
Abstract
Background
Terminal deletion of 6q27 produces a rare syndrome associated with unexplained mental retardation, hypotonia, epilepsy, and multiple malformations. Structural brain malformations are consistently observed, including agenesis of the corpus callosum, hydrocephalus, periventricular nodular heterotopia, polymicrogyria, and cerebellar malformations. Here we report a fetal risk assessment of a 27-year-old woman with mental retardation, hypotonia and dysmorphic features at 17 weeks of pregnancy.
Results
Cytogenetic analyses revealed an addition at chromosome 6qter in the mother. Haploinsufficiency of 6q27 to 6qter (1.3 Mb) and trisomy of the entire short arm of chromosome 18 (15.2 Mb) were found using a single nucleotide polymorphism based array. Results were confirmed by molecular cytogenetics and multiplex ligation-dependent probe amplification. The karyotype of the mother was 46,XX dic(6;18)(6pter → 6q27::18p10 → 18pter).arr [hg19]6q27(169,591,548-170,898,549) × 1,18p11.3p10(12,842-15,375,878) × 3.ish dic(6;18)(q27;p10)(RP11-614P3-,RP11-1035E2+,D18Z1+). Deletion of 6q27 was associated with the structural brain malformations, whereas trisomy of 18p had minor clinical effects. The unbalanced rearrangement of chromosome 6 and chromosome 18 was de novo and was not inherited by the developing fetus.
Conclusions
A rare rearrangement between 6q27 and 18p was identified, which led to a de novo 1.3 Mb deletion of 6q27 and a 15.2 Mb duplication of 18p in an adult with mental retardation, hypotonia, epilepsy, and multiple malformations.
Similar content being viewed by others


Background
Terminal deletion of 6q27, ranging from 0.4 Mb to 10.8 Mb, produces a rare syndrome with variable clinical features, including mental retardation, hypotonia, epilepsy, and multiple malformations [1-5]. With this syndrome, structural brain malformations are observed consistently, including agenesis of the corpus callosum (ACC), hydrocephalus, periventricular nodular heterotopia (PNH), polymicrogyria, and cerebellar malformations. Previous studies have determined that the critical deletion region for brain malformation is the terminal 1.7 Mb, which harbors four critical genes THBS2, PHF10, DLL1, and C6orf70 [4]. Trisomy18p, on the other hand, is tolerated in humans with mild clinical signs and symptoms. Trisomy 18p can result from unbalanced segmentation, balanced translocation, direct tandem duplication, or sSMC [6-7].
Here, we report a smallest 6q27 deletion with trisomy18p in a 27-year old patient at 17 weeks gestation. The patient presented with severe mental retardation (MR), hypotonia, and other malformations (Table 1). The goals of this report was to elucidate the genotype/phenotype correlation and test the hypothesis that the deletion results from unbalanced chromosome translocation and inheritance.
Case presentation
The patient was the product of the first pregnancy of non-consanguineous parents. Both parents and younger brother were healthy, and the family history lacked evidence of mental retardation (MR) and hypotonia. The patient was delivered at 41 weeks gestation. During the entire pregnancy, the mother of the patient felt sick and experienced emesis. Antiabortifacients were taken from 18 weeks of gestation until birth. The patient presented intra-uterine growth retardation; birth weight was approximately 1,500 g (>3SD), and birth length was 40 cm (>2SD); she also exhibited a weak cry and suckling. No apparent congenital abnormalities were noticed, although global developmental delays were observed.
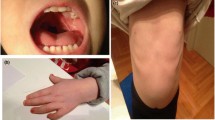
At age two, the patient had few single words and walked with an unstable gait. At age 27, evaluation revealed an IQ score of 46, hypotonia, prominent forehead, hypertelorism, long philtrum and, slow nystagmus, ataxic gait, joint laxity, bilateral clinodactyly, talipes cavus, scoliosis, pelvic obliquity, genu valgum on right, and femur length discrepancy of 1 cm (Figure 1A).

Clinical and iconography description of the patient. A) Present patients appearance and facial characteristics. B) Brain MRI images show brain structural abnormity: (a) T1-weighed sagittal section through the midline, showing corpus callosum hypoplasia; (b) T2-weighed axial section showing colpocephaly; (c) T2-weighed axial section showing asymmetric lateral ventricles; (d) T2-weighed axial section showing choroid fissure cysts on the left. C) Radiographic analysis show skeletal structure abnormity: (a) Thoracic vertebra showed T8-T10 scoliosis rightward; (b) pelvic obliquity.
Brain magnetic resonance imaging (MRI) showed the presence of ACC, colpocephaly, asymmetric lateral ventricles, and choroid fissure cysts on the left (Figure 1B). X-ray showed T8-T10 scoliosis and pelvic obliquity (Figure 1C).
Results
HumanCytoSNP-12 BeadChip for CNVs and SNP analyses
SNP-Array analysis of the present patient revealed a 1.3 Mb de novo deletion from 6q27 to 6qter and a 15.2 Mb de novo duplication including the entire short arm of chromosome 18. The proximal breakpoint in 6q27 mapped to (hg19:169,591,548 to 170,898,549 bp) (Figure 2) and 18p mapped to (hg19:12,842 to 15,375,878 bp) (Figure 3B).

Size, extent, and genomic content of deletions including 6q27 in cases comparable to the present one. All patients had structural brain abnormalities. Using this information for mapping of a critical region of brain malformations revealed C6orf70, PHF10, DLL1, and TBP as putative candidate genes for structural brain malformations.

Cytogenetical and molecular genetical description of the fetus. A) Cytogenetic analysis revealed a derivative chromosome 6 (blue arrow). B) SNP-Array shows deletion of 1.3 Mb at 6q27 to 6qter and a duplication of 15.2 Mb in the short arm of chromosome 18 (yellow arrow). C) Fluorescence in situ hybridization (FISH) 1) using BAC-probes RP11-614P3 (=6q27-Red), RP11-196G15 (=6p22.3-Orange), RP11-1035E2 (=18p11.32-Green) demonstrated the deletion of 6q27 and the duplication of 18p11.32; 2) using a centromeric probe D18Z1 (=cep18-Green) gave three signal and indicated that the derivative chromosome 6 is dicentric.
Cytogenetic and fluorescence in situ hybridization (FISH) analyses
Chromosome karyotype analyses showed 46,XX,add(6)(q27) (Figure 3A). Her parental and fetal karyotypes were normal. FISH confirmed the deletion of 6q27 and duplication of 18p including parts of the centromere (Figure 3C).
Multiplex ligation-dependent probe amplification (MLPA) and real-time qPCR analyses
MLPA P036-E2 was used to confirm the deletion of 6q27 and duplication of 18p (Figure 4A). Meanwhile, haploinsufficiency of the C6orf70 gene, related to the clinical phenotype of 6q27 deletion syndrome, was confirmed by qPCR (Figure 4B). From these data, the karyotype of the patient was revised to 46,XX,dic(6;18)(6pter → 6q27::18p10 → 18pter).arr [hg19]6q27(169,591,548-170,898,549) × 1,18p13p10(12,842-15,375,878) × 3.ish dic(6;18)(q27;p10)(RP11-614P3-,RP11-1036E2+,D18Z1+).The risk of recurrence for the further pregnancy was 50%.

MLPA and QF-PCR results. A) MLPA results with probe P036-E2. Probes targeting 18p11.32 (blue) were increased and the signals for 6q27 (red) were decreased, indicating unbalanced translocation with deletion of 6q27 and duplication of 18p; B) QF-PCR results with SYBR for gene C6orf70. PCR primer targeted exon 2,exon 12 and exon 18 of C6orf70 and showed the relative quantification ratios were 0.5741, 0.4519 and 0.5163, respectively, indicating the gene is heterozygous deletion.
Discussion
Here we report a patient with MR in the 17th week of pregnancy. Standard cytogenetic analyses revealed additional chromosomal material at the end of chromosome 6. SNP-Array analyses detected a 1.3 Mb deletion of 6q27-qter and a 15.2 Mb duplication of the entire short arm of chromosome 18. Result from the SNP-Array were confirmed by FISH and MLPA.
Deletion of 6q27, ranging from 0.4 Mb to 10.8 Mb, can result from unbalanced translocation or isolated deletion and produce a rare syndrome [1-5]. Partial terminal monosomy of 6q27 produces brain structural abnormalities, dysmorphic features, developmental delay, epilepsy, learning difficulties, and hypotonia. Patients with distal 6q27 deletions have been described, revealing variable brain phenotypes including agenesis of the corpus callosum (ACC), periventricular nodular heterotopias (PNH), cerebellar malformations, polymicrogyria, and hydrocephalus. Previous studies have shown that terminal 1.7 Mb of 6q27 harbors four genes, THBS2, PHF10, DLL1, and C6orf70 that play critical roles in morphogenesis of the nervous system during embryogenesis [4].
ACC is associated with many syndromes and shows significant variability ranging from complete absence or partial absence to hypoplasia of the corpus callosum. Twelve genomic loci are consistently associated with ACC, including the 6q terminal deletion [8]. Incidence of corpus callosum anomalies for 6q25-27 deletion is approximately 50% to 75%. The subject of this study presented with mild hypogenesis, a short, comma-shaped corpus callosum (Figure 1B). PNH lining the lateral ventricles due to defective neuronal migration and can by asymptomatic or present as small, unilateral or bilateral nodules, or as extensive agglomerates, with or without other brain abnormalities.
C6orf70 in the 6q27 region is a critical gene expressed during brain development in human and rodents. The gene product is a putative vesicle-associated protein that plays a major role in controlling neuronal migration. Haploinsufficiency or mutation of C6orf70 was reported to cause PNH [1], and incidence of PNH was 9 out of 12 patients of 6q25-27 deletion. Although PCR analyses confirmed the patient in this report had haploinsufficiency of C6orf70, no evidence of PNH was found, suggesting that haploinsufficiency of C6orf70 for PNH has variable phenotypic expression.
Other brain malformations, such as cerebellar malformation, polymicrogyria, and hydrocephalus, have also been reported in 6q27 deletion cases with variable penetrance and expression. Cerebellar malformations were reported in 10.8% and polymicrogyria in 6.7% of individuals. Interestingly, neither of these conditions were present in the patient characterized in this study, although colpocephaly, asymmetric lateral ventricles, and choroid fissure cysts on the left were present. These results may suggest that the same genes cause various different brain malformations through the same pathway. It is also possible that low resolution MRI may have missed subtle brain malformations.
Mental retardation and seizures are associated with brain malformation in 6q27 deletion patients. The TATA-binding protein (TBP) gene in 6q27 seemed the most likely causal mutation [3]. TATA-binding protein, a general transcription factor associates with aggregates in several polyglutamine disorders [9]. Although trisomy18p alone is tolerated in humans and presents as very mild mental retardation [6-7], it is reasonable to speculate that severe MR (IQ = 46) in the subject may result from the combination of TBP deletion and trisomy18p.
Hypotonia is a clinical phenotype that affects approximately half of 6q27 deletion patients. Brain malformation may cause this phenotype, and long-lasting repercussions of hypotonia without corrective procedures, include scoliosis, pelvic obliquity, genu valgum, and leg discrepancy. In this case study, chromosome 6 is dicentric, one centromere is from chromosome 6 and the other is from chromosome 18. When human chromosome are dicentrics or multicentrics, faulty alignments may result [10]. Three different ‘predominant’ activation patterns occur. When the distance between two centomeres is close, they are fused and operate together. When the distance between centromeres is 1.4 Mb to 13 Mb, both centromeres are active, and when the distance is over 15 Mb, only one is active [11]. In this case, the distance between the centromeres was estimated to be more than 15 Mb, indicating only one centromere was active.
Assessment of risk for the offspring was a major goal of this study. Results indicated no genetic abnormalities were present in the amniotic fluid, and the fetus was developmentally normal. The baby was born with no complications.
Conclusions
In conclusion, this study identified a rare rearrangement between 6q27 and 18p, which led to de novo 1.3 Mb deletion of 6q27 and 15.2 Mb duplication of 18p in an adult with mental retardation, hypotonia, epilepsy, and multiple malformations. Analyses revealed that the 6q27 deletion contributed to the majority of the patient’s clinical features.
Methods
HumanCytoSNP-12 BeadChip for CNVs and SNP analyses
DNA was analyzed with the Illumina Human CytoSNP-12 array to 100 kb resolution following the instructions provided in the Illumina Infinium® HD Assay Ultra manual. The iScan scanner was used to translate electronic signals into digital signals. Initial analyses and quality control were performed using Illumina GenomeStudio software. Copy number variation was determined using the Illumina KaryoStudio software.
Cytogenetic and FISH analyses
Chromosome preparations were obtained from cultured peripheral lymphocytes and amniotic fluid cells using standard cytogenetic protocols at 320-450 bands resolution. For FISH, probes RP11-614P3 (=6q27-Red), RP11-196G15 (=6p22.3-Orange), D18Z1 (=cep18-Green), RP11-1035E2 (=18p11.32-Green), (JiaHui, China) were used according to standard cytogenetic protocols.
MLPA and real-time qPCR analyses
P036-E2 probe mix contains a probe designed to detect deletions and duplications for every subtelomeric region. MLPA reactions were performed following the manufacturer’s instructions (Biometra Thermal Cycler, Westburg, Netherlands). Fragment separation was performed using the ABI-3130 sequencer (Applied Biosystems, Foster City, CA), and data was directly analyzed using Coffalyser software. Primers for the C6orf70 gene were designed for use in real-time qPCR analyses on an ABI 7900 HT fluorescence quantitative PCR instrument (Applied Biosystems, Foster City, CA). Primers sequences and PCR conditions are available by request.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Abbreviations
- ACC:
-
Agenesis of corpus callosum
- FISH:
-
Fluorescence in situ hybridization
- MLPA:
-
Multiplex ligation-dependent probe amplification
- MR:
-
Mental retardation
- SNP-Array:
-
Single nucleotid polymorphism based array
- PNH:
-
Periventricular nodular heterotopias
- sSMC:
-
Small supernumerary marker chromosomes
References
Conti V, Carabalona A, Pallesi-Pocachard E, Parrini E, Leventer RJ, Buhler E, McGillivray G, Michel FJ, Striano P, Mei D, Watrin F, Lise S, Pagnamenta AT, Taylor JC, Kini U, Clayton-Smith J, Novara F, Zuffardi O, Dobyns WB, Scheffer IE, Robertson SP, Berkovic SF, Represa A, Keays DA, Cardoso C, Guerrini R: Periventricular heterotopia in 6q terminal deletion syndrome: role of the C6orf70 gene. Brain 2013, 136:3378–3394.
Dupé V, Rochard L, Mercier S, Le Pétillon Y, Gicquel I, Bendavid C, Bourrouillou G, Kini U, Thauvin-Robinet C, Bohan TP, Odent S, Dubourg C, David V: NOTCH, a new signaling pathway implicated in holoprosencephaly. Hum Mol Genet 2011, 20:1122–1131.
Rooms L, Reyniers E, Scheers S, van Luijk R, Wauters J, Van Aerschot L, Callaerts-Vegh Z, D’Hooge R, Mengus G, Davidson I, Courtens W, Kooy RF: TBP as a candidate gene for mental retardation in patients with subtelomeric 6q deletions. Eur J Hum Genet 2006, 14:1090–1096.
Peddibhotla S, Nagamani SC, Erez A, Hunter JV, Holder JL Jr, Carlin ME, Bader PI, Perras HM, Allanson JE, Newman L, Simpson G, Immken L, Powell E, Mohanty A, Kang SH, Stankiewicz P, Bacino CA, Bi W, Patel A, Cheung SW: Delineation of candidate genes responsible for structural brain abnormalities in patients with terminal deletions of chromosome 6q27. Eur J Hum Genet 2014, doi:10.1038/ejhg.2014.51. [Epub ahead of print].
Striano P, Malacarne M, Cavani S, Pierluigi M, Rinaldi R, Cavaliere ML, Rinaldi MM, De Bernardo C, Coppola A, Pintaudi M, Gaggero R, Grammatico P, Striano S, Dallapiccola B, Zara F, Faravelli F: Clinical phenotype and molecular characterization of 6q terminal deletion syndrome: five new cases. Am J Med Genet A 2006, 140A:1944–1949.
Mabboux P, Brisset S, Aboura A, Pineau D, Koubi V, Joannidis S, Labrune P, Tachdjian G: Pure and complete trisomy 18p due to a supernumerary marker chromosome associated with moderate mental retardation. Am J Med Genet A 2007, 143A:727–733.
Albert S: A Catalogue of Unbalanced Chromosome Aberrations in Man. 2nd edition. New York: Walter de Gruyter & Co; 2001:715–782.
Mary CO’D, Graeme CM Black JC-S, Sherr EH, William BD: Identification of genomic loci contributing to agenesis of the corpus callosum. Am J Med Genet Part A 2010, 152A:2145–2159.
WMC VAN ROON-MOM SJREID, RLM FAULL, RG SNELL: TATA-binding protein in neurodegenerative didease. Neuroscience 2005, 133:863–872.
Sullivan BA, Willard HF: Stable dicentric X chromosomes with two functional centromeres. Nat Genet 1998, 20:227–228.
Ewers E, Yoda K, Hamid AB, Weise A, Manvelyan M, Liehr T: Centromere activity in dicentric small supernumerary marker chromosomes. Chromosome Res 2010, 18:555–562.
Acknowledgements
This study was funded by a Major Program of Ministry of Health (NO.WKJ2011-2-017), a Major Program of Wenzhou (NO.S20070027) and Population and Family Planning Commission Project of Zhejiang Province (NO.JSW2012-A001). We thank the patient and her family members for participating in this study. We thank Dr. Thomas Liehr for assistance in editorial review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ZLL and CC drafted the paper and participated in the molecular cytogenetic analyses. LHZ and CYY performed molecular analyses. LXL and XXQ performed cytogenetic analyses. TSH designed the study, evaluated the family, provided genetic counseling and approved the final manuscript. All authors read and approved the final manuscript.
An erratum to this article can be found at http://dx.doi.org/10.1186/s13039-016-0287-z.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zhou, L., Chen, C., Li, H. et al. Delineation variable genotype/phenotype correlations of 6q27 terminal deletion derived from dic(6;18)(q27;p10). Mol Cytogenet 7, 78 (2014). https://doi.org/10.1186/s13039-014-0078-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-014-0078-3