyotrophic lateral sclerosis. Molecular pathology of a patient with a SOD1 mutation. Neurology. 1997;49(6):1612–6.
Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY, et al. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol. 1996;55(4):481–90.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
Mori K, Weng S-M, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–8.
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68.
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–3.
Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–14.
Kim M, Sandford E, Gatica D, Qiu Y, Liu X, Zheng Y, et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife. 2016;26:5.
Roscic A, Baldo B, Crochemore C, Marcellin D, Paganetti P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem. 2011;119(2):398–407.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–95.
Lee JH, Tecedor L, Chen YH, Monteys AM, Sowada MJ, Thompson LM, et al. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron. 2015;85(2):303–15.
Fox JH, Connor T, Chopra V, Dorsey K, Kama JA, Bleckmann D, et al. The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington’s disease. Mol Neurodegener. 2010;5:26.
Pryor WM, Biagioli M, Shahani N, Swarnkar S, Huang W-C, Page DT, et al. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease. Sci Signal. 2014;7(349):ra103.
Wold MS, Lim J, Lachance V, Deng Z, Yue Z. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol Neurodegener. 2016;11(1):76.
Nascimento-Ferreira I, Nóbrega C, Vasconcelos-Ferreira A, Onofre I, Albuquerque D, Aveleira C, et al. Beclin 1 mitigates motor and neuropathological deficits in genetic mouse models of Machado-Joseph disease. Brain J Neurol. 2013;136(Pt 7):2173–88.
Nascimento-Ferreira I, Santos-Ferreira T, Sousa-Ferreira L, Auregan G, Onofre I, Alves S, et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain J Neurol. 2011;134(Pt 5):1400–15.
Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016;35(15):1656–76.
Webster CP, Smith EF, Grierson AJ, De Vos KJ. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases. 2018;9(5):399–408.
Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15(8):305–9.
Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22(8):407–17.
Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20(3):460–73.
Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. 2010;28(3):256–63.
Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow E-M, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18(21):4153–70.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–5.
Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118(2):777–88.
Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem. 2008;283(35):23731–8.
Orenstein SJ, Kuo S-H, Tasset I, Arias E, Koga H, Fernandez-Carasa I, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013;16(4):394–406.
Wang B, Cai Z, Tao K, Zeng W, Lu F, Yang R, et al. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy. 2016;12(8):1215–28.
Smith PD, Mount MP, Shree R, Callaghan S, Slack RS, Anisman H, et al. Calpain-regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J Neurosci Off J Soc Neurosci. 2006;26(2):440–7.
Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, et al. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science. 2009;323(5910):124–7.
Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev Cell. 2011;20(1):131–9.
Sharma M, Burré J, Bronk P, Zhang Y, Xu W, Südhof TC. CSPα knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 2012;31(4):829–41.
Uytterhoeven V, Lauwers E, Maes I, Miskiewicz K, Melo MN, Swerts J, et al. Hsc70–4 Deforms Membranes to Promote Synaptic Protein Turnover by Endosomal Microautophagy. Neuron. 2015;88(4):735–48.
Khaminets A, Behl C, Dikic I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016;26(1):6–16.
Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, et al. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci. 2013;126(4):939–52.
Farré J-C, Manjithaya R, Mathewson RD, Subramani S. PpAtg30 Tags Peroxisomes for Turnover by Selective Autophagy. Dev Cell. 2008;14(3):365–76.
Motley AM, Nuttall JM, Hettema EH. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012;31(13):2852–68.
Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522(7556):354–8.
Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015;522(7556):359–62.
Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191(5):933–42.
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298.
Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2(5):120080.
Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RLJ, et al. Broad activation of the ubiquitin–proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20(9):1726–37.
Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496(7445):372–6.
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–14.
Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–31.
Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15(8):887–900.
Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci. 2012;125(Pt 6):1488–99.
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100(7):4078–83.
Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2005;102(22):8024–9.
Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441(7097):1157–61.
Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162–6.
Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103(28):10793–8.
Chung SY, Kishinevsky S, Mazzulli JR, Graziotto J, Mrejeru A, Mosharov EV, et al. Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and α-Synuclein Accumulation. Stem Cell Rep. 2016;7(4):664–77.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet Lond Engl. 1989;1(8649):1269.
Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences: Mitochondrial dysfunction in Parkinson’s disease. EMBO J. 2012;31(14):3038–62.
Fiesel FC, Ando M, Hudec R, Hill AR, Castanedes-Casey M, Caulfield TR, et al. (Patho-)physiological relevance of PINK1-dependent ubiquitin phosphorylation. EMBO Rep. 2015;16(9):1114–30.
Hou X, Fiesel FC, Truban D, Castanedes Casey M, Lin W, Soto AI, et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy. 2018;14(8):1404–18.
Burchell VS, Nelson DE, Sanchez-Martinez A, Delgado-Camprubi M, Ivatt RM, Pogson JH, et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci. 2013 Sep;16(9):1257–65.
Bonello F, Hassoun SM, Mouton-Liger F, Shin YS, Muscat A, Tesson C, et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson’s disease. Hum Mol Genet. 2019;28:1645–60 Available from: https://www.ncbi.nlm.nih.gov/pubmed/30629163.
Smith GA, Jansson J, Rocha EM, Osborn T, Hallett PJ, Isacson O. Fibroblast Biomarkers of Sporadic Parkinson’s Disease and LRRK2 Kinase Inhibition. Mol Neurobiol. 2016;53(8):5161–77.
Moss DJH, Pardiñas AF, Langbehn D, Lo K, Leavitt BR, Roos R, et al. Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol. 2017;16(9):701–11.
Khalil B, El Fissi N, Aouane A, Cabirol-Pol MJ, Rival T, Liévens JC. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015;6:e1617.
Moore AS, Holzbaur ELF. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A. 2016;113(24):E3349–58.
Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015;60(1):7–20.
Heo JM, Ordureau A, Swarup S, Paulo JA, Shen K, Sabatini DM, et al. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci Adv. 2018;4(11):eaav0443.
Yamano K, Wang C, Sarraf SA, Münch C, Kikuchi R, Noda NN, et al. Endosomal Rab cycles regulate Parkin-mediated mitophagy. eLife. 2018;23:7.
Jimenez-Orgaz A, Kvainickas A, Nägele H, Denner J, Eimer S, Dengjel J, et al. Control of RAB7 activity and localization through the retromer-TBC1D5 complex enables RAB7-dependent mitophagy. EMBO J. 2017;37(2):235–54.
de Majo M, Topp SD, Smith BN, Nishimura AL, Chen H-J, Gkazi AS, et al. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol Aging. 2018;71:266.e1–266.e10.
Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22(3):401–12.
Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22(8):879–88.
Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356(6337):513–9.
Wang L, Pavlou S, Du X, Bhuckory M, Xu H, Chen M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol Neurodegener. 2019;14(1):2.
Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron. 2017;93(5):1015–34.
Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32.
Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11(9):1107–17.
Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain J Neurol. 2010;133(Pt 1):93–104.
Jiang T, Yu J-T, Zhu X-C, Zhang Q-Q, Cao L, Wang H-F, et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology. 2014;85:121–30.
Du J, Liang Y, Xu F, Sun B, Wang Z. Trehalose rescues Alzheimer’s disease phenotypes in APP/PS1 transgenic mice. J Pharm Pharmacol. 2013;65(12):1753–6.
Son SM, Shin H-J, Byun J, Kook SY, Moon M, Chang YJ, et al. Metformin Facilitates Amyloid-β Generation by β- and γ-Secretases via Autophagy Activation. J Alzheimers Dis JAD. 2016;51(4):1197–208.
Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9(9):1308–20.
Li Y, Guo Y, Wang X, Yu X, Duan W, Hong K, et al. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience. 2015;298:12–25.
Zhang X, Chen S, Song L, Tang Y, Shen Y, Jia L, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2014;10(4):588–602.
Ma TC, Buescher JL, Oatis B, Funk JA, Nash AJ, Carrier RL, et al. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci Lett. 2007;411(2):98–103.
Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282(8):5641–52.
Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain J Neurol. 2012;135(Pt 7):2169–77.
Suresh SN, Chavalmane AK, Pillai M, Ammanathan V, Vidyadhara DJ, Yarreiphang H, et al. Modulation of Autophagy by a Small Molecule Inverse Agonist of ERRα Is Neuroprotective. Front Mol Neurosci. 2018;11:109.
Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet. 2010;19(11):2144–53.
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4(5):295–305.
Hirano K, Fujimaki M, Sasazawa Y, Yamaguchi A, Ishikawa K-I, Miyamoto K, et al. Neuroprotective effects of memantine via enhancement of autophagy. Biochem Biophys Res Commun. 2019;518(1):161–70.
Naslavsky N, Caplan S. The enigmatic endosome - sorting the ins and outs of endocytic trafficking. J Cell Sci. 2018;131(13):jcs216499.
Neefjes J, Jongsma MML, Berlin I. Stop or Go? Endosome Positioning in the Establishment of Compartment Architecture, Dynamics, and Function. Trends Cell Biol. 2017;27(8):580–94.
Wandinger-Ness A, Zerial M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb Perspect Biol. 2014;6(11):a022616.
Burd C, Cullen PJ. Retromer: a master conductor of endosome sorting. Cold Spring Harb Perspect Biol. 2014;6(2):a016774.
van der Sluijs P, Hull M, Webster P, Mâle P, Goud B, Mellman I. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell. 1992;70(5):729–40.
Gorvel JP, Chavrier P, Zerial M, Gruenberg J. rab5 controls early endosome fusion in vitro. Cell. 1991;64(5):915–25.
Babbey CM, Ahktar N, Wang E, Chen CC-H, Grant BD, Dunn KW. Rab10 Regulates Membrane Transport through Early Endosomes of Polarized Madin-Darby Canine Kidney Cells. Mol Biol Cell. 2006;17(7):3156–75.
Proikas-Cezanne T, Gaugel A, Frickey T, Nordheim A. Rab14 is part of the early endosomal clathrin-coated TGN microdomain. FEBS Lett. 2006;580(22):5241–6.
Simpson JC, Griffiths G, Wessling-Resnick M, Fransen JAM, Bennett H, Jones AT. A role for the small GTPase Rab21 in the early endocytic pathway. J Cell Sci. 2004;117(Pt 26):6297–311.
Mesa R, Salomón C, Roggero M, Stahl PD, Mayorga LS. Rab22a affects the morphology and function of the endocytic pathway. J Cell Sci. 2001;114(Pt 22):4041–9.
Jovic M, Sharma M, Rahajeng J, Caplan S. The early endosome: a busy sorting station for proteins at the crossroads. Histol Histopathol. 2010;25(1):99–112.
Lawe DC, Chawla A, Merithew E, Dumas J, Carrington W, Fogarty K, et al. Sequential roles for phosphatidylinositol 3-phosphate and Rab5 in tethering and fusion of early endosomes via their interaction with EEA1. J Biol Chem. 2002;277(10):8611–7.
Kim WT, Chang S, Daniell L, Cremona O, Di Paolo G, De Camilli P. Delayed reentry of recycling vesicles into the fusion-competent synaptic vesicle pool in synaptojanin 1 knockout mice. Proc Natl Acad Sci U S A. 2002;99(26):17143–8.
Perera RM, Zoncu R, Lucast L, De Camilli P, Toomre D. Two synaptojanin 1 isoforms are recruited to clathrin-coated pits at different stages. Proc Natl Acad Sci U S A. 2006;103(51):19332–7.
Fasano D, Parisi S, Pierantoni GM, De Rosa A, Picillo M, Amodio G, et al. Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 2018;9(3):385.
Hasegawa T, Yoshida S, Sugeno N, Kobayashi J, Aoki M. DnaJ/Hsp40 Family and Parkinson’s Disease. Front Neurosci. 2018;11:743 [cited 2019 Mar 4]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5767785/.
Köroğlu Ç, Baysal L, Cetinkaya M, Karasoy H, Tolun A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord. 2013;19(3):320–4.
Follett J, Fox JD, Gustavsson EK, Kadgien C, Munsie LN, Cao LP, et al. DNAJC13 p.Asn855Ser, implicated in familial parkinsonism, alters membrane dynamics of sorting nexin 1. Neurosci Lett. 2019;706:114–22.
Freeman CL, Hesketh G, Seaman MNJ. RME-8 coordinates the activity of the WASH complex with the function of the retromer SNX dimer to control endosomal tubulation. J Cell Sci. 2014;127(Pt 9):2053–70.
Connor-Robson N, Booth H, Martin JG, Gao B, Li K, Doig N, et al. An integrated transcriptomics and proteomics analysis reveals functional endocytic dysregulation caused by mutations in LRRK2. Neurobiol Dis. 2019;127:512–26.
Patel D, Xu C, Nagarajan S, Liu Z, Hemphill WO, Shi R, et al. Alpha-synuclein inhibits Snx3-retromer-mediated retrograde recycling of iron transporters in S. cerevisiae and C. elegans models of Parkinson’s disease. Hum Mol Genet. 2018;27(9):1514–32.
Pal A, Severin F, Lommer B, Shevchenko A, Zerial M. Huntingtin–HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J Cell Biol. 2006;172(4):605–18.
Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29(2):166–73.
Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29(2):160–5.
Otomo A, Hadano S, Okada T, Mizumura H, Kunita R, Nishijima H, et al. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum Mol Genet. 2003;12(14):1671–87.
Lai C, Xie C, Shim H, Chandran J, Howell BW, Cai H. Regulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsin. Mol Brain. 2009;2:23.
Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157(1):277–86.
Tansey KE, Cameron D, Hill MJ. Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 2018;10(1):14.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30.
Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49(9):1373–84.
Balaji K, Mooser C, Janson CM, Bliss JM, Hojjat H, Colicelli J. RIN1 orchestrates the activation of RAB5 GTPases and ABL tyrosine kinases to determine the fate of EGFR. J Cell Sci. 2012;125(Pt 23):5887–96.
Peric A, Annaert W. Early etiology of Alzheimer’s disease: tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol (Berl). 2015;129(3):363–81.
Wen L, Tang F-L, Hong Y, Luo S-W, Wang C-L, He W, et al. VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J Cell Biol. 2011;195(5):765–79.
Reitz C, Tokuhiro S, Clark LN, Conrad C, Vonsattel J-P, Hazrati L-N, et al. SORCS1 Alters Amyloid Precursor Protein Processing and Variants May Increase Alzheimer’s Disease Risk. Ann Neurol. 2011;69(1):47–64.
Lane RF, Steele JW, Cai D, Ehrlich ME, Attie AD, Gandy S. Protein Sorting Motifs in the Cytoplasmic Tail of SorCS1 Control Generation of Alzheimer’s Amyloid-β Peptide. J Neurosci Off J Soc Neurosci. 2013;33(16):7099–107.
Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry. 2007;68(4):613–8.
Li J, Kanekiyo T, Shinohara M, Zhang Y, LaDu MJ, Xu H, et al. Differential Regulation of Amyloid-β Endocytic Trafficking and Lysosomal Degradation by Apolipoprotein E Isoforms. J Biol Chem. 2012;287(53):44593–601.
Nuutinen T, Huuskonen J, Suuronen T, Ojala J, Miettinen R, Salminen A. Amyloid-beta 1–42 induced endocytosis and clusterin/apoJ protein accumulation in cultured human astrocytes. Neurochem Int. 2007;50(3):540–7.
Hopkins CR. Intracellular routing of transferrin and transferrin receptors in epidermoid carcinoma A431 cells. Cell. 1983;35(1):321–30.
Ullrich O, Reinsch S, Urbé S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135(4):913–24.
Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5(2):121–32.
Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10(9):597–608.
Kouranti I, Sachse M, Arouche N, Goud B, Echard A. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr Biol CB. 2006;16(17):1719–25.
Takahashi S, Kubo K, Waguri S, Yabashi A, Shin H-W, Katoh Y, et al. Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. J Cell Sci. 2012;125(17):4049–57.
Li X, Standley C, Sapp E, Valencia A, Qin Z-H, Kegel KB, et al. Mutant Huntingtin Impairs Vesicle Formation from Recycling Endosomes by Interfering with Rab11 Activity. Mol Cell Biol. 2009;29(22):6106–16.
Richards P, Didszun C, Campesan S, Simpson A, Horley B, Young KW, et al. Dendritic spine loss and neurodegeneration is rescued by Rab11 in models of Huntington’s disease. Cell Death Differ. 2011;18(2):191–200.
Steinert JR, Campesan S, Richards P, Kyriacou CP, Forsythe ID, Giorgini F. Rab11 rescues synaptic dysfunction and behavioural deficits in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2012;21(13):2912–22.
Roberts RC, Peden AA, Buss F, Bright NA, Latouche M, Reilly MM, et al. Mistargeting of SH3TC2 away from the recycling endosome causes Charcot-Marie-Tooth disease type 4C. Hum Mol Genet. 2010;19(6):1009–18.
Woodruff G, Reyna SM, Dunlap M, Van Der Kant R, Callender JA, Young JE, et al. Defective Transcytosis of APP and Lipoproteins in human iPSC-derived neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016;17(3):759–73.
Sannerud R, Esselens C, Ejsmont P, Mattera R, Rochin L, Tharkeshwar AK, et al. Restricted Location of PSEN2/γ-Secretase Determines Substrate Specificity and Generates an Intracellular Aβ Pool. Cell. 2016;166(1):193–208.
Lazo OM, Gonzalez A, Ascaño M, Kuruvilla R, Couve A, Bronfman FC. BDNF regulates Rab11-mediated recycling endosome dynamics to induce dendritic branching. J Neurosci Off J Soc Neurosci. 2013;33(14):6112–22.
Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305(5692):1972–5.
Schmidt O, Teis D. The ESCRT machinery. Curr Biol. 2012;22(4):R116–20.
Vanlandingham PA, Ceresa BP. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem. 2009;284(18):12110–24.
Priya A, Kalaidzidis IV, Kalaidzidis Y, Lambright D, Datta S. Molecular insights into Rab7-mediated endosomal recruitment of core retromer: deciphering the role of Vps26 and Vps35. Traffic Cph Den. 2015;16(1):68–84.
Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30(17):3481–500.
Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303–7.
Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A. 2014;111(7):2626–31.
Gómez-Suaga P, Rivero-Ríos P, Fdez E, Blanca Ramírez M, Ferrer I, Aiastui A, et al. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum Mol Genet. 2014;23(25):6779–96.
MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77(3):425–39.
Clayton EL, Mizielinska S, Edgar JR, Nielsen TT, Marshall S, Norona FE, et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol (Berl). 2015;130(4):511–23.
Zhang Y, Schmid B, Nikolaisen NK, Rasmussen MA, Aldana BI, Agger M, et al. Patient iPSC-Derived Neurons for Disease Modeling of Frontotemporal Dementia with Mutation in CHMP2B. Stem Cell Rep. 2017;8(3):648–58.
Platt FM. Emptying the stores: lysosomal diseases and therapeutic strategies. Nat Rev Drug Discov. 2018;17(2):133–50.
Moreno-García A, Kun A, Calero O, Medina M, Calero M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front Neurosci. 2018;12:464.
Solovyeva VV, Shaimardanova AA, Chulpanova DS, Kitaeva KV, Chakrabarti L, Rizvanov AA. New Approaches to Tay-Sachs Disease Therapy. Front Physiol. 2018;9:1663.
Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, Barranger JA, et al. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N Engl J Med. 1987;316(10):570–5.
Ron I, Horowitz M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet. 2005;14(16):2387–98.
Schmitz M, Alfalah M, Aerts JMFG, Naim HY, Zimmer K-P. Impaired trafficking of mutants of lysosomal glucocerebrosidase in Gaucher’s disease. Int J Biochem Cell Biol. 2005;37(11):2310–20.
Braunstein H, Maor G, Chicco G, Filocamo M, Zimran A, Horowitz M. UPR activation and CHOP mediated induction of GBA1 transcription in Gaucher disease. Blood Cells Mol Dis. 2018;68:21–9.
Maor G, Rencus-Lazar S, Filocamo M, Steller H, Segal D, Horowitz M. Unfolded protein response in Gaucher disease: from human to Drosophila. Orphanet J Rare Dis. 2013;8:140.
Wei H, Kim S-J, Zhang Z, Tsai P-C, Wisniewski KE, Mukherjee AB. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum Mol Genet. 2008;17(4):469–77.
Klein AD, Mazzulli JR. Is Parkinson’s disease a lysosomal disorder? Brain J Neurol. 2018;141(8):2255–62.
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–61.
Perrin RJ, Woods WS, Clayton DF, George JM. Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J Biol Chem. 2000;275(44):34393–8.
Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, et al. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc Natl Acad Sci U S A. 2018;115(4):798–803.
Rocha EM, Smith GA, Park E, Cao H, Graham A-R, Brown E, et al. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain α-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid Redox Signal. 2015;23(6):550–64.
Mazzulli JR, Xu Y-H, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37–52.
Lee H-J, Khoshaghideh F, Patel S, Lee S-J. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci Off J Soc Neurosci. 2004;24(8):1888–96.
Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem. 2010;285(18):13621–9.
Bendikov-Bar I, Rapaport D, Larisch S, Horowitz M. Parkin-mediated ubiquitination of mutant glucocerebrosidase leads to competition with its substrates PARIS and ARTS. Orphanet J Rare Dis. 2014;9:86.
Ron I, Rapaport D, Horowitz M. Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease. Hum Mol Genet. 2010;19(19):3771–81.
Li H, Ham A, Ma TC, Kuo SH, Kanter E, Kim D, et al. Mitochondrial dysfunction and mitophagy defect triggered by heterozygous GBA mutations. Autophagy. 2019;15(1):113–30.
Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, et al. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 2013;17(6):941–53.
Rocha EM, Smith GA, Park E, Cao H, Brown E, Hayes MA, et al. Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis. 2015;82:495–503.
Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A. 2017;114(10):2699–704.
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible Conformational Conversion of α-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron. 2018;97(1):92–107.e10.
Davies SE, Hallett PJ, Moens T, Smith G, Mangano E, Kim HT, et al. Enhanced ubiquitin-dependent degradation by Nedd4 protects against α-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol Dis. 2014;64:79–87.
Butler JD, Vanier MT, Pentchev PG. Niemann-Pick C disease: cystine and lipids accumulate in the murine model of this lysosomal cholesterol lipidosis. Biochem Biophys Res Commun. 1993;196(1):154–9.
Higashi Y, Murayama S, Pentchev PG, Suzuki K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol (Berl). 1993;85(2):175–84.
Pentchev PG, Boothe AD, Kruth HS, Weintroub H, Stivers J, Brady RO. A genetic storage disorder in BALB/C mice with a metabolic block in esterification of exogenous cholesterol. J Biol Chem. 1984;259(9):5784–91.
te Vruchte D, Lloyd-Evans E, Veldman RJ, Neville DCA, Dwek RA, Platt FM, et al. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J Biol Chem. 2004;279(25):26167–75.
Vance JE. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 2006;580(23):5518–24.
Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277(5323):228–31.
Verot L, Chikh K, Freydière E, Honoré R, Vanier MT, Millat G. Niemann-Pick C disease: functional characterization of three NPC2 mutations and clinical and molecular update on patients with NPC2. Clin Genet. 2007;71(4):320–30.
Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14(11):1247–55.
Auer IA, Schmidt ML, Lee VM, Curry B, Suzuki K, Shin RW, et al. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer’s disease. Acta Neuropathol (Berl). 1995;90(6):547–51.
Jongsma MLM, Berlin I, Wijdeven RHM, Janssen L, Janssen GMC, Garstka MA, et al. An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell. 2016;166(1):152–66.
Cabukusta B, Neefjes J. Mechanisms of lysosomal positioning and movement. Traffic Cph Den. 2018;19(10):761–9.
Hollenbeck PJ, Swanson JA. Radial extension of macrophage tubular lysosomes supported by kinesin. Nature. 1990;346(6287):864–6.
Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol. 1998;141(1):51–9.
Overly CC, Lee KD, Berthiaume E, Hollenbeck PJ. Quantitative measurement of intraorganelle pH in the endosomal-lysosomal pathway in neurons by using ratiometric imaging with pyranine. Proc Natl Acad Sci U S A. 1995;92(8):3156–60.
Overly CC, Hollenbeck PJ. Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J Neurosci Off J Soc Neurosci. 1996;16(19):6056–64.
Lie PPY, Nixon RA. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis. 2019;122:94–105.
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307(5713):1282–8.
Su JH, Cummings BJ, Cotman CW. Identification and distribution of axonal dystrophic neurites in Alzheimer’s disease. Brain Res. 1993;625(2):228–37.
Cataldo AM, Hamilton DJ, Nixon RA. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res. 1994;640(1–2):68–80.
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64(2):113–22.
Terry RD, Gonatas NK, Weiss M. Ultrastructural studies in Alzheimer’s presenile dementia. Am J Pathol. 1964;44:269–97.
Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci U S A. 2015;112(28):E3699–708.
Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, et al. Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell. 2000;103(4):583–94.
Horiuchi D, Barkus RV, Pilling AD, Gassman A, Saxton WM. APLIP1, a kinesin binding JIP-1/JNK scaffold protein, influences the axonal transport of both vesicles and mitochondria in Drosophila. Curr Biol CB. 2005;15(23):2137–41.
Gowrishankar S, Wu Y, Ferguson SM. Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J Cell Biol. 2017;216(10):3291–305.
Cataldo AM, Peterhoff CM, Schmidt SD, Terio NB, Duff K, Beard M, et al. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J Neuropathol Exp Neurol. 2004;63(8):821–30.
Lee J-H, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015;12(9):1430–44.
Erie C, Sacino M, Houle L, Lu ML, Wei J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur J Neurosci. 2015;42(3):1941–51.
Ishikawa K-I, Saiki S, Furuya N, Imamichi Y, Tsuboi Y, Hattori N. p150glued deficiency impairs effective fusion between autophagosomes and lysosomes due to their redistribution to the cell periphery. Neurosci Lett. 2019;690:181–7.
Shires SE, Kitsis RN, Gustafsson ÅB. Beyond Mitophagy: The Diversity and Complexity of Parkin Function. Circ Res. 2017;120(8):1234–6.
Voigt A, Berlemann LA, Winklhofer KF. The mitochondrial kinase PINK1: functions beyond mitophagy. J Neurochem. 2016;139(Suppl 1):232–9.
Lee JJ, Sanchez-Martinez A, Zarate AM, Benincá C, Mayor U, Clague MJ, et al. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol. 2018;217(5):1613–22.
McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, et al. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018;27(2):439–449.e5.
Patel D, Witt SN. Sorting Out the Role of α-Synuclein in Retromer-Mediated Endosomal Protein Sorting. J Exp Neurosci. 2018;12:1179069518796215.
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285(17):13107–20.
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PloS One. 2010;5(4):e9979.
Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16(7):487–511.
Acknowledgements
N/A
Funding
This work was supported by the UK Dementia Research Institute which receives its funding from DRI Ltd., funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. GS is funded by an MRC Momentum Award (MC_PC_16030/1) and OP is funded by an MRC Momentum Award (MC_PC_16030/2).
Author information
Authors and Affiliations
Contributions
BM, DM, GS & OP wrote the manuscript. GS & OP edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
There are no ethical approval requirements for this review.
Consent for publication
All researchers consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Malik, B.R., Maddison, D.C., Smith, G.A. et al. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol Brain 12, 100 (2019). https://doi.org/10.1186/s13041-019-0504-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-019-0504-x