
Abstract
Alzheimer’s Disease (AD), the most prevalent neurodegenerative disease of aging, affects one in eight older Americans. Nearly all drug treatments tested for AD today have failed to show any efficacy. There is a great need for therapies to prevent and/or slow the progression of AD. The major challenge in AD drug development is lack of clarity about the mechanisms underlying AD pathogenesis and pathophysiology. Several studies support the notion that AD is a multifactorial disease. While there is abundant evidence that amyloid plays a role in AD pathogenesis, other mechanisms have been implicated in AD such as tangle formation and spread, dysregulated protein degradation pathways, neuroinflammation, and loss of support by neurotrophic factors. Therefore, current paradigms of AD drug design have been shifted from single target approach (primarily amyloid-centric) to developing drugs targeted at multiple disease aspects, and from treating AD at later stages of disease progression to focusing on preventive strategies at early stages of disease development. Here, we summarize current strategies and new trends of AD drug development, including pre-clinical and clinical trials that target different aspects of disease (mechanism-based versus non-mechanism based, e.g. symptomatic treatments, lifestyle modifications and risk factor management).
Similar content being viewed by others


Background
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease of aging, and it affects over 26 million people worldwide with this number continuously increasing [1,2,3]. Nearly all drug treatments tested for AD today have failed to show any efficacy. There is a great need for therapies to prevent and/or slow the progression of AD.
AD is a complex disease with multifactorial etiology. Early-onset AD, a rare form of disease, follows an autosomal-dominant pattern in a majority of cases with mutations identified in amyloid precursor protein (APP), presenilin (PS) 1 and 2. Late-onset AD, a sporadic form of disease, affects over 90% of patients with several genetic loci and risk factors identified by a combination of genetic studies and bioinformatic approaches. These discoveries have shaped our current understanding of AD pathogenesis, as well as the development of therapeutic targets and design of clinical trials. While some of identified genetic risk factors can be linked to the most popular amyloid cascade hypothesis and the Tau theory [3], several commonly shared pathways have been implicated in AD such as immune system dysfunction, lipid and cholesterol dyshomeostasis, and vesicle trafficking and protein degradation pathway dysregulation [4,5,6]. Other theories have also been suggested such as mitochondrial dysfunction, lack of intrinsic support of neurotrophic factors, pre-existing comorbid medical conditions including cerebrovascular diseases, diabetes and hypertension, as well as environmental exposure such as virus infection [7,8,9,10].
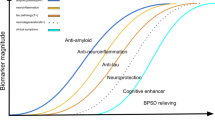
Here we focus on current AD therapeutic strategies which comprise of mechanism-based approaches including amyloid-beta (Aβ) clearance, tau protein deposits, apolipoprotein-E (ApoE) function, neuroprotection and neuroinflammation, as well as non-mechanism based approaches including symptomatic cognitive stimulation, AD prevention, lifestyle modifications and risk factor management including non-pharmacological interventions (Table 1: a summary list of therapeutic strategies discussed in this paper).
Mechanism-based approaches
Therapies targeted at amyloid
According to the amyloid cascade hypothesis, AD-related pathology typically begins with asymptomatic cerebral amyloidosis many years before the onset of clinical symptoms [11]. Accumulation of Aβ in the brain starts with monomeric Aβ leaving its reservoir in the cerebrospinal fluid (CSF) to form toxic aggregates followed by deposition on neuronal surface and synaptic terminals. Therefore, the majority of AD treatment strategies targeted at the amyloid cascade in the past 30 years or so has been focused on reducing Aβ generation through development of β- and γ-secretase inhibitors, accelerating Aβ clearance through active and passive immunotherapies, as well as preventing formation of toxic amyloid aggregates.
Reducing Aβ generation
Amyloid Aβ is derived from APP cleaved by two membrane-bound enzymes, β-secretase and γ-secretase complex. Therefore, modulation of these enzymes to inhibit Aβ production has been a major focus in developing AD therapies. The development of β-site APP cleaving enzyme 1 (BACE1) inhibitors was limited at the beginning due to difficulties in drug delivery. Later brain-penetrant BACE1 inhibitors have been developed with data showing promising efficacy at reducing Aβ in animal models [12, 13]. However, most BACE1 inhibitors tested today failed to survive beyond phase II/III clinical trials due to either lack of efficacy, or undesirable long-term side effects (Table 2: a summary list of AD drugs tested in clinical trials). For example, Merck halted its ongoing clinical trials of verubecestat (MK-8931) in mild to moderate AD patients [14,15,16], and most recently in people with prodromal AD (the APECS: β amyloid Production and Effects on Cognition Study; NCT01953601). However, despite the disappointing results from current clinical trials of BACE1 inhibitors, a recent study demonstrated that conditional knockout of BACE1 was capable of completely reversing pre-formed amyloid deposition and improving cognitive function in a mouse model with 5× Familial AD (FAD) transgenic background, suggesting sequential and gradual inhibition of BACE1 could be beneficial for AD patients [17]. It was pointed out that BACE1 is necessary to maintain optimal cognitive function and that the BACE1 inhibition is not without concerns [17]. More studies are needed to clarify the mechanism(s) of BACE inhibitors in AD, to determine optimal timing for BACE1 inhibition in adult AD patients, and to search for drug candidates without unwanted and off-target toxicities.
The γ-secretase complex is comprised of four subunits [18, 19], with presenilin (PS) exhibiting catalytic activities of γ -secretase [20,21,22,23,24,25,26,27,28]. There is a long list of γ-secretase substrates with APP and Notch among the most well-known due to their implications in AD and cancer [18, 29]. Substantial effort has been invested into developing small-molecule inhibitors of γ-secretase for AD therapies. Non-selective γ-secretase inhibitors led to a decrease in brain Aβ [30, 31], and reduced Notch signaling at the same time causing gastrointestinal (GI) symptoms and compromised immune system [32, 33]. Despite the concerns, non-selective γ-secretase inhibitors such as semagacestat were tested in clinical trials (see in Table 2; NCT00762411, NCT00594568, NCT01035138), but discontinued at Phase III stage because of lack of efficacy or even worsened cognitive performance, and patient intolerance due to severe off-target side effects like GI irritation and skin cancer [15, 18, 19, 34,35,36,37]. Selective γ-secretase inhibitors include Notch-sparing γ-secretase inhibitors and γ-secretase modulators. Gleevec, the abl kinase inhibitor was found to reduce Aβ production but spare Notch cleavage by γ-secretase in primary neuronal cells and animals [38]. Avagacestat was reported to inhibit APP processing more preferably than Notch cleavage [39]. However, Phase II clinical trial of Avagacestat was discontinued due to adverse side effects suggesting possible Notch inhibition just like Semagacestat ([35]; NCT00890890). A recently developed Notch-sparing γ-secretase inhibitor pinitol (NIC5–15) was derived from natural product and reported to have insulin sensitization property. It is currently in Phase II trial for the treatment of AD ([15, 40]; NCT01928420). Non-steroidal anti-inflammatory drugs (NSAIDs) were the first γ-secretase modulators shown to shift Aβ production from the aggregable form (Aβ42) to a more soluble form (Aβ38) [41]. One of these NSAIDs, R-flurbiprofen [42] failed to exhibit any efficacy in Phase III trial of mild AD subjects (NCT00105547, NCT00322036). A γ-secretase modulator EVP-0962 showed potency in preclinical stage but no efficacy in Phase II trial of mild cognitive impairment (MCI) or early AD subjects [15] (NCT01661673).
Accelerating Aβ clearance
On the other hand, immunotherapy via active and passive vaccines against Aβ has been utilized as a therapeutic approach in AD [43]. The clinical trials of active immunotherapy targeted at Aβ failed until now due to autoimmune responses. The first active immunization clinical study in mild to moderate AD patients was discontinued in Phase II because a small portion of cohort developed severe meningoencephalitis with the AN-1792 vaccine targeting full-length Aβ1–42 peptide (NCT00021723) [44]. Subsequent development of vaccines was tailored to target specific Aβ epitopes such as ACC-001 which targets an N-terminal Aβ1–7 peptide fragment [15, 45]. Currently 2 s-generation active immunotherapy vaccines have been progressed into clinical trials (Affitope AD02 terminated in Phase II study [46], NCT02008513; CAD106 [15, 47] still in Phase II/III study, NCT02565511).
Comparing to active vaccination, passive anti-Aβ immunization may be a more promising strategy for treating AD. Unfortunately, most currently available data in clinical trials fail to meet the primary endpoints. Bapineuzumab is an antibody targeted at the N-terminus of Aβ peptide acting on soluble Aβ. However, no beneficial effect was found with Bapineuzumab in Phase III clinical studies of mild-to-moderate AD subjects [43, 48] (NCT00667810; NCT00575055; NCT00574132). Similarly, Solanezumab targeted at Aβ16–24 epitope, which was reported to only recognize soluble instead of fibrillary Aβ, failed in Phase III trial of mild AD patients [43, 49] (NCT00905372; NCT00904683; NCT01900665). BAN2401 targeted at soluble Aβ protofibrils, recently completed a Phase II trial of 856 MCI or mild AD subjects with confirmed brain amyloid pathology [50] (NCT01767311) with positive results from the highest dosage treatment group (10 mg/kg bi-weekly). It was reported that the statistical significance was achieved on key efficacy endpoints after 18 months of high dose treatment on slowing clinical progression measured by the Alzheimer’s Disease Composite Score (ADCOMS) and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) score, as well as on reducing brain amyloid accumulation determined by amyloid-PET. However, those who interpret these results must be cautious as most of the ApoE4+ carriers were removed from the high-dose treatment group due to concerns of developing amyloid-related imaging abnormalities-edema (ARIA-E; based on AAIC 2018 Conference news). The full data set including the detailed demographic information of treatment versus placebo groups has yet to be shared with the scientific community.
Crenezumab (NCT03114657; NCT02670083), Gantenerumab (NCT03443973; NCT03444870), and Aducanumab (NCT02484547; NCT02477800) targeted both soluble and aggregated Aβ species (both oligomeric and fibrillar Aβ), are currently in Phase III trials in patients with prodromal, mild, and early AD, respectively. The results from preclinical and Phase 1b clinical trial studies regarding Aducanumab demonstrated that treatment reduced amyloid plaque levels in prodromal and mild AD patients in a dose-dependent manner. Additionally, cognitive results from clinical dementia rating-sum of the boxes scale (CDR-SB) and mini-mental status examination (MMSE) taken at the 54th week of treatment demonstrated that Aducanumab confers delayed cognitive decline. The main safety and tolerability concerns were ARIA [51] (NCT01677572).
Other anti-amyloidogenic compounds with diverse mechanisms of action
In addition to enzyme inhibitors and vaccines, there are other compounds developed with diverse mechanisms of action. ALZT-OP1, a combination therapy of two drugs with efficacy at preventing Aβ aggregation and neuroinflammation, is currently in Phase III clinical trial [43] (NCT02547818). GV-971, an oral sodium oligo-mannurarate with the ability to reduce Aβ toxicity in vitro, is in Phase III clinical trial of mild-to-moderate AD patients [43] (NCT02293915). Several low-molecular-weight anti-amyloidogenic drugs are currently in clinical investigations, e.g. Posiphen in Phase I/II trial for the treatment of MCI, AD and Parkinson’s disease (PD) [52] (NCT02925650), ELND005 in Phase II/III trial for treatment of AD type dementia [53] (NCT00934050), and ALZ-801 in Phase III clinical trial for AD [43].
The current belief in the field about the failure of anti-Aβ therapies indicates that late stage of interventions failed to provide full effects. Ongoing clinical trials of passive immunotherapy are focusing on targeting prodromal AD study cohorts with the goal to test anti-amyloid treatments in “correct” populations. This will be discussed in detail in the following section of “Therapies and Interventions for AD Prevention”. On the other hand, active Aβ immunotherapy as an effective way to prevent AD remains to be tested, and many safety concerns such as the severity of immune responses against the vaccine need to be investigated further.
Therapies targeted at tau
Tau is a microtubule-binding protein which forms neurofibrillary tangles (NFTs), another neuropathological hallmark of AD [54, 55]. Interestingly, previous reports suggest that tau deposit correlates better with cognitive decline than amyloid plaque does [56], and that Aβ-induced neuro-toxicity is mediated by tau hyper-phosphorylation through a toxic gain-of-function effect [57,58,59,60]. Because of many recent failures in anti-Aβ drug trials, therapies targeted at tau have come into focus in AD drug development field. Current therapeutic strategies are categorized into inhibiting tau aggregation, reducing hyper-phosphorylation or other toxic post-translational modifications of tau, as well as promoting tau clearance and preventing tau spread [61].
Tau stabilizers and aggregation inhibitors
Most tau stabilizing agents manifested undesirable toxic side effects, e.g. paclitaxel and epothilone D [15]. A recent Phase I clinical trial study of a tau stabilizer TPI 287 in mild-to-moderate AD, progressive supranuclear palsy (PSP), and corticobasal syndrome (CBS) patients, conveyed encouraging results with beneficial effects of TP1 287 on cognitive performance and/or nerve cells activity (based on CTAD 2017 conference news). The trial is still ongoing with more safety and exploratory clinical effects to be analyzed (NCT01966666).
In parallel, several tau aggregation inhibitors tested have failed in clinical trials due to either unwanted side effects or lack of efficacy, e.g. Rember™ in Phase II [62, 63] (NCT00684944; NCT00515333), and TRx0237 (LMTM) in three Phase III trials of patients with mild-to-moderate AD and behavioral variant frontotemporal dementia [64] (NCT01689246; NCT01689233; NCT02245568). An ongoing Phase II/III trial by TauRx, aiming to enroll people with all-cause dementia and AD from multiple sites, will compare a 6-month course of 4 mg of LMTM twice daily to a different kind of placebo. Primary outcomes include 18F- fludeoxyglucose positron emission tomography (FDG-PET) imaging and safety; secondary outcomes include structural magnetic resonance imaging (MRI), as well as measures of cognition and activities of daily living (ADL; NCT03539380).
Therapies targeted at tau post-translational modifications
Another aspect of tau targeted therapies focuses on toxic post-translational modifications of tau: 1) inhibiting tau hyper-phosphorylation kinases such as glycogen synthase kinase 3 beta (GSK3훽) and cyclin dependent kinase 5 (CDK5); 2) promoting the activity of tau dephosphorylation enzyme protein phosphate 2A (PP2A) [65]; 3) modulating tau acetylation and cis-transformation [66].
There have been no success in clinical trial studies of GSK3훽 inhibitors, e.g. lithium and valproate in Phase II trial of AD patients [67, 68] (NCT00088387), and NP-031112 (NP-12) in Phase IIb trial [69,70,71] (NCT01350362). Development of other GSK3훽 inhibitors like paullone has not progressed past the preclinical trials due to concerns with cytotoxic effects [72, 73]. Unfortunately, there are no tau kinase inhibitors other than GSK3 inhibitors that have entered clinical trials due to several limiting factors such as kinase specificities and unclear safety profiles. Most selective inhibitors of CDK5 with preclinical efficacy [74, 75] have yet to be tested in clinical trials due to uncertainty of how selective these inhibitors are over other CDK family members as well as poor understanding of safety risks associated with CDK5 inhibition in humans. The results of other inhibitors of tau kinases like JNK and DYRKIA were disappointing with either negative clinical outcome measures or severe adverse side effects [66].
Besides tau hyperphosphorylation, other tau post-translational modifications such as acetylation have been implicated in AD and related tauopathies [76,77,78]. Therapeutic approaches such as limiting tau K280/K281 acetylation [79] or reducing tau acetylation by protein deacetylase SIRT1 [80] could restore microtubule stability and/or ameliorate tau-associated neurodegeneration in animal models. Whether targeting tau acetylation could be a feasible therapeutic approach for AD or other tauopathies is yet to be tested in clinical studies. Furthermore, studies by Lu and colleagues demonstrated that cis-p-tau transformation causes gain-of-toxic conformational changes of tau leading to aggregation, and that a monoclonal antibody against cis-p-tau can block this phenomenon [81,82,83]. Another strategy is to inhibit O-GlcNAcase, an enzyme that strips sugars from tau. It is believed that O-GlcNAcylation either competes with phosphorylation for the same serine/threonine residues, or simply prevents tau molecules from cozying up to one another. In animal studies, O-GlcNAcase inhibitors suppress tau phosphorylation, prevent tangles, and boost neuronal survival [84, 85]. Further studies are needed to determine the clinical efficacy of these strategies.
Anti-tau immunotherapy
The basis for anti-tau immunotherapy stems upon the discovery of trans-cellular tau spread with evidence supported by studies in mouse models as well as in clinical settings [86,87,88,89]. The rationale behind the application of immunization against pathologic tau variants is to interrupt the uptake and propagation processes of abnormal tau [90]. Using high-affinity antibody against phosphorylated tau is an approach that hopefully would not disturb the function of physical tau from active immunization of phosphorylated tau [91].
Several studies using active immunization against phospho-tau peptides modulating tau pathology showed positive results in tauopathy or AD mouse models [92, 93]. Currently, two active vaccines (AADvac1 and ACI-35) are being tested in clinical trials with AD patients [15]. AADvac-1 is a synthetic peptide derived from tau for active immunization which is going into Phase II trials [94] (NCT02579252). ACI-35 is a liposome-based 16-amino acid, tetra-palmitoylated phospho-tau peptide being tested in people with mild to moderate AD during Phase I study [61] (ISRCTN13033912). However, several concerns about active immunization of tau remain to be addressed such as the development of immune responses to other regions of tau epitope also known as epitope spread, and the likelihood of irreversible active immunization processes [64].
Tau-targeted passive immunization is an exciting development in AD because it offers the possibility of halting the spread of tau pathology through the extracellular space. In some tauopathy mouse models, certain tau antibodies halt the progression of NFT pathology even after onset of tangle formation [95,96,97,98]. Currently, ABBV-8E12, an antibody against aggregated, extracellular tau, is in two Phase II trials targeting PSP patients and MCI subjects with positive amyloid scan (NCT03391765; NCT02880956). RO7105705, a pan-tau antibody targeted at N-terminus of all six isoforms of human tau, is now in Phase I trial which evaluates once weekly 8400 mg doses of the antibody in healthy controls and AD patients (NCT02820896; AAIC 2017 Conference News). Several other tau antibody candidates are heading toward Phase I trials. However, concerns of autoimmunity against endogenous tau just like Aβ immunotherapy remain to be addressed.
The link between tau pathology and cognitive dysfunction raises the hope that cognitive decline can be slowed or even halted by therapies targeted at tau. Strategies discussed in this section include stabilizing microtubules, reducing tau aggregation, inhibiting hyperphosphorylation or other toxic modifications of tau, as well as active and passive immunization against tau to promote clearance and interrupt transcellular spread. Other approaches that upregulate the clearance and degradation of tau through the ubiquitin/proteasome system and the autophagy/lysosome pathway would likely have favorable effects as well, which are not discussed in this section.
Therapies targeted at ApoE
The ApoE4 genotype is one of the strongest genetic risk factors for developing sporadic AD [99]. On the other hand, those with ApoE2 genotype demonstrate a lower risk of developing AD and a delayed age of onset regarding AD symptoms. Several Aβ-dependent [100,101,102] and Aβ-independent mechanisms [103, 104] have been proposed for ApoE in AD pathogenesis. Different therapeutic strategies targeted at ApoE were tested in vitro and/or in vivo preclinical animal models including blocking Aβ-ApoE interaction by small peptide fragments [105,106,107,108], manipulating ApoE levels (drug treatments to stimulate ApoE expression [109], or decrease ApoE expression [110]), viral delivery of ApoE2 [111, 112], ApoE antibodies [113, 114], structural modifiers [115,116,117], lipidation promoting compounds [118,119,120,121] and ApoE mimetic peptides [122,123,124,125].
Blocking the interaction between ApoE and Aβ by a peptide fragment of Aβ reduced brain amyloid accumulation, ameliorated memory deficits [105, 107], and reduced brain insoluble tau levels [126] in AD transgenic mouse models. Interestingly, ApoE immunotherapy can achieve similar effects on decreasing amyloid burden [113, 114]. One possible mechanism of action is blocking the ApoE-Aβ interaction by anti-ApoE antibodies. When given at pre-plaque stage [113] or after plaque deposition [114], an anti-mouse ApoE antibody decreased amyloid plaque load and improved brain functional connectivity and cognition in an APP transgenic mouse model. No overt adverse side effects such as changes in total cholesterol or cerebral amyloid angiopathy load were observed with anti-ApoE therapy in mice [113, 114]. It should be noted that severe dyslipidemia associated with ApoE deficiency [127] needs to be carefully examined before moving forward into clinical applications.
Regulation of ApoE quantity is one of the main therapeutic approaches tested for AD. However, conflicting results have been reported from clinical studies for AD biomarkers comparing ApoE levels of AD patients with normal individuals [128,129,130,131]. Interestingly in animal models, decreasing expression of ApoE3 or ApoE4, or increasing expression of ApoE2 lowered brain amyloid deposition [111, 112]. Numerous animal studies have also been performed to evaluate the therapeutic potential of compounds that increase brain ApoE levels [109, 118, 120, 121, 132,133,134,135,136,137,138,139,140,141]. For example, oral administration of bexarotene, an agonist of retinoid X receptors (RXRs) which positively regulates ApoE transcription, was found to increase brain ApoE, reduce Aβ deposition and improve cognitive function in an AD transgenic mouse model [109]. However, a later report failed to replicate the beneficial effects of bexarotene in mouse models [137]. When juxtaposed with the adverse side effects of bexarotene including hepatic failure and lack of efficacy in reducing brain amyloid load from a Phase II clinical trial of AD patients (NCT01782742), the enthusiasm for this drug diminished [142].
On the other hand, effects of viral delivery of human ApoE assessed in APP transgenic mice showed reduced amyloid plaque load by ApoE2 and increased plaque load by ApoE4 [111, 112]. Similar effects on mouse Aβ levels were observed in ApoE4 targeted replacement (TR) mice without human APP transgenic background [143]. Overall, results from animal studies suggest that gene therapy increasing ApoE2 expression might be beneficial [144]. Further studies are critical to determine gene therapy strategies for ApoE4 conditions, e.g. gene silencing versus increasing expression [144].
Another line of approach targeted at ApoE in AD is through the modification of ApoE properties such as ApoE structural modifiers and lipidation-promoting agents. It was reported that the interaction between residues Arg61 and Glu255 of ApoE4 confers abnormal structural conformation associated with ApoE4-induced neurotoxicity [145, 146]. Therefore, a potential therapeutic approach is to modify the pathological structure of ApoE4. For example, a recent report using human neurons derived from induced pluripotent stem cells of ApoE4 subjects, demonstrated that treatment with a small-molecule structure corrector of ApoE4 reduced levels of ApoE4 fragments, increased numbers of GABAergic neurons, as well as reduced production and/or secretion of p-tau, Aβ40 and Aβ42 [117].
Moreover, lipidation of ApoE significantly affects its function [147] and differential properties of lipidation between ApoE isoforms have been reported. ApoE4 is found to be poorly lipidated in humans [148] and in ApoE mice [149, 150], and promoting ApoE4 lipidation through activation of nuclear receptor pathways (the LXR/RXR-ABCA1 axis) could be a therapeutic strategy [140, 151, 152]. However, one potential concern is the likelihood of increasing ApoE4 levels and exacerbating the detrimental effects of ApoE4 [104]. Future work is needed to explore how ApoE lipidation and function are regulated by brain lipid homeostasis [153].
It should be noted that the ApoE4 genotype is an important determinant of therapeutic responses in AD clinical trials [154]. For example, Phase III trials of bapinuezumab showed significant differences in treatment responses between ApoE4+ and ApoE4− AD patients [155]. Similarly, data from the Cardiovascular Health Cognition Study indicated that protective effects of nonsteroidal anti-inflammatory drugs (NSAIDs) against AD development, can only be seen in 65 years or older ApoE4+ individuals [156]. More importantly, the risk for developing AD was significantly higher in ApoE4+ women than in ApoE4+ men [157,158,159]. Collectively, the potential impact of ApoE4 genotype and its interaction with sex need to be carefully considered when designing clinical trials to evaluate therapeutic efficacy in AD.
Neuroprotective therapies
In AD, synaptic dysfunction caused by a combination of factors including accumulation of toxic aggregates, age-related processes, and neuroinflammation is one of disease’s hallmarks. While the majority of AD therapeutic efforts advanced in clinical trials have been focused on targeting amyloid and tau, neuroprotective strategies are developed to target at degenerative mechanisms triggered by or involving factors mentioned above.
Neurotrophins and their receptor-based therapies
Neurotrophins (NTs) and their receptor-based therapies in AD have been explored for years because of the pleiotropic actions of NTs and receptor signaling [160,161,162,163,164,165,166]. However, suboptimal pharmacological profiles of NTs such as short plasma half-lives, poor oral bioavailability and BBB permeability, as well as limited brain tissue diffusion, limit their clinical application [167,168,169]. Alternatively, gene transfer technology has been utilized in developing NTs-based AD therapy.
A phase I clinical trial of implantation of genetically modified autologous fibroblasts expressing nerve growth factor (NGF) into the basal forebrain area of eight mild AD patients demonstrated efficacy at improving the cognitive decline rate and increasing brain metabolic activity measured by serial 18-fluorodeoxyglucose PET scans [170] (NCT00017940). Autopsied brains confirmed sustained expression and activity of viral-delivered NGF [171]. In addition, it was observed that axons sprouted toward the local source of NGF and cell hypertrophy [172]. Subsequent Phase II trial recruited 49 mild to moderate AD subjects who were randomly assigned to receive intracerebral injections of AAV2-NGF or sham surgery. While AAV2-NGF delivery was well-tolerated, it did not affect clinical outcomes, or selected AD biomarkers 2 years after NGF delivery [173] (NCT00876863). It was argued that these results were inconclusive due to a small sample size. In addition, whether viral delivery achieved the targeted expression of NGF in treatment group needs to be evaluated.
NT receptor-based therapies focus on targeting p75, Tropomyosin Receptor Kinase A (TrkA) and TrkB receptors. The goal of p75-targeted therapeutic strategies is to develop small molecules that can inhibit degenerative signaling of p75 [174, 175], and promote survival even in the absence of NTs [175, 176]. A brain-penetrant small molecule, called LMA11A-31, counteracts the toxicity of Aβ and tau to prevent synaptic dysfunction, spine loss, neurite degeneration, microglia activation, and cognitive deficits in animal models [177, 178]. This compound reversed degeneration in 12-month-old AD mice [179], and repaired age-related loss of cholinergic neurons in wild-type mice (CTAD 2017 meeting report). In a Phase I trial, single or multiple doses of LM11A-31 were well tolerated by young or old volunteers with no adverse events reported. An ongoing Phase IIa trial targeted at mild to moderate AD patients will evaluate the effects of LM11A-31 at two dosages (NCT03069014).
Previous conflicting reports implicate the complexity of targeting TrkA for AD therapies, e.g. results showing positive effects of TrkA activation in cellular and mouse models of AD [180, 181] versus data showing beneficial effects of TrkA inhibition [182, 183]. A series of small-molecule ligands have been developed that either bind to and activate TrkB alone, or both TrkB and TrkC as well as downstream effectors of the signaling pathways, demonstrating some beneficial effects in vitro and in wildtype animals [175, 184, 185]. Systemic injection of a TrkB agonist 7,8-dihydroxyflavone (7,8-DHF) reduced brain amyloid load, prevented synaptic loss, and rescued cognitive deficits in 5XFAD mice [186,187,188]. However, later work failed to show efficacy at reducing pathology or improving cognitive deficits in the APP/PS1 transgenic mouse model with concerns regarding limited bioavailability of 7,8-DHF [189]. Together, NTs and receptor-based therapies have some potential for synergistic intracellular signaling [190], and the feasibility to combat multifaceted pathological mechanisms of AD but these strategies needs to be carefully evaluated before clinical applications.
Therapies targeted at Neuroinflammation and oxidative stress
The role of neuroinflammation and involvement of microglia in AD pathogenesis have been increasingly recognized and supported by a large amount of evidence including genetic studies, data mining and multiscale network analysis [191,192,193,194]. The microglial priming model proposes that during pre-symptomatic stage of AD, microglia is activated by proinflammatory mediators [192], and subsequently astrocytes acquire a proinflammatory phenotype amplifying neuronal damage [195,196,197,198,199]. The glial dysfunction may be independent from the presence of Aβ and tau at early stages of disease, leading to synaptic dysfunction and neuronal death [200, 201]. Therefore, molecules that restore physiological function of microglia and astrocytes may offer new directions for AD therapy.
Different strategies are developed at modulating immune cell function in neuroinflammation such as reducing gene expression of cytokines, inhibiting cytokine release and preventing cytokines binding to their receptors [202]. Molecules such as minocycline with anti-inflammatory properties have been shown to reduce cytokine release from astrocytes, and thereby rescue cognitive deficits in AD mouse models [203, 204]. Interestingly, it was found that inhibiting tau phosphorylation kinases such as GSK3β could also achieve satisfactory results of modulating neuroinflammation in animal models [205]. Reduced oxidative injury is another neuroprotective approach. For example, inhibition of cyclooxygenase-2 and inducible nitric oxide synthase has positive outcomes based on in vitro and in vivo animal studies [206,207,208]. While mitochondrial enhancers like Dimebon (latrepirdine) failed to show efficacy in clinical trials [209] (NCT 00912288), effort continues at searching for more effective agents targeted mitochondrial dysfunction with the hope to restore synaptic and neuronal function [210, 211].
Another strategy targeted at microglial function is through experimental manipulations to promote microglial encapsulation of amyloid plaques and reduce axonal dystrophy [212]. Several groups have shown that passive immunization of anti-Aβ antibody [51, 212, 213], or anti-ApoE antibody [113] increases recruitment of microglia around amyloid plaques in AD mouse models. Genetic deletion of the chemokine receptor CX3CR1 in microglia can also enhance formation of microglial barrier around amyloid plaques [212], and thereby reduce plaque load [214, 215]. Other strategies of neutralizing CX3CR1 include antibodies targeted at the receptor or its ligand, or small molecule antagonists such as AZD8797 [216]. However, therapies targeted at CX3CR1 are not without concerns. The likelihood of exacerbating tau hyper-phosphorylation [217, 218], as well as disrupting bacterial clearance by peripheral immune system with systemic suppression of CX3CR1 signaling [219], suggest the importance of searching for approaches to target CX3CR1 signaling specifically in plaque-associated microglia and to minimize potential systemic side effects [220]. Intriguingly, recent reports described that LED-based light flicker stimuli at 40 Hz could promote microglial recruitment, thereby reducing brain amyloid levels in AD transgenic mouse models [221,222,223].
Studies have linked viral infection with an antimicrobial innate immune response [224, 225], and regulation of AD risk genes [226]. In alignment with these studies, a Phase II trial has been initiated to investigate the efficacy of treating mild AD patients who test positive for serum antibodies for herpes simplex virus 1 or 2, with an anti-viral drug valacyclovir (NCT03282916). The ApoE genotype was taken into consideration during this trial design. The outcome measures include ADAS-cog and ADCS-ADL scores, as well as Aβ and tau burden measured by PET and CSF studies. In addition, changes in cortical thinning on structural MRI, olfactory identification deficits, and antiviral antibody titers from baseline to 78 weeks, will be evaluated. This study will directly address whether virus infections may be etiologic or contribute to the pathology of AD.
Non-mechanism based approaches
Symptomatic cognitive enhancers
Symptomatic therapies aiming at cognitive enhancement have been focused on modulating cholinergic and glutamatergic function: the cholinesterase inhibitors (ACHEIs) and a N-methyl-D-aspartate (NMDA) receptor antagonist (memantine). ACHEIs decrease the degradation of acetylcholine released from cholinergic neurons, thereby increasing synaptic transmission. Evidence suggests that ACHEIs moderately improve cognitive and global function status of mild to moderate AD patients [227]. However, the efficacy wanes with long-term treatment due to side effects such as weight loss and syncope [227]. Memantine blocks over-excited NMDA receptors to prevent glutamate release, thereby inhibiting neurotoxicity [228]. It also inhibits and reverses tau hyper-phosphorylation with only mild adverse effects [229]. A combination therapy of memantine with ACHEIs (Namzaric) has been approved for treatment of moderate to severe AD patients [230].
Therapies targeted at other neurotransmitter systems are currently under study. Phase II trials of two compounds: the alpha-7 nicotinic acid agonist, encenicline [231, 232], and the serotonin 5-HT6 antagonist, idalopirdine [233] showed positive results on primary outcome measures (NCT01019421). However, a recent review of results from three Phase III trials of idalopiridine (NCT01955161, NCT02006641, and NCT02006654) suggests no benefit on cognitive function with treatment [234]. Clinical trial of histamine H3 receptor antagonists failed to show consistent benefits [235, 236] (GSK239512, NCT01009255; ABT288, NCT01018875). A Phase II clinical trial of monoamine oxidase (MAO) B inhibitor rasagiline in people with mild to moderate AD is currently ongoing to evaluate its effects on brain metabolism [237] (NCT02359552). Two Phase II clinical trials of ladostigil, a combination of a cholinesterase inhibitor and a MAO B inhibitor, failed to achieve beneficial effects on primary endpoint measures [238,239,240,241] (NCT01429623; NCT01354691). However, one trial that targeted delaying MCI conversion to AD, showed a trend towards benefits on selective cognitive tests and brain MRI measure (NCT00000173).
Therapies targeted at synaptic function have also been tested including protein kinase C epsilon (PKCξ) activators, Fyn kinase inhibitors and phosphodiesterase (PDE) inhibitors. Agents activating PKCξ like bryostatin have been tested based on results showing that reduced PKCξ activity is correlated with impaired synaptic and cognitive functions in AD animal models [242]. Moreover, Fyn kinase has been linked to synaptic function and AD pathogenesis. Fyn plays an important role in synaptic plasticity by regulating trafficking of the NMDA glutamate receptor subunits NR2A and NR2B [243,244,245,246]. Fyn deficiency mice presented blunted long-term potentiation (LTP) and impaired contextual fear memory function [243, 247]. On the other hand, Fyn has been found to mediate Aβ toxicity through interaction between oligomeric Aβ and the metabotropic glutamate receptor mGluR5 at the post-synaptic plasma membrane [248,249,250]. Fyn also phosphorylates dendritic tau [249,250,251]. A preclinical study reported that saracatinib (AZD0530), a Src family kinase inhibitor with high potency for Fyn and Src kinase, rescued synaptic dysfunction and spatial memory impairment in AD transgenic mice [248]. A Phase Ib study of AZD0530 in 24 subjects of mild to moderate AD indicated a reasonable safety and tolerance profile, as well as good BBB penetration [252] (NCT01864655). A Phase II study is currently ongoing in patients of mild AD diagnosis confirmed by amyloid imaging to compare two doses of AZD0530 (100 mg and 125 mg daily for 12 months) versus placebo and the influence of ApoE genotype was taken into consideration during trial design (NCT02167256).
The therapeutic implications of PDE inhibitors as cognitive enhancers in AD have been explored. As key enzymes hydrolyzing secondary messenger cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), PDEs play important roles in regulating signaling pathways critical in brain function. The major challenges of developing PDE inhibitors for AD therapy are narrow dose-response ranges and selection of isoform specific inhibitors. Out of 11 family members, inhibitors targeted at PDE3, 4, 5 and 9 have been tested in clinical trials for AD [253, 254]. Several clinical trial studies of a PDE3 inhibitor cilostazol demonstrated beneficial effects on cognitive function in MCI and AD patients [255,256,257] (NCT02491268). An ongoing Phase IV trial is studying the effects of cilostazol on subcortical white matter hyperintensities in AD subjects (NCT01409564). For inhibitors of PDE4: HT-0712 (NCT02013310), roflumilast (NCT02051335; NCT01433666; ISRCTN96013814), and BPN 14770 (NCT02648672; NCT02840279), and inhibitors of PDE9: BI 409306 (NCT01343706; NCT02337907) and PF-004447943 (NCT00988598), available results are limited to determine their clinical efficacy at this point [253, 254].
Therapies and interventions for AD prevention
Similar to stroke prevention, AD prevention becomes the second major wave of effort in the field of neurology. In the paper, we divide the effort on AD prevention into secondary and primary prevention interventions. The key concept of secondary prevention is to initiate the mechanism-based interventions with the hope to treat underlying pathophysiology in order to prevent cognitive symptoms from ever developing [258]. Different from secondary prevention trials, primary prevention trials seek to decrease modifiable risks for AD [259] with strategies including lifestyle interventions, co-morbidity treatments, supplemental and multi-domain interventions.
Secondary prevention interventions
Since 2011, global collaborative efforts have embarked on secondary prevention trials. The five large trials include: 1) the API Autosomal-Dominant AD (ADAD), 2) API APOE4 Trial, 3) the DIAN-Trials Unit (DIAN-TU), 4) the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease (A4) trial, and 5) the TOMMORROW Trial. API, DIAN, and A4 have already formed an umbrella group called the Collaboration for Alzheimer’s Prevention (CAP) to maintain regular dialogue about study design and outcome validation. Four of the five trials above have agreed to continue testing the amyloid hypothesis and are actively working together to make sure that the cognitive outcomes are comparable and meaningful [260]. The questions to be addressed include guidelines for safe monitoring of subjects for ARIA [261], timing of interventions for maximal benefit of prevention [260], and innovative cognitive outcomes for approval of investigational drugs for early AD with post-approval monitoring [262].
Primary prevention interventions
Primary prevention targeted at specific lifestyle interventions often includes management of cardiovascular disease or metabolic risk factors, changes in diet and exercise, cognitive stimulation or training, and social engagement [260].
Hypertension and hyperlipidemia were two major cardiovascular risk factors targeted in AD and dementia prevention. The Systolic Hypertension in Europe (Syst-Eur) trial, also called vascular dementia project, demonstrated a significant benefit for stroke with reduction of systolic blood pressure by at least 20 mmHg to a goal of below 150 mmHg [263]. The intervention was found to reduce the incidence of dementia by 50%, with AD dementia included as a subcategory. It was calculated that out of 1000 persons treated for hypertension for 5 years, 19 cases of dementia could be prevented. A recent study of participants in the Framingham Heart Study indicates that the incidence of dementia has declined over the course of three decades, which paralleled with observed improvement in cardiovascular health over time [264]. On the other hand, hyperlipidemia as a potential risk factor for AD remains controversial. Several trials have carried out to evaluate the effects of statins in AD. Two trials considered to meet inclusion criteria by the Cochrane review were the Heart Protection Study (HPS) trial and the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER) trial [265] (NCT00939822). However, neither of these two studies demonstrated any beneficial effects of statins on AD prevention or cognitive decline [266, 267].
Diabetes has been another focus of AD prevention. The most prominent trial was the multi-site randomized study called the Action to Control Cardiovascular Risk in Diabetes Trial with Memory in Diabetes (ACCORD-MIND) sub-study, which showed that the group underwent intensive glycemic control of hemoglobin A1c less than 6%, had greater total brain volume measured by MRI, but no differences in the cognitive scores compared to the group of standard of care with hemoglobin A1c at the range between 7 and 7.9% [268] (NCT00182910).
On the other hand, the observation of insulin resistance in AD forms the foundation of evaluating effects of insulin and insulin-sensitizing agents [269, 270]. Early pilot studies of insulin treatment in MCI and AD subjects showed beneficial effects on cognitive function [271,272,273] (NCT00438568). An ongoing Phase II/III clinical trial in amnestic MCI and mild AD subjects (SNIFF: Study of Nasal Insulin to Fight Forgetfulness) will determine the effects of intranasal insulin on cognitive decline, brain volume loss, and changes in CSF biomarkers (NCT01767909). Insulin-sensitizing agents such as metformin [274] and peroxisome proliferator-activated receptor gamma (PPARγ) agonists like pioglitazone [275], have been advanced into clinical trials of AD (NCT02432287; NCT00982202). Future follow up evaluations of clinical efficacy are needed.
Non-pharmacological interventions like diet, exercise, cognitive training, and vitamin supplement have also been studied in AD prevention trials. The most promising diet intervention has been the Mediterranean diet, rich in fruits and vegetables, combined with olive oil and fish. The Three-City (3C) Study suggests that participants who adhered to the Mediterranean diet had a slower rate of decline on the mini-mental status examination (MMSE), but not the other cognitive tests [276]. Studies have also shown that the Mediterranean-DASH Intervention for Neurodegenerative Delay (MIND) diet may reduce the risk of Alzheimer’s by up to 50%, and the protective effects persist till later time points even when diet recommendations were not followed rigorously [277, 278]. An ongoing preventive trial targeted at 65 years or older overweight individuals without cognitive impairment but with suboptimal diet habits will determine the effects of the MIND diet on cognitive decline and brain neurodegeneration over a 3-year period (NCT02817074).
There have been many studies investigating the association between physical activity and AD. An inverse association has been implicated despite moderate quality of evidence based on literature review of prospective observational and intervention studies [279]. However, current evidence is insufficient to provide detailed recommendations regarding specific physical exercise linked to AD prevention, e.g. type, frequency, intensity and duration of exercise [279]. Overall, it is indicated that physical activities combined with social and cognitive stimulation or diet modification may be more beneficial at reducing risks of AD [279]. For example, in a recent randomized controlled trial known as Fitness for the Aging Brain Study (FABS), subjects with memory complaints but no dementia, who underwent a 6-month exercise program had modest cognitive improvement at 18 months (0.26 points increase on the ADAS-cog score), whereas the control group declined 1.04 points [280] (ACTRN12605000136606). When combined with the Mediterranean diet, physical activity was associated with a significant reduction of AD incidence. For those with a high score for both healthy diet and physical activity, the hazard ratio for AD was 0.65 [281]. Besides physical activity, the effects of cognitive training have also been evaluated. The most robust study to date is the Advanced Cognitive Training for Independent and Vital Elderly (ACTIVE) trial, which provided the strongest evidence for beneficial effects of cognitive intervention in the prevention of cognitive impairment [282] (NCT00298558).
In the past decade or so, more attention has been brought to the use of supplements such as vitamin E, Gingko biloba and omega-3 fatty acids for AD prevention. Based on a recent systematic review, there is no evidence suggesting any beneficial effects of vitamin E on preventing MCI conversion into AD, or improving cognitive function of MCI or AD patient [283]. However, results from a single study of combined vitamin E and memantine therapy in AD subjects suggest that vitamin E may delay functional decline in these patients by 19% per year over the 4-year time period [284, 285] (NCT00235716).
The clinical studies of Ginkgo biloba extract, also known as EGb 761, showed no beneficial effects in AD prevention. The Ginkgo Evaluation of Memory (GEM) study of 3069 cognitively normal subjects aged 75 or older and 482 subjects with MCI showed that Ginkgo biloba did not prevent cognitive decline in normal aging or MCI subjects during 6 years of follow up [286, 287] (NCT00010803). Another trial called GuidAge had the same results in a group of 2854 subjects with subjective memory complaints who were given the same Ginkgo biloba extract for 5 years [288] (NCT00276510).
Omega-3 fatty acids have a complex association with AD, as noted in a recent systematic review, which found that seven out of the 11 observational studies had positive findings, but none of the four clinical trials had any benefit for prevention or treatment of dementia [289]. An example of a clinical trial that has shown the benefits of omega-3 fatty acids is the Memory Improvement with Docasahexaenoic Acid Study (MIDAS) [290] (NCT00278135). Future studies are needed to determine the effects of omega-3 fatty acids in larger clinical trials, and possible multimodal interventions for AD prevention.
Finally, multidomain interventions targeting vascular and lifestyle risk factors as previously described have been tested in AD prevention trials: 1) the Prevention of Dementia by Intensive Vascular Care study (PreDIVA), 2) the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER; NCT01041989) [291], 3) the Multidomain Alzheimer Preventive Trial (MAPT), and 4) the Multimodal Preventive trial for AD (MIND-ADMINI; NCT03249688). These ongoing multi-centered trials emphasize the importance of international collaboration and standardization of study design. They are unique in AD clinical research because these are the first randomized, controlled trials looking into a combination of treatments.
Future directions for AD drug development
As research in AD progresses, knowledge of underlying AD pathogenesis will guide future drug development effort. In the past two decades or so, we have learned that amyloid may not be the only critical step or the only mechanism of action to be targeted in AD. Tau-modifying agents have been developed, as well as many other agents discussed in this article as summarized in Table 2, which open the door for combination therapy like the case in cancer, cardiovascular and infectious disease treatment [260] Table 2.
The importance of utilizing biomarkers to help diagnosis of preclinical AD has been increasingly recognized in the field [292]. AD biomarkers include key proteins that reflect AD pathology such as amyloid and tau, as well as biomarkers of neuronal injury and regional patterns of abnormalities detected by various imaging modalities that provide indirect evidence of disease development and progression [293, 294]. AD clinical trials may incorporate biomarkers, so that in trial design precise timing of interventions can be elucidated, as well as subgroups of patients for intervention be determined and outcome measures be monitored. For example, Tau PET imaging is now emerging on the scene and could help further refine the preclinical stages of AD, as well as serve as an important outcome measure in AD prevention trials. The ultimate goal is to develop an algorithm of utilizing multiple biomarkers to predict the probability of conversion or progression to dementia in at-risk individuals [293].
Recent failures of several AD clinical trials make us realize that when AD progresses to certain point, excessive neurodegeneration becomes irreversible and aberrant neural networks cannot be repaired by simply reducing amyloid burden or oxidative stress. Current effort has been shifted to AD prevention in the early stages of disease [260, 295]. The recent report of beneficial effects on slowing down disease progression by anti-Aβ antibody BAN2401 in prodromal AD subjects suggests the feasibility of initiating disease-modifying treatments as early as possible. Many other preventative measures as discussed in this article, such as neuroprotection, cognitive enhancement and lifestyle modifications, may synergistically contribute to slow the progression of AD, when intervention(s) begins at the early stages of the disease processes [296]. Other major focuses are multi-targeted drug development and drug repositioning in AD [43].
Conclusions
AD is a complex and multifactorial disease for which the mechanisms remain to be fully elucidated. The lack of success in the Aβ-centric single target approach is compelling evidence that the paradigm of AD drug design needs to be shifted. As new insights into AD pathogenesis and progression are gained, developing or repurposing drugs with the capacity to target different aspects of the disease pathogenesis at once become promising in AD therapies. In addition, the development of novel biomarkers and imaging tools in AD has advanced dramatically in the past decade. However, the application of these tools in clinical trial practice is yet to be fully optimized. Finally, we are entering the era of “big data”. The concept of precision medicine has been introduced into AD field with the goal to utilize patient-centered approaches focusing on early screening for risks and detection of pathophysiology. Using customized multi-targeted and biomarker-guided strategies, we can achieve both effective and safe preventive therapies, based on the disease characteristics of each individual patient.
Abbreviations
- 3C Study:
-
The three city study
- 7,8-DHF:
-
Flavone 7,8-dihydroxyflavone
- A4 Trial:
-
Anti-amyloid treatment in asymptomatic alzheimer’s disease trial
- ACCORD-MIND:
-
Action to control cardiovascular risk in diabetes trial with memory in diabetes
- ACHEI:
-
Cholinesterase inhibitors
- ACTIVE:
-
Advanced cognitive training for independent and vital elderly
- AD:
-
Alzheimer’s disease
- ADAD:
-
API autosomal-dominant AD
- ADCOMS:
-
Alzheimer’s disease composite score
- ADCS-ADL:
-
Alzheimer’s disease cooperative study – activities of daily living
- APECS:
-
Amyloid-b production and effects on cognition study
- APH-1:
-
Anterior pharynx defective 1
- ApoE:
-
Apolipoprotein E
- APP:
-
Amyloid Precursor Protein (APP)
- ARIA:
-
Amyloid-related imaging abnormalities
- ARIA-E:
-
Amyloid-related imaging abnormalities edema
- Aβ:
-
Amyloid-beta
- BACE-1:
-
b-site APP cleaving enzyme 1
- BBB:
-
Blood brain barrier
- CAP:
-
Collaboration for Alzheimer’s prevention
- CBS:
-
Corticobasal syndrome
- CDK5:
-
Cyclin dependent kinase 5
- CDR-SB:
-
Clinical dementia rating sum of boxes
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DIAN-TU:
-
DIAN trials unit
- FABS:
-
Fitness for the aging brain study
- FAD:
-
Familial Alzheimer’s disease
- FDG-PET:
-
Flurodeoxyglucose positron emission tomography
- FINGER:
-
Finnish geriatric intervention study to prevent cognitive impairment and disability
- GEM:
-
Ginkgo evaluation of memory
- GI:
-
Gastrointestinal
- HPS:
-
Heart protection study
- LTD:
-
Long term depression
- LTP:
-
Long term potentiation
- MAO:
-
Monoamine Oxidase
- MAPT:
-
Multidomain alzheimer preventive trial
- MCI:
-
Mild cognitive impairment
- MIDAS:
-
Memory Improvement with Docasahexaenoic Acid Study
- MIND Diet:
-
Mediterranean-DASH intervention for neurodegenerative delay diet
- MIND-ADMINI:
-
Multimodal preventative trial for AD
- MMSE:
-
Mini mental state examination
- MT:
-
Microtubule
- NCT:
-
Nicastrin
- NFT:
-
Neurofibrillary tangles
- NGF:
-
Nerve growth factor
- NIC5–-15:
-
Pinitol
- NMDA:
-
N-methyl-D-aspartate
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- NT:
-
Neurotrophins
- PEN-2:
-
Presenilin enhancer 2
- PHF:
-
Paired helical filaments
- PK:
-
Protein kinase
- PP:
-
Protein phosphatase
- PPARγ:
-
Proliferator-activated receptor gamma
- PreDIVA:
-
Prevention of dementia by intensive vascular care study
- PROSPER:
-
Prospective study of pravastatin in the elderly at risk
- PS:
-
Presenilin
- PSP:
-
Progressive supranuclear palsy
- RXR:
-
Retinoid X receptor
- SNIFF:
-
Study of nasal insulin to fight forgetfulness
- Syst-Eur Trial:
-
Systolic hypertension in europe trial
- Tg:
-
Transgenic
- Trk:
-
Tropomyosin receptor kinase
References
Alzheimer's Association. 2012 Alzheimer's disease facts and figures. Alzheimers Dement. 2012;8:131–68.
Minino AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep. 2011;59:1–126.
Thies W, Bleiler L. Alzheimer's disease facts and figures. Alzheimers Dement. 2013;2013(9):208–45.
Freudenberg-Hua Y, Li W, Davies P. The role of genetics in advancing precision medicine for Alzheimer's disease-a narrative review. Front Med. 2018;5:108.
Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421–30.
Verheijen J, Sleegers K. Understanding Alzheimer disease at the Interface between genetics and transcriptomics. Trends Genet. 2018;34:434–47.
Sorbi S. Molecular genetics of Alzheimer's disease. Aging (Milano). 1993;5:417–25.
Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM. Alzheimer's disease. Lancet. 2016;388:505–17.
Bradley WG. Alzheimer's disease: theories of causation. Adv Exp Med Biol. 1990;282:31–8.
Armstrong RA. What causes alzheimer's disease? Folia Neuropathol. 2013;51:169–88.
Morris JC. Early-stage and preclinical Alzheimer disease. Alzheimer Dis Assoc Disord. 2005;19:163–5.
Kennedy ME, Stamford AW, Chen X, Cox K, Cumming JN, Dockendorf MF, Egan M, Ereshefsky L, Hodgson RA, Hyde LA, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer's disease patients. Sci Transl Med. 2016;8:363ra150.
May PC, Willis BA, Lowe SL, Dean RA, Monk SA, Cocke PJ, Audia JE, Boggs LN, Borders AR, Brier RA, et al. The potent BACE1 inhibitor LY2886721 elicits robust central Abeta pharmacodynamic responses in mice, dogs, and humans. J Neurosci. 2015;35:1199–210.
May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, Boggs LN, Monk SA, Mathes BM, Mergott DJ, Watson BM, et al. Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci. 2011;31:16507–16.
Hung SY, Fu WM. Drug candidates in clinical trials for Alzheimer's disease. J Biomed Sci. 2017;24:47.
Moussa CE. Beta-secretase inhibitors in phase I and phase II clinical trials for Alzheimer's disease. Expert Opin Investig Drugs. 2017;26:1131–6.
Hu X, Das B, Hou H, He W, Yan R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J Exp Med. 2018;215(3):927–40.
De Strooper B, Iwatsubo T, Wolfe MS. Presenilins and gamma-secretase: structure, function, and role in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006304.
Zhang X, Li Y, Xu H, Zhang YW. The gamma-secretase complex: from structure to function. Front Cell Neurosci. 2014;8:427.
Barton AJ, Crook BW, Karran EH, Brown F, Dewar D, Mann DM, Pearson RC, Graham DI, Hardy J, Hutton M, et al. Alteration in brain presenilin 1 mRNA expression in early onset familial Alzheimer's disease. Neurodegeneration. 1996;5:213–8.
Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–13.
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–90.
Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol. 2000;2:461–2.
Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci U S A. 2000;97:6138–43.
Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–94.
Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, et al. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–21.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–70.
Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, Yankner BA. Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nat Cell Biol. 2000;2:463–5.
Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J Alzheimers Dis. 2011;25:3–28.
Dovey HF, John V, Anderson JP, Chen LZ, de Saint AP, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2001;76:173–81.
Lanz TA, Wood KM, Richter KE, Nolan CE, Becker SL, Pozdnyakov N, Martin BA, Du P, Oborski CE, Wood DE, et al. Pharmacodynamics and pharmacokinetics of the gamma-secretase inhibitor PF-3084014. J Pharmacol Exp Ther. 2010;334:269–77.
Searfoss GH, Jordan WH, Calligaro DO, Galbreath EJ, Schirtzinger LM, Berridge BR, Gao H, Higgins MA, May PC, Ryan TP. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional gamma-secretase inhibitor. J Biol Chem. 2003;278:46107–16.
Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, Engstrom L, Pinzon-Ortiz M, Fine JS, Lee HJ, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;279:12876–82.
Albright CF, Dockens RC, Meredith JE Jr, Olson RE, Slemmon R, Lentz KA, Wang JS, Denton RR, Pilcher G, Rhyne PW, et al. Pharmacodynamics of selective inhibition of gamma-secretase by avagacestat. J Pharmacol Exp Ther. 2013;344:686–95.
Coric V, van Dyck CH, Salloway S, Andreasen N, Brody M, Richter RW, Soininen H, Thein S, Shiovitz T, Pilcher G, et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol. 2012;69:1430–40.
Doody RS, Aisen PS, Iwatsubo T. Semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013;369:1661.
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013;369:341–50.
Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, Bornmann WG, Clarkson B, Xu H, Greengard P. Gleevec inhibits beta-amyloid production but not notch cleavage. Proc Natl Acad Sci U S A. 2003;100:12444–9.
Gillman KW, Starrett JE Jr, Parker MF, Xie K, Bronson JJ, Marcin LR, McElhone KE, Bergstrom CP, Mate RA, Williams R, et al. Discovery and evaluation of BMS-708163, a potent, selective and orally bioavailable gamma-secretase inhibitor. ACS Med Chem Lett. 2010;1:120–4.
Folch J, Petrov D, Ettcheto M, Abad S, Sanchez-Lopez E, Garcia ML, Olloquequi J, Beas-Zarate C, Auladell C, Camins A. Current research therapeutic strategies for Alzheimer's disease treatment. Neural Plast. 2016;2016:8501693.
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001;414:212–6.
Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH, Tarenflurbil Phase 3 Study G. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302:2557–64.
Bachurin SO, Bovina EV, Ustyugov AA. Drugs in clinical trials for Alzheimer's disease: the major trends. Med Res Rev. 2017;37:1186–225.
Lemere CA. Developing novel immunogens for a safe and effective Alzheimer's disease vaccine. Prog Brain Res. 2009;175:83–93.
Pasquier F, Sadowsky C, Holstein A, Leterme Gle P, Peng Y, Jackson N, Fox NC, Ketter N, Liu E, Ryan JM, Team ACCS. Two phase 2 multiple ascending-dose studies of Vanutide Cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer's disease. J Alzheimers Dis. 2016;51:1131–43.
Winblad B, Graf A, Riviere ME, Andreasen N, Ryan JM. Active immunotherapy options for Alzheimer's disease. Alzheimers Res Ther. 2014;6:7.
Davtyan H, Bacon A, Petrushina I, Zagorski K, Cribbs DH, Ghochikyan A, Agadjanyan MG. Immunogenicity of DNA- and recombinant protein-based Alzheimer disease epitope vaccines. Hum Vaccin Immunother. 2014;10:1248–55.
Abushouk AI, Elmaraezy A, Aglan A, Salama R, Fouda S, Fouda R, AlSafadi AM. Bapineuzumab for mild to moderate Alzheimer's disease: a meta-analysis of randomized controlled trials. BMC Neurol. 2017;17:66.
Laske C. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer's disease. N Engl J Med. 2014;370:1459.
Tucker S, Moller C, Tegerstedt K, Lord A, Laudon H, Sjodahl J, Soderberg L, Spens E, Sahlin C, Waara ER, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-beta protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis. 2015;43:575–88.
Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2016;537:50–6.
Maccecchini ML, Chang MY, Pan C, John V, Zetterberg H, Greig NH. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-beta peptide and tau levels: target engagement, tolerability and pharmacokinetics in humans. J Neurol Neurosurg Psychiatry. 2012;83:894–902.
Sinha S, Du Z, Maiti P, Klarner FG, Schrader T, Wang C, Bitan G. Comparison of three amyloid assembly inhibitors: the sugar scyllo-inositol, the polyphenol epigallocatechin gallate, and the molecular tweezer CLR01. ACS Chem Neurosci. 2012;3:451–8.
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130.
Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–54.
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–81.
Fath T, Eidenmuller J, Brandt R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J Neurosci. 2002;22:9733–41.
Leschik J, Welzel A, Weissmann C, Eckert A, Brandt R. Inverse and distinct modulation of tau-dependent neurodegeneration by presenilin 1 and amyloid-beta in cultured cortical neurons: evidence that tau phosphorylation is the limiting factor in amyloid-beta-induced cell death. J Neurochem. 2007;101:1303–15.
Shahani N, Subramaniam S, Wolf T, Tackenberg C, Brandt R. Tau aggregation and progressive neuronal degeneration in the absence of changes in spine density and morphology after targeted expression of Alzheimer's disease-relevant tau constructs in organotypic hippocampal slices. J Neurosci. 2006;26:6103–14.
Tackenberg C, Brandt R. Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci. 2009;29:14439–50.
Panza F, Solfrizzi V, Seripa D, Imbimbo BP, Lozupone M, Santamato A, Zecca C, Barulli MR, Bellomo A, Pilotto A, et al. Tau-centric targets and drugs in clinical development for the treatment of Alzheimer's disease. Biomed Res Int. 2016;2016:3245935.
Baddeley TC, McCaffrey J, Storey JM, Cheung JK, Melis V, Horsley D, Harrington CR, Wischik CM. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer's disease. J Pharmacol Exp Ther. 2015;352:110–8.
Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, Kook KA, Harrington CR. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. J Alzheimers Dis. 2015;44:705–20.
Bakota L, Brandt R. Tau biology and tau-directed therapies for Alzheimer's disease. Drugs. 2016;76:301–13.
Pei JJ, Bjorkdahl C, Zhang H, Zhou X, Winblad B. p70 S6 kinase and tau in Alzheimer's disease. J Alzheimers Dis. 2008;14:385–92.
Jia Q, Deng Y, Qing H. Potential therapeutic strategies for Alzheimer's disease targeting or beyond beta-amyloid: insights from clinical trials. Biomed Res Int. 2014;2014:837157.
Hampel H, Ewers M, Burger K, Annas P, Mortberg A, Bogstedt A, Frolich L, Schroder J, Schonknecht P, Riepe MW, et al. Lithium trial in Alzheimer's disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70:922–31.
Tariot PN, Aisen PS. Can lithium or valproate untie tangles in Alzheimer's disease? J Clin Psychiatry. 2009;70:919–21.
del Ser T, Steinwachs KC, Gertz HJ, Andres MV, Gomez-Carrillo B, Medina M, Vericat JA, Redondo P, Fleet D, Leon T. Treatment of Alzheimer's disease with the GSK-3 inhibitor tideglusib: a pilot study. J Alzheimers Dis. 2013;33:205–15.
Martinez A. Preclinical efficacy on GSK-3 inhibitors: towards a future generation of powerful drugs. Med Res Rev. 2008;28:773–96.
Sereno L, Coma M, Rodriguez M, Sanchez-Ferrer P, Sanchez MB, Gich I, Agullo JM, Perez M, Avila J, Guardia-Laguarta C, et al. A novel GSK-3beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–67.
Natarajan P, Priyadarshini V, Pradhan D, Manne M, Swargam S, Kanipakam H, Bhuma V, Amineni U. E-pharmacophore-based virtual screening to identify GSK-3beta inhibitors. J Recept Signal Transduct Res. 2016;36:445–58.
Tell V, Hilgeroth A. Recent developments of protein kinase inhibitors as potential AD therapeutics. Front Cell Neurosci. 2013;7:189.
Mushtaq G, Greig NH, Anwar F, Al-Abbasi FA, Zamzami MA, Al-Talhi HA, Kamal MA. Neuroprotective mechanisms mediated by CDK5 inhibition. Curr Pharm Des. 2016;22:527–34.
Wen Y, Yu WH, Maloney B, Bailey J, Ma J, Marie I, Maurin T, Wang L, Figueroa H, Herman M, et al. Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008;57:680–90.
Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 2015;21:1154–62.
Sohn PD, Tracy TE, Son HI, Zhou Y, Leite RE, Miller BL, Seeley WW, Grinberg LT, Gan L. Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener. 2016;11:47.
Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li Y, Zhou Y, Le D, Lo I, Ponnusamy R, et al. Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes Tauopathy-related memory loss. Neuron. 2016;90:245–60.
Trzeciakiewicz H, Tseng JH, Wander CM, Madden V, Tripathy A, Yuan CX, Cohen TJ. A dual pathogenic mechanism links tau acetylation to sporadic Tauopathy. Sci Rep. 2017;7:44102.
Min SW, Sohn PD, Li Y, Devidze N, Johnson JR, Krogan NJ, Masliah E, Mok SA, Gestwicki JE, Gan L. SIRT1 deacetylates tau and reduces pathogenic tau spread in a mouse model of Tauopathy. J Neurosci. 2018;38:3680–8.
Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, Li C, Herbert MK, Qiu J, Monuteaux M, Driver J, et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun. 2017;8:1000.
Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, Yao Y, Lin YM, Driver JA, Sun Y, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523:431–6.
Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, Zhou XZ. Potential of the antibody against cis-phosphorylated tau in the early diagnosis, treatment, and prevention of Alzheimer disease and brain injury. JAMA Neurol. 2016;73:1356–62.
Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–58.
Liu F, Shi J, Tanimukai H, Gu J, Gu J, Grundke-Iqbal I, Iqbal K, Gong CX. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease. Brain. 2009;132:1820–32.
Flight MH. Neurodegenerative disease: tau immunotherapy targets transcellular propagation. Nat Rev Drug Discov. 2013;12:904.
Goedert M, Eisenberg DS, Crowther RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci. 2017;40:189–210.
Goedert M, Spillantini MG. Propagation of tau aggregates. Mol Brain. 2017;10:18.
Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–51.
Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80:402–14.
Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol. 2015;2:278–88.
Chukwu JE, Pedersen JT, Pedersen LO, Volbracht C, Sigurdsson EM, Kong XP. Tau antibody structure reveals a molecular switch defining a pathological conformation of the tau protein. Sci Rep. 2018;8:6209.
Rajamohamedsait H, Rasool S, Rajamohamedsait W, Lin Y, Sigurdsson EM. Prophylactic active tau immunization leads to sustained reduction in both tau and amyloid-beta pathologies in 3xTg mice. Sci Rep. 2017;7:17034.
Gruninger F. Invited review: drug development for tauopathies. Neuropathol Appl Neurobiol. 2015;41:81–96.
Dai CL, Chen X, Kazim SF, Liu F, Gong CX, Grundke-Iqbal I, Iqbal K. Passive immunization targeting the N-terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J Neural Transm. 2015;122:607–17.
Dai CL, Hu W, Tung YC, Liu F, Gong CX, Iqbal K. Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated tau-induced pathology in 3 × Tg-AD mice. Alzheimers Res Ther. 2018;10:13.
Dai CL, Tung YC, Liu F, Gong CX, Iqbal K. Tau passive immunization inhibits not only tau but also Abeta pathology. Alzheimers Res Ther. 2017;9:1.
Malia TJ, Teplyakov A, Ernst R, Wu SJ, Lacy ER, Liu X, Vandermeeren M, Mercken M, Luo J, Sweet RW, Gilliland GL. Epitope mapping and structural basis for the recognition of phosphorylated tau by the anti-tau antibody AT8. Proteins. 2016;84:427–34.
Mayeux R. Epidemiology of neurodegeneration. Annu Rev Neurosci. 2003;26:81–104.
Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57.
Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–86.
Li J, Kanekiyo T, Shinohara M, Zhang Y, LaDu MJ, Xu H, Bu G. Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J Biol Chem. 2012;287:44593–601.
Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006312.
Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Kuszczyk MA, Sanchez S, Pankiewicz J, Kim J, Duszczyk M, Guridi M, Asuni AA, Sullivan PM, Holtzman DM, Sadowski MJ. Blocking the interaction between apolipoprotein E and Abeta reduces intraneuronal accumulation of Abeta and inhibits synaptic degeneration. Am J Pathol. 2013;182:1750–68.
Liu Q, Wu WH, Fang CL, Li RW, Liu P, Lei P, Hu J, Sun X, Zheng YZ, Zhao YF, Li YM. Mapping ApoE/Abeta binding regions to guide inhibitor discovery. Mol BioSyst. 2011;7:1693–700.
Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, Wisniewski T. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:18787–92.
Yang J, Ji Y, Mehta P, Bates KA, Sun Y, Wisniewski T. Blocking the apolipoprotein E/amyloid-beta interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice. J Alzheimers Dis. 2011;24:269–85.
Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–6.
Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular a beta clearance. Neuron. 2009;64:632–44.
Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, Paul SM. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102:1211–6.
Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, Scheel M, Spires-Jones T, Arbel-Ornath M, Betensky R, et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 2013;5:212ra161.
Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Kim J, Stewart FR, Basak JM, Holtzman DM. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med. 2012;209:2149–56.
Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, Mahan TE, Lefton KB, Zhang TJ, Dearborn JT, Kim J, et al. Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J Neurosci. 2014;34:7281–92.
Brodbeck J, McGuire J, Liu Z, Meyer-Franke A, Balestra ME, Jeong DE, Pleiss M, McComas C, Hess F, Witter D, et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J Biol Chem. 2011;286:17217–26.
Chen HK, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, McGuire JG, Pleiss MA, Ji ZS, Balestra ME, Walker DW, et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012;287:5253–66.
Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, Yoon SY, Yuan H, Li G, Miller ZA, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018;24:647–57.
Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J, Wellington CL. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285:34144–54.
Fitz NF, Cronican A, Pham T, Fogg A, Fauq AH, Chapman R, Lefterov I, Koldamova R. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci. 2010;30:6862–72.
Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer's disease. J Biol Chem. 2005;280:4079–88.
Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol Cell Neurosci. 2007;34:621–8.
Laskowitz DT, Lei B, Dawson HN, Wang H, Bellows ST, Christensen DJ, Vitek MP, James ML. The apoE-mimetic peptide, COG1410, improves functional recovery in a murine model of intracerebral hemorrhage. Neurocrit Care. 2012;16:316–26.
Laskowitz DT, McKenna SE, Song P, Wang H, Durham L, Yeung N, Christensen D, Vitek MP. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma. 2007;24:1093–107.
Vitek MP, Christensen DJ, Wilcock D, Davis J, Van Nostrand WE, Li FQ, Colton CA. APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer's disease transgenics. Neurodegener Dis. 2012;10:122–6.
Wang H, Durham L, Dawson H, Song P, Warner DS, Sullivan PM, Vitek MP, Laskowitz DT. An apolipoprotein E-based therapeutic improves outcome and reduces Alzheimer's disease pathology following closed head injury: evidence of pharmacogenomic interaction. Neuroscience. 2007;144:1324–33.
Liu S, Breitbart A, Sun Y, Mehta PD, Boutajangout A, Scholtzova H, Wisniewski T. Blocking the apolipoprotein E/amyloid beta interaction in triple transgenic mice ameliorates Alzheimer's disease related amyloid beta and tau pathology. J Neurochem. 2014;128:577–91.
Mak AC, Pullinger CR, Tang LF, Wong JS, Deo RC, Schwarz JM, Gugliucci A, Movsesyan I, Ishida BY, Chu C, et al. Effects of the absence of apolipoprotein e on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol. 2014;71:1228–36.
Heywood WE, Galimberti D, Bliss E, Sirka E, Paterson RW, Magdalinou NK, Carecchio M, Reid E, Heslegrave A, Fenoglio C, et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol Neurodegener. 2015;10:64.
Martinez-Morillo E, Hansson O, Atagi Y, Bu G, Minthon L, Diamandis EP, Nielsen HM. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer's disease patients and controls. Acta Neuropathol. 2014;127:633–43.
Simon R, Girod M, Fonbonne C, Salvador A, Clement Y, Lanteri P, Amouyel P, Lambert JC, Lemoine J. Total ApoE and ApoE4 isoform assays in an Alzheimer's disease case-control study by targeted mass spectrometry (n=669): a pilot assay for methionine-containing proteotypic peptides. Mol Cell Proteomics. 2012;11:1389–403.
Talwar P, Sinha J, Grover S, Agarwal R, Kushwaha S, Srivastava MV, Kukreti R. Meta-analysis of apolipoprotein E levels in the cerebrospinal fluid of patients with Alzheimer's disease. J Neurol Sci. 2016;360:179–87.
Boehm-Cagan A, Michaelson DM. Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J Neurosci. 2014;34:7293–301.
Burns MP, Vardanian L, Pajoohesh-Ganji A, Wang L, Cooper M, Harris DC, Duff K, Rebeck GW. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J Neurochem. 2006;98:792–800.
Casali BT, Corona AW, Mariani MM, Karlo JC, Ghosal K, Landreth GE. Omega-3 fatty acids augment the actions of nuclear receptor agonists in a mouse model of Alzheimer's disease. J Neurosci. 2015;35:9173–81.
Escribano L, Simon AM, Gimeno E, Cuadrado-Tejedor M, Lopez de Maturana R, Garcia-Osta A, Ricobaraza A, Perez-Mediavilla A, Del Rio J, Frechilla D. Rosiglitazone rescues memory impairment in Alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology. 2010;35:1593–604.
Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–93.
LaClair KD, Manaye KF, Lee DL, Allard JS, Savonenko AV, Troncoso JC, Wong PC. Treatment with bexarotene, a compound that increases apolipoprotein-E, provides no cognitive benefit in mutant APP/PS1 mice. Mol Neurodegener. 2013;8:18.
Skerrett R, Pellegrino MP, Casali BT, Taraboanta L, Landreth GE. Combined liver X receptor/peroxisome proliferator-activated receptor gamma agonist treatment reduces amyloid beta levels and improves behavior in amyloid precursor protein/Presenilin 1 mice. J Biol Chem. 2015;290:21591–602.
Tachibana M, Shinohara M, Yamazaki Y, Liu CC, Rogers J, Bu G, Kanekiyo T. Rescuing effects of RXR agonist bexarotene on aging-related synapse loss depend on neuronal LRP1. Exp Neurol. 2016;277:1–9.
Tai LM, Koster KP, Luo J, Lee SH, Wang YT, Collins NC, Ben Aissa M, Thatcher GR, LaDu MJ. Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem. 2014;289:30538–55.
Vanmierlo T, Rutten K, Dederen J, Bloks VW, van Vark-van der Zee LC, Kuipers F, Kiliaan A, Blokland A, Sijbrands EJ, Steinbusch H, et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging. 2011;32:1262–72.
Cummings JL, Zhong K, Kinney JW, Heaney C, Moll-Tudla J, Joshi A, Pontecorvo M, Devous M, Tang A, Bena J. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer's disease. Alzheimers Res Ther. 2016;8:4.
Hu J, Liu CC, Chen XF, Zhang YW, Xu H, Bu G. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol Neurodegener. 2015;10:6.
Yamazaki Y, Painter MM, Bu G, Kanekiyo T. Apolipoprotein E as a therapeutic target in Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs. 2016;30:773–89.
Zhong N, Scearce-Levie K, Ramaswamy G, Weisgraber KH. Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimers Dement. 2008;4:179–92.
Zhong N, Weisgraber KH. Understanding the association of apolipoprotein E4 with Alzheimer disease: clues from its structure. J Biol Chem. 2009;284:6027–31.
Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer's disease: accidental encounters or partners? Neuron. 2014;81:740–54.
Hanson AJ, Bayer-Carter JL, Green PS, Montine TJ, Wilkinson CW, Baker LD, Watson GS, Bonner LM, Callaghan M, Leverenz JB, et al. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol. 2013;70:972–80.
Tai LM, Bilousova T, Jungbauer L, Roeske SK, Youmans KL, Yu C, Poon WW, Cornwell LB, Miller CA, Vinters HV, et al. Levels of soluble apolipoprotein E/amyloid-beta (Abeta) complex are reduced and oligomeric Abeta increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J Biol Chem. 2013;288:5914–26.
Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, Kim J, Eimer WA, Estus S, Rebeck GW, et al. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287:41774–86.
Tai LM, Mehra S, Shete V, Estus S, Rebeck GW, Bu G, LaDu MJ. Soluble apoE/Abeta complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol Neurodegener. 2014;9:2.
Vance JE, Hayashi H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim Biophys Acta. 1801;2010:806–18.
Zhu L, Zhong M, Elder GA, Sano M, Holtzman DM, Gandy S, Cardozo C, Haroutunian V, Robakis NK, Cai D. Phospholipid dysregulation contributes to ApoE4-associated cognitive deficits in Alzheimer's disease pathogenesis. Proc Natl Acad Sci U S A. 2015;112:11965–70.
Hanson AJ, Craft S, Banks WA. The APOE genotype: modification of therapeutic responses in Alzheimer's disease. Curr Pharm Des. 2015;21:114–20.
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:322–33.
Szekely CA, Breitner JC, Fitzpatrick AL, Rea TD, Psaty BM, Kuller LH, Zandi PP. NSAID use and dementia risk in the cardiovascular health study: role of APOE and NSAID type. Neurology. 2008;70:17–24.
Altmann A, Tian L, Henderson VW, Greicius MD, Alzheimer's Disease Neuroimaging Initiative I. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol. 2014;75:563–73.
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease Meta analysis consortium. JAMA. 1997;278:1349–56.
Nyarko JNK, Quartey MO, Pennington PR, Heistad RM, Dea D, Poirier J, Baker GB, Mousseau DD. Profiles of beta-amyloid peptides and key secretases in brain autopsy samples differ with sex and APOE epsilon4 status: impact for risk and progression of Alzheimer disease. Neuroscience. 2018;373:20–36.
Abram M, Wegmann M, Fokuhl V, Sonar S, Luger EO, Kerzel S, Radbruch A, Renz H, Zemlin M. Nerve growth factor and neurotrophin-3 mediate survival of pulmonary plasma cells during the allergic airway inflammation. J Immunol. 2009;182:4705–12.
Bergmann I, Reiter R, Toyka KV, Koltzenburg M. Nerve growth factor evokes hyperalgesia in mice lacking the low-affinity neurotrophin receptor p75. Neurosci Lett. 1998;255:87–90.
Dyck PJ, Peroutka S, Rask C, Burton E, Baker MK, Lehman KA, Gillen DA, Hokanson JL, O'Brien PC. Intradermal recombinant human nerve growth factor induces pressure allodynia and lowered heat-pain threshold in humans. Neurology. 1997;48:501–5.
Malcangio M, Lessmann V. A common thread for pain and memory synapses? Brain-derived neurotrophic factor and trkB receptors. Trends Pharmacol Sci. 2003;24:116–21.
Pezet S, Malcangio M, McMahon SB. BDNF: a neuromodulator in nociceptive pathways? Brain Res Brain Res Rev. 2002;40:240–9.
Sah DW, Ossipo MH, Porreca F. Neurotrophic factors as novel therapeutics for neuropathic pain. Nat Rev Drug Discov. 2003;2:460–72.
Shu XQ, Mendell LM. Neurotrophins and hyperalgesia. Proc Natl Acad Sci U S A. 1999;96:7693–6.
Pardridge WM. Neurotrophins, neuroprotection and the blood-brain barrier. Curr Opin Investig Drugs. 2002;3:1753–7.
Poduslo JF, Curran GL. Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res Mol Brain Res. 1996;36:280–6.
Saltzman WM, Mak MW, Mahoney MJ, Duenas ET, Cleland JL. Intracranial delivery of recombinant nerve growth factor: release kinetics and protein distribution for three delivery systems. Pharm Res. 1999;16:232–40.
Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R, Patel P, Blesch A, Vahlsing HL, Ho G, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11:551–5.
Rafii MS, Baumann TL, Bakay RA, Ostrove JM, Siffert J, Fleisher AS, Herzog CD, Barba D, Pay M, Salmon DP, et al. A phase1 study of stereotactic gene delivery of AAV2-NGF for Alzheimer's disease. Alzheimers Dement. 2014;10:571–81.
Tuszynski MH, Yang JH, Barba D, U HS, Bakay RA, Pay MM, Masliah E, Conner JM, Kobalka P, Roy S, Nagahara AH. Nerve growth factor gene therapy: activation of neuronal responses in Alzheimer disease. JAMA Neurol. 2015;72:1139–47.
Rafii MS, Tuszynski MH, Thomas RG, Barba D, Brewer JB, Rissman RA, Siffert J, Aisen PS, Team ANS. Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with Alzheimer disease: a randomized clinical trial. JAMA Neurol. 2018;75(7):834–41.
Longo FM, Massa SM. Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat Rev Drug Discov. 2013;12:507–25.
Massa SM, Xie Y, Yang T, Harrington AW, Kim ML, Yoon SO, Kraemer R, Moore LA, Hempstead BL, Longo FM. Small, nonpeptide p75NTR ligands induce survival signaling and inhibit proNGF-induced death. J Neurosci. 2006;26:5288–300.
Tep C, Lim TH, Ko PO, Getahun S, Ryu JC, Goettl VM, Massa SM, Basso M, Longo FM, Yoon SO. Oral administration of a small molecule targeted to block proNGF binding to p75 promotes myelin sparing and functional recovery after spinal cord injury. J Neurosci. 2013;33:397–410.
Knowles JK, Simmons DA, Nguyen TV, Vander Griend L, Xie Y, Zhang H, Yang T, Pollak J, Chang T, Arancio O, et al. Small molecule p75NTR ligand prevents cognitive deficits and neurite degeneration in an Alzheimer's mouse model. Neurobiol Aging. 2013;34:2052–63.
Nguyen TV, Shen L, Vander Griend L, Quach LN, Belichenko NP, Saw N, Yang T, Shamloo M, Wyss-Coray T, Massa SM, Longo FM. Small molecule p75NTR ligands reduce pathological phosphorylation and misfolding of tau, inflammatory changes, cholinergic degeneration, and cognitive deficits in AbetaPP(L/S) transgenic mice. J Alzheimers Dis. 2014;42:459–83.
Simmons DA, Knowles JK, Belichenko NP, Banerjee G, Finkle C, Massa SM, Longo FM. A small molecule p75NTR ligand, LM11A-31, reverses cholinergic neurite dystrophy in Alzheimer's disease mouse models with mid- to late-stage disease progression. PLoS One. 2014;9:e102136.
Aboulkassim T, Tong XK, Tse YC, Wong TP, Woo SB, Neet KE, Brahimi F, Hamel E, Saragovi HU. Ligand-dependent TrkA activity in brain differentially affects spatial learning and long-term memory. Mol Pharmacol. 2011;80:498–508.
Scarpi D, Cirelli D, Matrone C, Castronovo G, Rosini P, Occhiato EG, Romano F, Bartali L, Clemente AM, Bottegoni G, et al. Low molecular weight, non-peptidic agonists of TrkA receptor with NGF-mimetic activity. Cell Death Dis. 2012;3:e389.
Wang J, Hancock MK, Dudek JM, Bi K. Cellular assays for high-throughput screening for modulators of Trk receptor tyrosine kinases. Curr Chem Genomics. 2008;1:27–33.
Zhang Q, Descamps O, Hart MJ, Poksay KS, Spilman P, Kane DJ, Gorostiza O, John V, Bredesen DE. Paradoxical effect of TrkA inhibition in Alzheimer's disease models. J Alzheimers Dis. 2014;40:605–17.
Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q, Massa SM, Longo FM, Katz DM. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci. 2012;32:1803–10.
Simmons DA, Belichenko NP, Yang T, Condon C, Monbureau M, Shamloo M, Jing D, Massa SM, Longo FM. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington's disease. J Neurosci. 2013;33:18712–27.
Devi L, Ohno M. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2012;37:434–44.
Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A. 2010;107:2687–92.
Zhang Z, Liu X, Schroeder JP, Chan CB, Song M, Yu SP, Weinshenker D, Ye K. 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2014;39:638–50.
Zhou W, Li X, Huang D, Zhou W, Li T, Song W. No significant effect of 7,8-dihydroxyflavone on APP processing and Alzheimer-associated phenotypes. Curr Alzheimer Res. 2015;12:47–52.
Volinsky N, Kholodenko BN. Complexity of receptor tyrosine kinase signal processing. Cold Spring Harb Perspect Biol. 2013;5:a009043.
Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14:463–77.
Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–24.
Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer's disease. Acta Neuropathol. 2013;126:461–77.
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–20.
Mathur R, Ince PG, Minett T, Garwood CJ, Shaw PJ, Matthews FE, Brayne C, Simpson JE, Wharton SB, Function MRCC, Ageing Neuropathology Study G. A reduced astrocyte response to beta-amyloid plaques in the ageing brain associates with cognitive impairment. PLoS One. 2015;10:e0118463.
Mrak RE, Griffinbc WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2001;22:915–22.
Paradisi S, Sacchetti B, Balduzzi M, Gaudi S, Malchiodi-Albedi F. Astrocyte modulation of in vitro beta-amyloid neurotoxicity. Glia. 2004;46:252–60.
Thal DR. The role of astrocytes in amyloid beta-protein toxicity and clearance. Exp Neurol. 2012;236:1–5.
Tuppo EE, Arias HR. The role of inflammation in Alzheimer's disease. Int J Biochem Cell Biol. 2005;37:289–305.
Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–74.
Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer's disease. Neurobiol Aging. 2013;34:1051–9.
von Bernhardi R, Cornejo F, Parada GE, Eugenin J. Role of TGFbeta signaling in the pathogenesis of Alzheimer's disease. Front Cell Neurosci. 2015;9:426.
Garwood CJ, Cooper JD, Hanger DP, Noble W. Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front Psychiatry. 2010;1:136.
Parachikova A, Vasilevko V, Cribbs DH, LaFerla FM, Green KN. Reductions in amyloid-beta-derived neuroinflammation, with minocycline, restore cognition but do not significantly affect tau hyperphosphorylation. J Alzheimers Dis. 2010;21:527–42.
Green HF, Nolan YM. GSK-3 mediates the release of IL-1beta, TNF-alpha and IL-10 from cortical glia. Neurochem Int. 2012;61:666–71.
Hu J, Ferreira A, Van Eldik LJ. S100beta induces neuronal cell death through nitric oxide release from astrocytes. J Neurochem. 1997;69:2294–301.
Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7.
Malinski T. Nitric oxide and nitroxidative stress in Alzheimer's disease. J Alzheimers Dis. 2007;11:207–18.
Swerdlow RH, Burns JM, Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta. 1842;2014:1219–31.
Bharadwaj PR, Bates KA, Porter T, Teimouri E, Perry G, Steele JW, Gandy S, Groth D, Martins RN, Verdile G. Latrepirdine: molecular mechanisms underlying potential therapeutic roles in Alzheimer's and other neurodegenerative diseases. Transl Psychiatry. 2013;3:e332.
Shevtsova EF, Vinogradova DV, Kireeva EG, Reddy VP, Aliev G, Bachurin SO. Dimebon attenuates the Abeta-induced mitochondrial permeabilization. Curr Alzheimer Res. 2014;11:422–9.
Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun. 2015;6:6176.
Wang A, Das P, Switzer RC 3rd, Golde TE, Jankowsky JL. Robust amyloid clearance in a mouse model of Alzheimer's disease provides novel insights into the mechanism of amyloid-beta immunotherapy. J Neurosci. 2011;31:4124–36.
Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–62.
Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosis. J Neurosci. 2010;30:17091–101.
Cederblad L, Rosengren B, Ryberg E, Hermansson NO. AZD8797 is an allosteric non-competitive modulator of the human CX3CR1 receptor. Biochem J. 2016;473:641–9.
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31.
Lopez-Lopez A, Gelpi E, Lopategui DM, Vidal-Taboada JM. Association of the CX3CR1-V249I variant with neurofibrillary pathology progression in late-onset Alzheimer's disease. Mol Neurobiol. 2018;55:2340–9.
Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–8.
Condello C, Yuan P, Grutzendler J. Microglia-Mediated Neuroprotection, TREM2, and Alzheimer's Disease: Evidence From Optical Imaging. Biol Psychiatry. 2018;83:377–87.
Singer AC, Martorell AJ, Douglas JM, Abdurrob F, Attokaren MK, Tipton J, Mathys H, Adaikkan C, Tsai LH. Noninvasive 40-Hz light flicker to recruit microglia and reduce amyloid beta load. Nat Protoc. 2018;13:1850–68.
Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, Mathys H, Seo J, Kritskiy O, Abdurrob F, et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature. 2016;540:230–5.
Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, Mathys H, Seo J, Kritskiy O, Abdurrob F, et al. Author correction: gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature. 2018;562:E1.
Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, Gyorgy B, Breakefield XO, Tanzi RE, Moir RD. Alzheimer's disease-associated beta-amyloid is rapidly seeded by Herpesviridae to protect against brain infection. Neuron. 2018;99:56–63 e53.
Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. 2016;8:340ra372.
Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, Sano M, Liang WS, Beckmann ND, Price ND, et al. Multiscale analysis of independent Alzheimer's cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 2018;99:64–82 e67.
Buckley JS, Salpeter SR. A risk-benefit assessment of dementia medications: systematic review of the evidence. Drugs Aging. 2015;32:453–67.
Johnson JW, Kotermanski SE. Mechanism of action of memantine. Curr Opin Pharmacol. 2006;6:61–7.
Olivares D, Deshpande VK, Shi Y, Lahiri DK, Greig NH, Rogers JT, Huang X. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer's disease, vascular dementia and Parkinson's disease. Curr Alzheimer Res. 2012;9:746–58.
Patel L, Grossberg GT. Combination therapy for Alzheimer's disease. Drugs Aging. 2011;28:539–46.
Deardorff WJ, Shobassy A, Grossberg GT. Safety and clinical effects of EVP-6124 in subjects with Alzheimer's disease currently or previously receiving an acetylcholinesterase inhibitor medication. Expert Rev Neurother. 2015;15:7–17.
Thomsen MS, Hansen HH, Timmerman DB, Mikkelsen JD. Cognitive improvement by activation of alpha7 nicotinic acetylcholine receptors: from animal models to human pathophysiology. Curr Pharm Des. 2010;16:323–43.
Wilkinson D, Windfeld K, Colding-Jorgensen E. Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer's disease (LADDER): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2014;13:1092–9.
Atri A, Frolich L, Ballard C, Tariot PN, Molinuevo JL, Boneva N, Windfeld K, Raket LL, Cummings JL. Effect of Idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: three randomized clinical trials. JAMA. 2018;319:130–42.
Grove RA, Harrington CM, Mahler A, Beresford I, Maruff P, Lowy MT, Nicholls AP, Boardley RL, Berges AC, Nathan PJ, Horrigan JP. A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer's disease. Curr Alzheimer Res. 2014;11:47–58.
Haig GM, Pritchett Y, Meier A, Othman AA, Hall C, Gault LM, Lenz RA. A randomized study of H3 antagonist ABT-288 in mild-to-moderate Alzheimer's dementia. J Alzheimers Dis. 2014;42:959–71.
Youdim MB. The path from anti Parkinson drug selegiline and rasagiline to multifunctional neuroprotective anti Alzheimer drugs ladostigil and m30. Curr Alzheimer Res. 2006;3:541–50.
Bansal Y, Silakari O. Multifunctional compounds: smart molecules for multifactorial diseases. Eur J Med Chem. 2014;76:31–42.
Weinreb O, Amit T, Bar-Am O, Yogev-Falach M, Youdim MB. The neuroprotective mechanism of action of the multimodal drug ladostigil. Front Biosci. 2008;13:5131–7.
Weinreb O, Amit T, Bar-Am O, Youdim MB. Ladostigil: a novel multimodal neuroprotective drug with cholinesterase and brain-selective monoamine oxidase inhibitory activities for Alzheimer's disease treatment. Curr Drug Targets. 2012;13:483–94.
Youdim MB. Multi target neuroprotective and neurorestorative anti-Parkinson and anti-Alzheimer drugs ladostigil and m30 derived from rasagiline. Exp Neurobiol. 2013;22:1–10.
Hongpaisan J, Sun MK, Alkon DL. PKC epsilon activation prevents synaptic loss, Abeta elevation, and cognitive deficits in Alzheimer's disease transgenic mice. J Neurosci. 2011;31:630–43.
Grant SG, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–10.
Kojima N, Ishibashi H, Obata K, Kandel ER. Higher seizure susceptibility and enhanced tyrosine phosphorylation of N-methyl-D-aspartate receptor subunit 2B in fyn transgenic mice. Learn Mem. 1998;5:429–45.
Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2001;276:693–9.
Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–57.
Isosaka T, Hattori K, Kida S, Kohno T, Nakazawa T, Yamamoto T, Yagi T, Yuasa S. Activation of Fyn tyrosine kinase in the mouse dorsal hippocampus is essential for contextual fear conditioning. Eur J Neurosci. 2008;28:973–81.
Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, Robinson SA, Gunther EC, van Dyck CH, Nygaard HB, Strittmatter SM. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. 2015;77:953–71.
Nygaard HB, van Dyck CH, Strittmatter SM. Fyn kinase inhibition as a novel therapy for Alzheimer's disease. Alzheimers Res Ther. 2014;6:8.
Um JW, Strittmatter SM. Amyloid-beta induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion. 2013;7:37–41.
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–97.
Nygaard HB, Wagner AF, Bowen GS, Good SP, MacAvoy MG, Strittmatter KA, Kaufman AC, Rosenberg BJ, Sekine-Konno T, Varma P, et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer's disease. Alzheimers Res Ther. 2015;7:35.
Prickaerts J, Heckman PRA, Blokland A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer's disease. Expert Opin Investig Drugs. 2017;26:1033–48.
Garcia-Osta A, Cuadrado-Tejedor M, Garcia-Barroso C, Oyarzabal J, Franco R. Phosphodiesterases as therapeutic targets for Alzheimer's disease. ACS Chem Neurosci. 2012;3:832–44.
Arai H, Takahashi T. A combination therapy of donepezil and cilostazol for patients with moderate Alzheimer disease: pilot follow-up study. Am J Geriatr Psychiatry. 2009;17:353–4.
Ihara M, Nishino M, Taguchi A, Yamamoto Y, Hattori Y, Saito S, Takahashi Y, Tsuji M, Kasahara Y, Takata Y, Okada M. Cilostazol add-on therapy in patients with mild dementia receiving donepezil: a retrospective study. PLoS One. 2014;9:e89516.
Taguchi A, Takata Y, Ihara M, Kasahara Y, Tsuji M, Nishino M, Stern D, Okada M. Cilostazol improves cognitive function in patients with mild cognitive impairment: a retrospective analysis. Psychogeriatrics. 2013;13:164–9.
Sperling RA, Jack CR Jr, Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med. 2011;3:111cm133.
Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer's disease: an analysis of population-based data. Lancet Neurol. 2014;13:788–94.
Hsu D, Marshall GA. Primary and secondary prevention trials in Alzheimer disease: looking Back, Moving Forward. Curr Alzheimer Res. 2017;14:426–40.
Sperling RA, Jack CR Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association research roundtable workgroup. Alzheimers Dement. 2011;7:367–85.
Kozauer N, Katz R. Regulatory innovation and drug development for early-stage Alzheimer's disease. N Engl J Med. 2013;368:1169–71.
Forette F, Seux ML, Staessen JA, Thijs L, Birkenhager WH, Babarskiene MR, Babeanu S, Bossini A, Gil-Extremera B, Girerd X, et al. Prevention of dementia in randomised double-blind placebo-controlled systolic hypertension in Europe (Syst-Eur) trial. Lancet. 1998;352:1347–51.
Satizabal C, Beiser AS, Seshadri S. Incidence of dementia over three decades in the Framingham heart study. N Engl J Med. 2016;375:93–4.
McGuinness B, Craig D, Bullock R, Passmore P. Statins for the prevention of dementia. Cochrane Database Syst Rev. 2009;(2):CD003160. https://doi.org/10.1002/14651858.CD003160.pub2.
Heart Protection Study Collaborative G. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22.
Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM, Ford I, Gaw A, Hyland M, Jukema JW, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–30.
Launer LJ, Miller ME, Williamson JD, Lazar RM, Gerstein HC, Murray AM, Sullivan M, Horowitz KR, Ding J, Marcovina S, et al. Effects of intensive glucose lowering on brain structure and function in people with type 2 diabetes (ACCORD MIND): a randomised open-label substudy. Lancet Neurol. 2011;10:969–77.
Dineley KT, Jahrling JB, Denner L. Insulin resistance in Alzheimer's disease. Neurobiol Dis. 2014;72(Pt A):92–103.
Freiherr J, Hallschmid M, Frey WH 2nd, Brunner YF, Chapman CD, Holscher C, Craft S, De Felice FG, Benedict C. Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs. 2013;27:505–14.
Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38.
Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, Plymate SR, Cherrier MM, Schellenberg GD, Frey WH 2nd, Craft S. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis. 2008;13:323–31.
Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, Fishel MA, Plymate SR, Breitner JC, DeGroodt W, et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008;70:440–8.
Yarchoan M, Arnold SE. Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes. 2014;63:2253–61.
Perez MJ, Quintanilla RA. Therapeutic actions of the Thiazolidinediones in Alzheimer's disease. PPAR Res. 2015;2015:957248.
Feart C, Samieri C, Rondeau V, Amieva H, Portet F, Dartigues JF, Scarmeas N, Barberger-Gateau P. Adherence to a Mediterranean diet, cognitive decline, and risk of dementia. JAMA. 2009;302:638–48.
Morris MC, Tangney CC, Wang Y, Sacks FM, Barnes LL, Bennett DA, Aggarwal NT. MIND diet slows cognitive decline with aging. Alzheimers Dement. 2015;11:1015–22.
Morris MC, Tangney CC, Wang Y, Sacks FM, Bennett DA, Aggarwal NT. MIND diet associated with reduced incidence of Alzheimer's disease. Alzheimers Dement. 2015;11:1007–14.
Stephen R, Hongisto K, Solomon A, Lonnroos E. Physical activity and Alzheimer's disease: a systematic review. J Gerontol A Biol Sci Med Sci. 2017;72:733–9.
Lautenschlager NT, Cox KL, Flicker L, Foster JK, van Bockxmeer FM, Xiao J, Greenop KR, Almeida OP. Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA. 2008;300:1027–37.
Scarmeas N, Luchsinger JA, Schupf N, Brickman AM, Cosentino S, Tang MX, Stern Y. Physical activity, diet, and risk of Alzheimer disease. JAMA. 2009;302:627–37.
Rebok GW, Ball K, Guey LT, Jones RN, Kim HY, King JW, Marsiske M, Morris JN, Tennstedt SL, Unverzagt FW, et al. Ten-year effects of the advanced cognitive training for independent and vital elderly cognitive training trial on cognition and everyday functioning in older adults. J Am Geriatr Soc. 2014;62:16–24.
Farina N, Llewellyn D, Isaac MG, Tabet N. Vitamin E for Alzheimer's dementia and mild cognitive impairment. Cochrane Database Syst Rev. 2017;1:CD002854.
Dysken MW, Guarino PD, Vertrees JE, Asthana S, Sano M, Llorente M, Pallaki M, Love S, Schellenberg GD, McCarten JR, et al. Vitamin E and memantine in Alzheimer's disease: clinical trial methods and baseline data. Alzheimers Dement. 2014;10:36–44.
Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, Love S, Schellenberg GD, McCarten JR, Malphurs J, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA. 2014;311:33–44.
DeKosky ST, Williamson JD, Fitzpatrick AL, Kronmal RA, Ives DG, Saxton JA, Lopez OL, Burke G, Carlson MC, Fried LP, et al. Ginkgo biloba for prevention of dementia: a randomized controlled trial. JAMA. 2008;300:2253–62.
Williamson JD, Vellas B, Furberg C, Nahin R, Dekosky ST. Comparison of the design differences between the Ginkgo evaluation of memory study and the GuidAge study. J Nutr Health Aging. 2008;12:73S–9S.
Vellas B, Coley N, Ousset PJ, Berrut G, Dartigues JF, Dubois B, Grandjean H, Pasquier F, Piette F, Robert P, et al. Long-term use of standardised Ginkgo biloba extract for the prevention of Alzheimer's disease (GuidAge): a randomised placebo-controlled trial. Lancet Neurol. 2012;11:851–9.
Fotuhi M, Mohassel P, Yaffe K. Fish consumption, long-chain omega-3 fatty acids and risk of cognitive decline or Alzheimer disease: a complex association. Nat Clin Pract Neurol. 2009;5:140–52.
Yurko-Mauro K, McCarthy D, Rom D, Nelson EB, Ryan AS, Blackwell A, Salem N Jr, Stedman M, Investigators M. Beneficial effects of docosahexaenoic acid on cognition in age-related cognitive decline. Alzheimers Dement. 2010;6:456–64.
Ngandu T, Lehtisalo J, Solomon A, Levalahti E, Ahtiluoto S, Antikainen R, Backman L, Hanninen T, Jula A, Laatikainen T, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385:2255–63.
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–9.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–9.
Panza F, Solfrizzi V, Imbimbo BP, Logroscino G. Amyloid-directed monoclonal antibodies for the treatment of Alzheimer's disease: the point of no return? Expert Opin Biol Ther. 2014;14:1465–76.
Hane FT, Robinson M, Lee BY, Bai O, Leonenko Z, Albert MS. Recent Progress in Alzheimer's disease research, part 3: diagnosis and treatment. J Alzheimers Dis. 2017;57:645–65.
Acknowledgements
We thank Margaret Zhong, Larry Zhang, Hannah Escow and Caroline Wang for critical reading of the manuscript.
Funding
DC is supported by NIH R01 (1R01AG048923) and RF1 (1RF1AG054014), by Department of Veteran Affairs BLR&D (1I01BX003380) and RR&D (1I01RX002290), as well as by New York State SCI Foundation. JC is supported by NNSFC (81771162) and RF1 (1RF1AG054014 for DC). PJ is supported by NNSFC (81771162).
Availability of data and materials
All supporting data analyzed in this study is included in this article.
Author information
Authors and Affiliations
Contributions
DC constructed the idea, participated in literature search and interpretation, as well as in manuscript writing. JC participated in literature search and manuscript writing. PJ participated in manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cao, J., Hou, J., Ping, J. et al. Advances in developing novel therapeutic strategies for Alzheimer’s disease. Mol Neurodegeneration 13, 64 (2018). https://doi.org/10.1186/s13024-018-0299-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-018-0299-8