
Abstract
Background
Chronic myelogenous leukemia (CML) is a pluripotent hematopoietic stem cell disorder caused by the fusion of the BCR and ABL1 genes. Quantitative RT-PCR (qRT-PCR) is a routinely performed screening technique to identify BCR-ABL1 fusion genes, but a limitation of this method is its inability to recognize novel fusions that have not been previously characterized. Next-generation sequencing (NGS) is an effective and sensitive detection method for the determination of novel BCR-ABL1 fusion genes as well as previously characterized ones. The oncoprotein tyrosine kinase BCR-ABL1 is a constitutively active kinase involved in the activation of a number of signaling pathways, and it has been the therapeutic target for tyrosine kinase inhibitors (TKIs) such as imatinib. Reports have presented opposing viewpoints about the effect of the disrupted Src homology 3 (SH3) domain on TKI efficacy.
Findings
We here report that using NGS we identified a novel BCR-ABL1 fusion gene with breakpoints in the BCR intron 14 and the ABL1 intron 2, leading to partial deletion of its SH3 domain. In the present case, the patient received targeted therapy with the TKI imatinib at 400 mg/day and no adverse reaction was reported. The patient eventually entered remission with decreased proliferation of karyocytes and granulocytes. We also identified mutations in genes, including TP53, FLT3, ASXL1, SETBP1, CEBPA and CBL, that seemed to have an influence on the outcome of TKI therapy targeting the BCR-ABL1 protein.
Conclusions
Together with previously reported results, it is clear that the genetic heterogeneity of CML patients significantly affects the presentation of the disease and its progression and therefore should inform the design of the therapeutic strategy.
Similar content being viewed by others

Background
CML, a clonal hematopoietic stem cell disorder, is characterized by the fusion of the Abelson gene (ABL1) on chromosome 9q34 with the breakpoint cluster region (BCR) gene on chromosome 22q11.2, which is known as the Philadelphia translocation [1]. This molecular rearrangement results in formation of the BCR-ABL1 oncogene. Its translation product, oncoprotein BCR-ABL1, shows enhanced tyrosine kinase activity and plays a critical role in the transformation of hematopoietic stem cells through activation of a number of signaling pathways [2, 3]. According to the locations of the breakpoints in the BCR and ABL1 genes, fusion genes are divided into many known species, such as e13-a2, e14-a2, e19-a2 and e1-a2 [4, 5], as well as other uncharacterized fusion genes. Routine screening procedures, such as multiplex qRT-PCR assays, are designed to detect previously characterized BCR-ABL1 fusion transcripts and, thus, have limited ability to detect novel ones. That problem can be resolved with the application of NGS technology since it can identify these novel mutations undetectable by routine screening procedures [6,7,8], as well as those previously characterized. Thus NGS plays an important role in genetic diagnostics and is helpful for a better understanding of the cancer genome.
An article [9] entitled “A novel BCR-ABL1 fusion gene identified by next-generation sequencing in chronic myeloid leukemia” has been published recently. Here, we report that we have also identified this novel BCR-ABL1 fusion gene in another patient using NGS technology. We also report that this patient carries a different set of genetic mutations than those that impacted the outcome of TKI imatinib treatment in the Lyu et al. report [9]. Comparison of these studies demonstrates that genetic heterogeneity can be a key influencing factor in the therapeutic resolution of CML.
Results
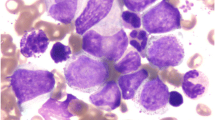
Our patient is a 62-year-old male who presented at our hospital in February 2016 with intermittent nasal bleeding that had exceed 1 month in duration. After hospitalization, we determined the patient had a significantly elevated platelet level that increased the risk of bleeding and thrombosis to a life-threatening level. No superficial lymph nodes were detected anywhere in the body. The patient was diagnosed with CML through blood and bone marrow examinations. Peripheral blood smear analysis indicated elevated levels of total white blood cells (WBCs, 55.24 g/L), neutrophils (34.58 g/L), thrombocytes (2597 g/L), and a normal level of hemoglobin (103 g/L). Bone marrow aspiration analysis revealed the active proliferation of bone marrow nucleated cells (BMNCs) and elevated proportions of eosinophils and basophils. Granulocytes accounted for 88% of WBCs due to the excessive proliferation of band granulocytes and segmented granulocytes (Table 1 and Fig. 1a). We also observed a decreased level of lymphocytes with normal morphology.

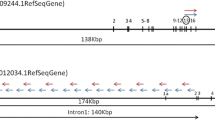
Summary of FISH and molecular studies. a Image of bone marrow aspiration (400x) showing hypercellularity with elevated level of myeloblasts, eosinophils, and basophils. b FISH analysis. Separated green and red signals indicate probe-targeted sequences located on different chromosomes in a normal nucleus. Yellowish signal formed from the colocalization of green and red fluorescent signals indicates the fusion of BCR and ABL1 genes. c The breaking point (or fusion junction) and flanking sequences from BCR Intron 14 and ABL1 intron 2. d BCR-ABL1 cDNA sequence around the fusion junction and related chromatogram are shown. The junctions are indicated with arrows. e Image of bone marrow aspiration after imatinib targeted therapy. f FISH analysis after imatinib targeted therapy
Bone marrow karyotype analysis showed a phenotype of 46,XY,t(9;22)(q34;q11.2) (Data not shown). Fluorescence in situ hybridization (FISH) analysis was then used to detect the fusion between BCR and ABL1 genes, which were demonstrated as dots of yellowish fluorescent signals formed from the colocalization of the green (BCR) and red (ABL1) fluorescent signals. We found at least one yellowish fluorescent dot per cell in 44% of cells, representing the tumor cells with BCR-ABL1 fusions (Fig. 1b). However, fluorescent qRT-PCR failed to detect the previously characterized BCR-ABL1 fusion transcripts p190 (e1-a2), p210 (e13-a2 and e14-a2) and p230 (e19-a2). To further clarify the existence of a BCR-ABL1 fusion in this patient, we conducted whole genome sequencing (WGS) analysis on a NGS platform. We detected a BCR-ABL1 fusion gene with novel breakpoints in BCR intron 14 and ABL1 intron 2 (Fig. 1c), confirming the fusion of BCR exon 14 (e14) and ABL1 exon 3 (a3). The corresponding BCR-ABL1 hybrid mRNA was eventually identified by RT-PCR with a pair of custom primers targeting e14 and a3, followed by Sanger sequencing (Fig. 1d). After 7 days of imatinib treatment, the disease was under control with an improved platelet count and the patient discharged. As an outpatient, he then continued treatment with imatinib at 400 mg/day, accompanied by sodium bicarbonate tablets at 3.0 g/day and allopurinol tablets at 0.3 g/day, with regular follow-up visits. After 4 months, we measured a significant decrease in bone marrow karyocyte proliferation with the reduced number of granulocytes now accounting for 67% of WBCs in this CML patient (Table 1, Fig. 1e). We saw a great improvement in disease progression – the patient achieved both hematologic and molecular remission (Fig. 1e–f).
Discussion
Philadelphia translocation, formed by the junction of BCR and ABL1 genes, has been proven to be involved in the carcinogenesis of CML. In this work, we have identified a novel BCR-ABL1 fusion gene by NGS, together with other co-existing mutations, indicating that genetic heterogeneity is associated with the response to imatinib treatment for this CML case and may require optimization of the personalized therapeutic schedule targeting CML.
The introduction of small molecule TKIs has contributed to marked improvements in the therapeutic outcomes of CML by forcefully blocking phosphorylation by the BCR-ABL1 oncoprotein and inhibiting its cell signal transduction activity [10,11,12]. Imatinib is a tyrosine-kinase inhibitor used in the treatment of multiple cancers and was the first TKI to receive approval by the Food and Drug Administration for the treatment of patients with Philadelphia chromosome-positive (Ph+) CML [13, 14]. After detecting the novel BCR-ABL1 fusion gene in our patient, imatinib was used at 400 mg/day as a targeted treatment. It has been reported that the SH3-SH2 (Src homology 3-Src homology 2) domain in the ABL protein plays a crucial role in regulating its tyrosine kinase activity [15]. The function of partial deletions of the SH3 domain, which is encoded by ABL1 exons 2 and 3, remains controversial. The report from Lyu et al. showed that their patient was intolerant to a normal dosage of imatinib, indicating an interaction between this unusual therapy outcome and the incomplete SH3 domain caused by the deletion of ABL1 exon 2 [9]. Our case differed from that of Lyu et al. in that our patient with the e14-a3 BCR-ABL1 fusion gene was not refractory to or intolerant of imatinib treatment. The patient achieved both hematologic and molecular remission after 4 months of imatinib treatment. A previous study [16] reported that the STAT5 signaling pathway induced by the ABL1 SH3 domain plays a critical role in the anti-apoptotic activity and cell cycle progression involved in BCR-ABL1 leukemogenesis. Thus the truncation of the SH3 domain caused by deletion of ABL exon 2 may result in the reduction of leukemogenesis. CML patients with an e13a3 fusion gene were found to have a good response to imatinib, and could achieve complete hematologic and cytogenetic remission [17].
Similar to previous results, we also detected nonsynonymous mutations in leukemic genes such as TP53 (c.C215G: p.P72R) and FLT3 (c.C680T: p.T227M) through NGS. Furthermore, we also found mutations in ASXL1 (c.T2444C:p.L815P), SETBP1 (c.G664A: p.A222T) (c.G3301A: p.V1101I) (c.C3388A: p.P1130T), CEBPA (c.570_571insCACCCG: p.H191delinsHPH) and CBL (c.C1858T:p.L620F) that co-existed with the BCR-ABL1 fusion in our patient. ASXL1 mutations are common in myeloid neoplasms, including myelodysplastic syndrome (MDS) [18, 19], chronic myelomonocytic leukemia (CMML) [20, 21], primary myelofibrosis [18, 22], and acute myeloid leukemia (AML) [19, 23]. SETBP1 mutations have been identified in atypical chronic myeloid leukemia (aCML), which is a rare disorder of hematopoietic stem cells and shares clinical and laboratory features with CML but lacks the BCR-ABL fusion gene [24]. Other strongly linked hematological malignancies, such as chronic neutrophilic leukemia (CNL), CMML, unclassified MDS, myeloproliferative neoplasms (MPNs), and secondary acute myelocytic leukemia (AML) evolving from MDS [25,26,27,28,29], are also related to SETBP1. Despite the fact that mutations in both ASXL1 and SETBP1 are generally associated with an adverse prognosis [20, 21, 26, 30], our patient’s symptoms seemed not to be related to his mutations in these genes. CCAAT enhancer binding protein α (C/EBPα), a general inhibitor of cell proliferation and a tumor suppressor [31] plays a pivotal role in early granulocyte development. C/EBPα is one of the crucial transcription factors for myeloid cell development and has been found to be involved in hematopoietic differentiation. The mutation of its coding gene CEBPA results in dysregulation of transcription, translation or post-translational modifications. These disruptions cause disorders of differentiation and over proliferation of immature hematopoietic cells [32, 33]. In our patient, the outcome of imatinib therapy for CML suggests that his disease was not significantly affected by what we can consider to be ancillary mutations. Considering the results reported by Lyu et al. [9], it can be concluded that the variety of genetic mutations among individual CML patients may lead to different treatment outcomes of TKI therapies targeted for BCR-ABL1. More research is needed to illuminate the interactions between these uncommon mutations and the variety of BCR-ABL1 fusion genes in CML.
Conclusions
We report this case to demonstrate that by NGS we have detected the same BCR-ABL1 fusion that disrupts the SH3 domain, as Lyu et al. [9]. Meanwhile, we also found numerous other mutations in genes such as TP53, FLT3, ASXL1, SETBP1, CEBPA and CBL, suggesting that CML may be more highly heterogeneous than previously appreciated. Our findings show that such genetic heterogeneity may significantly affect treatment outcomes and should therefore inform the therapeutic strategy. Since these conclusions remain speculative, more studies should be performed to characterize the various interactions between BCR-ABL1 gene rearrangements and mutations in other oncogenes.
Methods
Detection of BCR-ABL fusion by FISH analysis
To validate the presence of BCR-ABL1 fusion, we performed FISH analysis with dual color, single fusion probes on the patient’s bone marrow aspiration sample using the BCR-ABL FISH Probe kit (Jinpujia Medical, Beijing, China) according to the manufacturer’s instructions. DNA probes targeting the BCR (chromosome 22q11.2) and ABL1 (chromosome 9q34) genes were labeled with green and red fluorescent dye, respectively. In normal cells, two green signals and two red signals were separated, representing that two probe-targeted sequences were located on different chromosomes. The presence of yellowish signal dots indicated the fusion events that resulted from the colocalization of BCR-targeting green fluorescent signals and ABL1-targeting red signals. The percent of cells with BCR-ABL1 fusions was counted and the cutoff value for the BCR-ABL1 fusion was set at 3% in our hospital.
Detection of BCR-ABL1 gene rearrangement by one-step RT-PCR
Routine fluorescence one-step RT-PCR was carried out to detect BCR-ABL1 fusion transcripts. RNA from patient bone marrow aspiration samples was extracted using an RNeasy Kit (Qiagen, CA, USA), following the protocol provided by the manufacturer. RNA was purified by DNase I (Ambion, Applied Biosystems, TX, USA) digestion and was then subjected to one-step RT-PCR by a Leukemia Related Fusion Gene Detection Kit for BCR-ABL p210, p190, or p230 (Yuanqi Bio-Pharmaceutical, Shanghai, China). In each PCR process, a total volume of 25 μl reaction solution contains 3 μl template RNA, 2 μl multiplex Enzyme and 20 μl multiplex RT-PCR buffer. Amplification and detection were performed on a 7300 Real Time PCR System (ABI, USA). PCR procedure parameters were as follows: reverse transcription at 42 °C for 30 min, inactivation at 94 °C for 5 min, followed by 40 cycles of fluorescence detection at 94 °C for 15 s, and annealing at 60 °C for 60 s.
Whole genome sequencing in a NGS platform
A genomic DNA (gDNA) library was constructed for sequencing following protocols of the TruSeq Nano DNA Library Preparation Kit (Illumina, San Diego, CA). Adaptors were ligated to library fragments sheared by Covaris (Covaris, Woburn, MA, USA) and were then subjected to PCR amplification. The quantitation and abundance determination of PCR amplicons were performed on Qubit 3.0 Fluorometer (Life Technologies, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, USA), respectively. WGS was performed on HiSeq X (Illumina, San Diego, CA), with the use of Illumina bcl2fastq software version 2.15 for base calling analysis.
Abbreviations
- AML:
-
Acute myeloid leukemia
- C/EBPα:
-
CCAAT enhancer binding protein α
- CML:
-
Chronic myelogenous leukemia
- CMML:
-
Chronic myelomonocytic leukemia
- CNL:
-
Chronic neutrophilic leukemia
- FISH:
-
Fluorescence in situ hybridization
- MDS:
-
Myelodysplastic syndrome
- MPNs:
-
Myeloproliferative neoplasms
- NGS:
-
Next-generation sequencing
- SH3:
-
Src homology 3
- TKI:
-
Tyrosine kinase inhibitor
- WBCs:
-
White blood cells
- WGS:
-
Whole genome sequencing
References
Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2016 update on diagnosis, therapy, and monitoring. Am J Hematol. 2016;91(2):252–65.
Sattler M, Griffin JD. Molecular mechanisms of transformation by the BCR-ABL oncogene. Semin Hematol. 2003;40(2 Suppl 2):4–10.
Melo JV, Deininger MW. Biology of chronic myelogenous leukemia--signaling pathways of initiation and transformation. Hematol Oncol Clin North Am. 2004;18(3):545–68. vii-viii.
Mauro MJ, Druker BJ. STI571: targeting BCR-ABL as therapy for CML. Oncologist. 2001;6(3):233–8.
Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88(7):2375–84.
Nakagawa H, Wardell CP, Furuta M, Taniguchi H, Fujimoto A. Cancer whole-genome sequencing: present and future. Oncogene. 2015;34(49):5943–50.
Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46.
Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–55.
Lyu X, Yang J, Wang X, Hu J, Liu B, Zhao Y, et al. A novel BCR-ABL1 fusion gene identified by next-generation sequencing in chronic myeloid leukemia. Mol Cytogenet. 2016;9:47.
Huang X, Cortes J, Kantarjian H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer. 2012;118(12):3123–7.
O'Brien S, Berman E, Moore JO, Pinilla-Ibarz J, Radich JP, Shami PJ, et al. NCCN Task Force report: tyrosine kinase inhibitor therapy selection in the management of patients with chronic myelogenous leukemia. J Natl Compr Canc Netw. 2011;9 Suppl 2:S1–25.
Druker BJ, Lydon NB. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest. 2000;105(1):3–7.
Mace ML, Dahl J, Jabbour EJ. Which tyrosine-kinase inhibitor to use first in chronic phase chronic myelogenous leukemia? Expert Opin Pharmacother. 2015;16(7):999–1007.
Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am J Hematol. 2014;89(5):547–56.
Colicelli J. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signal. 2010;3(139):re6.
Paz YMC, Arevalo M, Leone PE. B3/A3 rearrangement in a patient with chronic myeloid leukemia. Leuk Lymphoma. 2003;44(2):375–6.
Snyder DS, McMahon R, Cohen SR, Slovak ML. Chronic myeloid leukemia with an e13a3 BCR-ABL fusion: benign course responsive to imatinib with an RT-PCR advisory. Am J Hematol. 2004;75(2):92–5.
Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A, et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia. 2011;25(7):1200–2.
Boultwood J, Perry J, Pellagatti A, Fernandez-Mercado M, Fernandez-Santamaria C, Calasanz MJ, et al. Frequent mutation of the polycomb-associated gene ASXL1 in the myelodysplastic syndromes and in acute myeloid leukemia. Leukemia. 2010;24(5):1062–5.
Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N, et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145(6):788–800.
Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428–36.
Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adelaide J, Rey J, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009;23(11):2183–6.
Carbuccia N, Trouplin V, Gelsi-Boyer V, Murati A, Rocquain J, Adelaide J, et al. Mutual exclusion of ASXL1 and NPM1 mutations in a series of acute myeloid leukemias. Leukemia. 2010;24(2):469–73.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–51.
Damm F, Itzykson R, Kosmider O, Droin N, Renneville A, Chesnais V, et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27(6):1401–3.
Laborde RR, Patnaik MM, Lasho TL, Finke CM, Hanson CA, Knudson RA, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013;27(10):2100–2.
Thol F, Suchanek KJ, Koenecke C, Stadler M, Platzbecker U, Thiede C, et al. SETBP1 mutation analysis in 944 patients with MDS and AML. Leukemia. 2013;27(10):2072–5.
Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C, et al. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia. 2013;27(9):1852–60.
Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45(8):942–6.
Wang L, Du F, Zhang HM, Wang HX. Evaluation of a father and son with atypical chronic myeloid leukemia with SETBP1 mutations and a review of the literature. Braz J Med Biol Res. 2015;48(7):583–7.
Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3(2):89–101.
Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci U S A. 1997;94(2):569–74.
Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21(6):853–63.
Acknowledgements
The authors acknowledge that Shanghai Yuanqi Bio-Pharmaceutical Co., Ltd. provided valuable assistance in technical support.
Funding
The study was funded and supported by the National Natural Science Foundation of China (Grant No. 81570116 and 81300414).
Availability of data and material
The datasets supporting the conclusions of this article are included within the article. More details are available upon request.
Authors' contributions
FZ and HM collected the patient history and statistics, analyzed data, and were major contributors in writing the manuscript. RJ and YH collected the patient history and managed the clinical trials. HM managed the clinical trials, formulated the research program and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Ethics approval and consent to participate
This study was approved by the ethics committee of Union Hospital at the Tongji Medical College, Huazhong University of Science and Technology.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, F., Jin, R., Hu, Y. et al. A novel BCR-ABL1 fusion gene with genetic heterogeneity indicates a good prognosis in a chronic myeloid leukemia case. Mol Cytogenet 10, 19 (2017). https://doi.org/10.1186/s13039-017-0322-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-017-0322-8