
Abstract
Background
Duchenne muscular dystrophy (DMD) is the most common genetic muscle disease in human. We aimed to describe the genotype distribution in a large cohort of Chinese DMD patients and their delayed loss of ambulation by glucocorticoid (GC) treatments. This is to facilitate protocol designs and outcome measures for the emerging DMD clinical trials.
Results
A total of 1163 patients with DMD were recruited and genotyped. Genotype variations were categorized as large deletions, large duplications, and small mutations. Large deletions were further analyzed for those amenable to exon-skipping therapies. Participants aged 5 years or older were grouped into GC-treated and GC-naïve groups. Clinical progression among different genotypes and their responses to GC treatments were measured by age at loss of ambulation (LOA). Among the mutation genotypes, large deletions, large duplications, and small mutations accounted for 68.79%, 7.14%, and 24.07%, respectively. The mean age at diagnosis was 4.59 years; the median ages at LOA for the GC-naïve, prednisone/prednisolone-treated, and deflazacort-treated groups were 10.23, 12.02, and 13.95 years, respectively. The “deletion amenable to skipping exon 44” subgroup and the nonsense-mutation subgroup had older ages at LOA than the “other deletions” subgroup. Subgroups were further analyzed by both genotypes and GC status. All genotypes showed significant beneficial responses to GC treatment. Deletions amenable to skipping exon 44 showed a lower hazard ratio (0.155). The mean age at death was 18.57 years in this DMD group.
Conclusion
Genotype variation influences clinical progression in certain DMD groups. Beneficial responses to GC treatment were observed among all DMD genotypes. Compared with other genotypes, deletions amenable to skipping exon 44 had a lower hazard ratio, which may indicate a stronger protective effect of GC treatments on this subgroup. These data are valuable for designing future clinical trials, as clinical outcomes may be influenced by the genotypes.
Similar content being viewed by others


Background
Duchenne muscular dystrophy (DMD) is a genetic neuromuscular disorder caused by mutations in the dystrophin gene. Clinical symptoms of DMD include progressive muscle weakness in early childhood resulting in the loss of independent ambulation (LOA) before the age of 12 [1]. Often, in Becker muscular dystrophy (BMD), which is the mild allelic form with in-frame mutations of the dystrophin gene, LOA occurs after the age of 16 [1]. With the increased availability of the standard of care and glucocorticoid (GC) treatments, the average age at LOA in DMD has been delayed until after the age of 14 [2].
The recent discoveries of new therapeutic approaches to DMD have resulted in several clinical trials being conducted globally, participated by Chinese patients. We built a national registry in collaboration with the Treatment of Neuromuscular Diseases (TREAT-NMD) Network in 2012 (www.dmd-registry.com) and reported the initial genetic characterization of dystrophinopathies in China [3].
In this study, we recruited a large cohort and conducted more detailed genetic analyses on patients from nine neuromuscular centers in China. We performed detailed clinical studies of different genotypes, including those that are currently, or predicted to be, amenable to exon-skipping therapies. We compared the ages at LOA and the duration of survival to provide data for designing future clinical trials.
Results
Patient ascertainment and enrollment
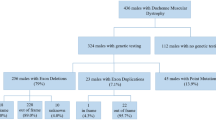
The enrollment process of this study and the patients’ genotype distribution are described in Fig. 1. We initially recruited 1548 Chinese patients with DMD. Among these participants, accurate genetic analysis results to confirm the diagnosis of DMD were not available for 173 patients, 8 patients were diagnosed using clinical features and muscle biopsy results without genetic analyses, 87 showed atypical phenotypes, and 117 had no valid follow-up data. All these patients were excluded from the study.

Participant ascertainment and genotype distribution
Statistical analyses were conducted on the data collected from the remaining 1163 participants who were genetically and clinically diagnosed. The mean age of the 1163 participants was 8.9 years (range, 0.1 to 33.7 years). All 1163 participants were male of Chinese descent. A total of 104 patients were lost to follow-up. Data analysis was based on the last valid follow-up data.
Genotype distribution of dystrophin gene mutations
Among all mutations, large deletions, large duplications, and small mutations accounted for 68.79% (800/1163), 7.14% (83/1163), and 24.07% (280/1163), respectively (Fig. 1). One patient had a combination of exon 51 deletion and exon 64–79 duplication. Of the 280 small mutations, 174 were nonsense mutations. We further divided large deletions into those with exon 3–7 deletion, those that were amenable to exon-skipping therapies (exons 44, 45, 51, and 53), and the “other deletions” subgroup, which included those deletions that could not be assigned to the aforementioned specific exon deletion patterns and were later used as the reference group for comparisons of clinical progression and responses to GC treatments. The detailed deletion patterns and the number of patients amenable to current and potential exon-skipping therapies are provided in Table 1. The patient with a single exon 52 deletion, amenable to skipping both exon 51 and exon 53, was assigned to the deletions amenable to exon 53 skipping subgroup.
Clinical progression of patients with different DMD genotypes
Age at diagnosis
The mean age at diagnosis was 4.59 years for the entire group of patients with DMD. Patients with nonsense mutations were older (5.03 years) at diagnosis than those in the “other deletions” subgroup (4.36 years), maybe suggesting a milder phenotype. No significant difference was observed among the other mutation subgroups compared with the “other deletions” subgroup. Age at diagnosis for those in the “exon 44 amenable skipping” subgroup was 4.68 years, whereas the ages of those in the “deletions amenable to skipping exon 45, skipping exon 51, and skipping exon 53” subgroups were 4.84 years, 4.41 years, and 4.35 years, respectively. Interestingly, we found that patients tended to be diagnosed earlier the more recently they were born. Approximately 48% of the patients in this study who were diagnosed before the age of 3 years had high CK and/or elevated transaminases during their routine medical examination for kindergarten enrollment, which is a widespread practice in China. In view of the recent availability of molecular diagnostic tools, we further analyzed the age at diagnosis of patients before and after 2010. In this study, 550 patients were born before 2010, and their mean age at diagnosis was 5.7 years. The other 613 patients born in or after 2010 had an average age of 3.5 years at diagnosis.
Age at LOA among the different Duchenne muscular dystrophy genotypes
During the study period, 290 patients experienced LOA. The number of participants with LOA in each mutation subgroup and the median age at LOA are shown in Table 2. Kaplan–Meier plots of the mutation groups are shown in Fig. 2. The “other deletions” subgroup had a median age at LOA of 11.03 years (n = 315; 95% CI, 10.29 to 11.77 years). Patients with deletions amenable to skipping exon 44 and nonsense mutations were significant older at LOA than those in the “other deletions” subgroup (p = 0.029 and p = 0.045, respectively). Subgroups with younger ages at LOA (but not statistically significant) included the “deletions amenable to skipping exon 45, exon 51, and exon 53” subgroups (Table 2). All eight exon 3–7 deletion patients had maintained the independent ability to ambulate at the last follow-up.

Age at LOA in the different DMD mutation subgroups. The black line indicates the “other deletions” subgroup with deletions not amenable to skipping of exons 44, 45, 51, or 53, as well as exon 3–7 deletion. The red line indicates the nonsense-mutation subgroup. The blue line indicates the “deletions amenable to skipping of exon 44” subgroup. The Navajo white line indicates the “deletions amenable to skipping of exon 45” subgroup. The cyan line refers to the “deletions amenable to skipping of exon 51” subgroup. The purple line indicates the “deletions amenable to skipping of exon 53” subgroup. The brown line indicates the exon 3–7 deletion subgroup. The “other deletions” subgroup was the reference group, and participants with deletions amenable to skipping of exon 44, exon 45, exon 51, exon 53, deletion of exon 3–7, and nonsense mutations were compared with the reference group. All participants in each mutation group were included. LOA loss of ambulation, DMD Duchenne muscular dystrophy
Mean age at death
Among all patients, 22 died during the study period. The mean age at death in this group of patients was 18.57 years (range, 13.3–33.4 years). Eleven deceased patients had experienced a pulmonary infection and died of respiratory failure, six patients died of cardiac failure, and five patients died of unknown causes. Among the 290 non-ambulatory patients in our study, only two used intermittent nocturnally assisted ventilation.
Since the participants of this cohort were much younger than those in previous studies, the number of patients who died during the study was quite small; the length of survival and Kaplan–Meier plots of different mutation groups were not further analyzed.
Differential response to GC treatment
Treatment with different GCs
Of the 979 participants older than 5 years, 530 received some form of GC treatment, 123 received an intermittent period, and 326 were GC-naïve. Among the 653 patients with known GC exposure, 71 were switched from prednisone/prednisolone to deflazacort. The most common doses were 0.9 mg/kg/day for deflazacort and 0.3 to 0.75 mg/kg/day for prednisone/prednisolone daily. Four patients received 1.5 mg/kg dosing (10 days “on” and 20 days “off”) of prednisone/prednisolone, and nine patients were administered a dose of 0.75 mg/kg/day every other day in this study. The most common side effects were cushingoid features, weight gain, growth delay, cataract, intraocular hypertension, and behavioral changes as previously reported [3].
Clinical responses to glucocorticoid treatment among the different genotypes
Among all participants aged 5 years or older, the median age at LOA was 10.23 years for the GC-naïve group, 12.02 years for patients treated with prednisone/prednisolone (hazard ratio [HR] = 0.40, p < 0.001), and 13.95 years for patients treated with deflazacort (HR = 0.06, p < 0.001). Both GC-treated groups displayed later LOA compared to the GC-naïve group (Table 2).
We further compared the responses to GC treatment among the different mutation subgroups (Table 3). A significantly older median age at LOA was observed among all mutation subgroups. An HR of 1 was assigned to GC treatment for less than 1 month, as it was used as a reference in the Cox regression model, and the p values in Table 3 represent the comparisons between the GC-treated versus GC-untreated patients in each row. Deletions amenable to skipping exon 44 showed a lower HR (0.155). Patients with deletions amenable to skipping of exons 45, 51, and 53, who seemed to have more severe phenotypes in this study, demonstrated a different response to GC treatment with a higher HR (Table 3).
Discussion
Natural history plays a paramount role in rare disease clinical trials and drug development. The United States Food and Drug Administration recommended that in single-arm interventional trials for rare diseases, natural history studies should be used as an external control [4]. In the current study, we described the genotype distribution of a cohort of Chinese patients with DMD and their responses to GC treatment. The clinical progression of the different genotype groups was measured by age at LOA.
Although detecting disease onset is an important part of the natural history of DMD, measuring the precise age at symptom onset is difficult because the delay in motor development is insidious and the symptoms progress slowly during the first few years of life. Approximately half the patients in this study who were diagnosed before the age of 3 years had high CK and/or elevated transaminases during their routine medical examination for kindergarten enrollment, which is a widespread practice in China. The average age at diagnosis of DMD is approximately 4 to 5 years worldwide [5,6,7]. The mean age at diagnosis was 4.59 years in our study, which is similar to the age of 4.43 years reported by the Parent Project Muscular Dystrophy [6]. The mean age at diagnosis in Italy was 3.4 years overall [8] and 3.5 years in patients born in or after 2010 in our study. These data indicated that patients born more recently tend to be diagnosed earlier, possibly because of the increased awareness of DMD and the availability of diagnostic tools. Several provinces in China have now included DMD in newborn screening to provide earlier diagnosis [9].
The distribution of DMD genotypes is similar worldwide. Our study showed that 75.9% of the probands had out-of-frame large deletions and duplications (multi-exon or single exon) and 24.1% had small mutations. These data corroborated the results of the TREAT-NMD DMD Global Database (80% large mutations and 20% small mutations) [10], which agrees with our previously finding [3]. The proportion of small mutations was slightly higher in this study, which could have been caused by the increased recruitment efforts to implement an ongoing clinical trial in China (PTC124-GD-041-DMD) studying patients with nonsense mutations in the dystrophin gene.
The benefits of GC treatment shown on the time function test, muscle strength, and forced vital capacity of patients with DMD were first reported in 1974 [11]. Further studies have confirmed the advantage of this treatment [12], and it has been recommended as a part of the standard care for DMD [13, 14]. As previously reported, treatment with deflazacort delayed the age at LOA by 1 year compared with treatment with prednisone/prednisolone [2, 15]. In the current study, we observed a nearly 2-year delay in LOA in the deflazacort group compared with the prednisone/prednisolone group. This is similar to the results of the NorthStar Database [16, 17].
Correlations between age at LOA and DMD genotype subgroups have been reported by previous studies [18, 19]. Exon 3–7 and single exon 45 deletions (amenable to exon 8 and exon 44 skipping) maintained longer ambulation than the other mutation subgroups. The milder phenotype was proposed to be due to endogenous exon skipping resulting in retaining partial dystrophin [20,21,22]. In our study, the age at LOA for those with deletions eligible for exon 44 skipping and those with nonsense mutations was significantly greater than that of patients in the “other deletions” subgroup. No patient with exon 3–7 deletion lost independent ambulation during the study period. Patients in the “deletions amenable to exon 45, 51, and 53 skipping” subgroups were younger at LOA, as also previously reported [23]. Therefore, genotype differences clearly influenced the clinical progression of DMD. Three mutation subgroups, i.e., the “nonsense mutations,” “exons 3–7 deletion,” and “deletions amenable to exon 44 skipping” had significantly milder clinical progression than the other subgroups. Some of these patients had such mild phenotype that they were excluded from the current study because of their “atypical” phenotype at the initial recruitment.
GC treatment has been proven beneficial for DMD. Our current study showed that all genotype subgroups benefited from GC treatment by significantly delaying LOA. Treatment with deflazacort appears to be more beneficial in delaying LOA compared with prednisone in all genotypes. However, it is possible that different mutation subgroups could show differential HR to GC treatment. Compared with other genotypes, deletions amenable to skipping exon 44 had a lower hazard ratio, which may indicate a stronger protective effect of GC treatments on this subgroup. Subgroups with severe phenotypes, such as the “deletions amenable to skipping of exon 45 and exon 51” subgroups, had a higher HR than the “deletions amenable to skipping of exon 44” regarding the response to GC treatments, which might be due to the rapid disease progression from an early age and the lack of sufficient time for the GC treatment to work. However, more evidence is needed to confirm this speculation.
With improvements in medical technology and the implementation of the standard of care, most DMD individuals are now living into their 30s and even 40s in Japan and some European countries [14, 24, 25]. Multidisciplinary intervention, including pulmonary, orthopedic, cardiac, and rehabilitation care, is currently the most effective way to enable long-term survival for patients with DMD. Currently, cultural and economic barriers have prevented parents from accepting the use of a ventilator for patients with DMD in China. Among the 290 non-ambulatory patients in our study, only two patients used intermittent nocturnal assisted ventilation. This reluctance to use assisted ventilation, coupled with the systemic difficulty in implementing the standard of care, may have contributed to the shorter survival of our study cohort. Since 2015, we have initiated a “one city, one doctor” project to promote awareness and implement the standard of care for patients with DMD in China [26]. We have seen improvements in the long-term care and survival of this group of patients.
Conclusion
In summary, this study provides detailed genotype characterization of a large cohort of Chinese patients with DMD. Detailed clinical progression within each genotype was studied by measuring age at diagnosis, age at LOA, and length of survival. We identified significant differences in clinical progression among the different DMD genotypes. The uniform positive responses to GCs confirmed the benefits of these treatments regardless of the DMD genotypes. However, the use of deflazacort clearly conferred larger clinical benefits than the use of prednisone. These data constitute an important knowledge base for designing future clinical trial protocols, as patients’ clinical outcomes may be influenced by their DMD genotypes.
Methods
Protocol approvals, registrations, and consent
This study was approved by our institutional ethics committee (No. 2015003). Nine major neuromuscular centers participated in this study (see author affiliations). Participants were recruited between March 1, 2015, and August 31, 2019. They were enrolled and followed up every year. Written or online informed consent was obtained from patients or their legal guardians. All data were entered and analyzed anonymously.
Recruitment criteria
Inclusion criteria were confirmed DMD diagnosis by genetic analysis and no participation in any clinical trial. The patients had to have shown phenotypic evidence of DMD before the age of 5, including progressive muscle weakness (proximal > distal), Gowers' sign, calf pseudohypertrophy, characteristic waddling gait, and elevated serum creatine kinase (CK).
Two groups of patients were excluded from this study since they had atypical clinical phenotypes, i.e., those who did not receive GC treatment and were able to maintain independent ambulation beyond the age of 12 and those who received GC treatment and were ambulatory beyond the age of 16. Participants who did not have valid follow-up data or could not be followed up for more than one year were also excluded.
Genotype assignments
Patients were categorized according to their genetic mutations: Large deletions (equal to or larger than one exon length), large duplications (equal to or larger than one exon length), and small mutations (including small deletions/insertions, single base-pair mutations such as missense/nonsense mutations and splice site mutations). For DMD mutation studies, single- or multi-exon out-of-frame deletions and duplications were confirmed using multiplex ligation-dependent probe amplification; then, second-generation sequencing was performed to identify small mutations. The genetic mutation results were confirmed by submission of a genetic report to the Duchenne Registry genetic counselors. Large deletions were further divided into deletions amenable to exon-skipping therapies at the exon 44, 45, 51, and 53 sites and to a group with exon 3–7 deletions. A patient with a single exon 52 deletion, amenable to skipping of both exon 51 and 53, was assigned to the “deletions amenable to exon 53 skipping” subgroup because of the emergence of golodirsen and viltolarsen treatments. Large deletions that could not be assigned to the aforementioned subgroups were included in the “other deletions” subgroup.
Monitoring the clinical progression of different genotypes
Participants were evaluated at outpatient visits every year. Those who had difficulty attending outpatient visits underwent follow-up by telephone or e-mail. We measured clinical progression by recording the age at LOA and the age at the time of death (length of survival). LOA was defined as a need for continuous wheelchair use. We collected these data from the patients' guardians during regular follow-ups.
Glucocorticoid treatments and clinical responses
In China, GC treatments for DMD usually start when the patient is between the age of 4 and 6 years before the rapid decline phase starts. Participants aged 5 years or older were assigned into three groups according to their exposure to GC: The GC-treated group, including those who received continuous GC treatment for 12 months or longer; GC-naïve group, including those who never received GC treatment or were only treated for less than 1 month; Intermittent group, including those with GC exposure that lasted longer than one month but less than 12 months. The patients in the GC group were further grouped into those who received prednisone/prednisolone and those who received deflazacort. Patients who switched between deflazacort and prednisone/prednisolone were assigned according to which treatment lasted longer. Clinical responses to these treatments among the different genotypes were measured by the age at LOA and length of survival.
Statistical analyses
Time-to-event analyses of LOA were performed to analyze the differences between groups with age (years) as the time variable and LOA as the event. The median age at LOA and corresponding 95% confidence intervals (CIs) were estimated by plotting empirical Kaplan–Meier curves for each group defined by the mutation type and by GC treatment administered while the patient was ambulatory. Cox proportional hazard models were used to estimate and compare the age-related risks of LOA. Covariates, including DMD mutations and GC drug (deflazacort or prednisone/prednisolone) treatments, were recorded. The significance level was set at p < 0.05. All analyses were conducted using the SPSS software package (SPSS 20 Inc., Chicago, IL, USA).
Availability of data and materials
The datasets used during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BMD:
-
Becker muscular dystrophy
- CGDR:
-
Chinese Genetic Disease Registry
- CI:
-
Confidence intervals
- CK:
-
Creatine kinase
- DMD:
-
Duchenne muscular dystrophy
- HR:
-
Hazard ratio
- LOA:
-
Loss of ambulation
- SPSS:
-
SPSS software
References
Brandsema JF, Darras BT. Dystrophinopathies. Semin Neurol. 2015;35:369–84.
Bello L, Gordish-Dressman H, Morgenroth LP, Henricson EK, Duong T, Hoffman EP, et al. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology. 2015;85:1048–55.
Ma P, Zhang S, Zhang H, Fang S, Dong Y, Zhang Y, et al. Comprehensive genetic characteristics of dystrophinopathies in China. Orphanet J Rare Dis. 2018;13:109.
Wu J, Wang C, Toh S, Pisa FE, Bauer L. Use of real-world evidence in regulatory decisions for rare diseases in the United States—current status and future directions. Pharmacoepidemiol Drug Saf. 2020;29:1213–8.
Bushby KM, Hill A, Steele JG. Failure of early diagnosis in symptomatic Duchenne muscular dystrophy. Lancet. 1999;353:557–8.
Wong SH, McClaren BJ, Archibald AD, Weeks A, Langmaid T, Ryan MM, et al. A mixed methods study of age at diagnosis and diagnostic odyssey for Duchenne muscular dystrophy. Eur J Hum Genet. 2015;23:1294–300.
Soim A, Smith MG, Kwon JM, Mann JR, Thomas S, Ciafaloni E, et al. Is there a delay in diagnosis of duchenne muscular dystrophy among preterm-born males? J Child Neurol. 2018;33:537–45.
D’Amico A, Catteruccia M, Baranello G, Politano L, Govoni A, Previtali SC, et al. Diagnosis of Duchenne Muscular Dystrophy in Italy in the last decade: critical issues and areas for improvements. Neuromuscul Disord. 2017;27:447–51.
Ke Q, Zhao ZY, Griggs R, Wiley V, Connolly A, Kwon J, et al. Newborn screening for Duchenne muscular dystrophy in China: follow-up diagnosis and subsequent treatment. World J Pediatr. 2017;13:197–201.
Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395–402.
Drachman DB, Toyka KV, Myer E. Prednisone in Duchenne muscular dystrophy. Lancet. 1974;2(7894):1409–12.
McDonald CM, Henricson EK, Abresch RT, Duong T, Joyce NC, Hu F, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. 2018;391:451–61.
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93.
Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17:251–67.
Griggs RC, Miller JP, Greenberg CR, Fehlings DL, Pestronk A, Mendell JR, et al. Efficacy and safety of deflazacort vs. prednisone and placebo for Duchenne muscular dystrophy. Neurology. 2016;87:2123–31.
Ricotti V, Ridout DA, Scott E, Quinlivan R, Robb SA, Manzur AY, et al. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry. 2013;84:698–705.
Joseph S, Wang C, Bushby K, Guglieri M, Horrocks I, Straub V, et al. Fractures and linear growth in a nationwide cohort of boys with Duchenne muscular dystrophy with and without glucocorticoid treatment: results from the UK NorthStar Database. JAMA Neurol. 2019;76:701–9.
Van den Bergen JC, Ginjaar HB, Niks EH, Aartsma-Rus A, Verschuuren JJ. Prolonged ambulation in Duchenne patients with a mutation amenable to exon 44 skipping. J Neuromuscul Dis. 2014;1:91–4.
Bello L, Morgenroth LP, Gordish-Dressman H, Hoffman EP, McDonald CM, Cirak S. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology. 2016;87:401–9.
Dwianingsih EK, Malueka RG, Nishida A, Itoh K, Lee T, Yagi M, et al. A novel splicing silencer generated by DMD exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J Hum Genet. 2014;59:423–9.
Wang RT, Barthelemy F, Martin AS, Douine ED, Eskin A, Lucas A, et al. DMD genotype correlations from the Duchenne Registry: endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat. 2018;39:1193–202.
Okubo M, Noguchi S, Hayashi S, Nakamura H, Komaki H, Matsuo M, et al. Exon skipping induced by nonsense/frameshift mutations in DMD gene results in Becker muscular dystrophy. Hum Genet. 2020;139:247–55.
Pane M, Mazzone ES, Sormani MP, Messina S, Vita GL, Fanelli L, et al. 6 Minute walk test in Duchenne MD patients with different mutations: 12 month changes. PLoS ONE. 2014;9:e83400.
Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Colvin MK, et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018;17:445–55.
Saito T, Kawai M, Kimura E, Ogata K, Takahashi T, Kobayashi M, et al. Study of Duchenne muscular dystrophy long-term survivors aged 40 years and older living in specialized institutions in Japan. Neuromuscul Disord. 2017;27:107–14.
Xu EX. Professor Shi-Wen Wu: One City, One Doctor-building up the national DMD registry network. Ann Transl Med. 2015;3:204.
Acknowledgements
We thank the boys, men, and their families for their cooperation with the study, as well as all members of the supporting staff of all the departments. Thanks are also due to those individuals who were instrumental in the conduct of this study and the collection of data. We thank Dr. Ching H. Wang for critically revising the manuscript for intellectual content.
Funding
This work was supported by Research and Development of Chinese Capital Clinical Characteristic Applications (Grant No. Z151100004015025) and the National Natural Science Foundation of China (Grant No. 81930121).
Author information
Authors and Affiliations
Consortia
Contributions
SZ contributed to the study design, analyzed and interpreted data, and drafted the manuscript. DQ contributed to the study design, analyzed and interpreted data, and performed statistical analyses. LW contributed to the study enrolment and critically revised the manuscript. ML contributed to the study enrolment, analyzed and interpreted data, and revised the manuscript. LS contributed to the study design, the study enrolment, helped plan statistical analyses, and revised the manuscript. CW contributed to the study design and the study enrolment. CL contributed to the study enrolment and revised the manuscript. XZ contributed to the study enrolment and analyzed and interpreted data. SH contributed to the study enrolment and revised the manuscript. MM contributed to the study enrolment and revised the manuscript. SW contributed to study design, performed statistical analyses, analysed and interpreted data, and critically revised the manuscript for intellectual content. All authors participated in the preparation, review, and revision of the report, which has been approved by each author. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by ethics committee of General Hospital of the Chinese People's Armed Police Forces (No.2015003). Written or online informed consent was obtained from patients or their legal guardians.
Consent for publication
Consents for publication were obtained from the patients and/or their parents or their legal guardians.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, S., Qin, D., Wu, L. et al. Genotype characterization and delayed loss of ambulation by glucocorticoids in a large cohort of patients with Duchenne muscular dystrophy. Orphanet J Rare Dis 16, 188 (2021). https://doi.org/10.1186/s13023-021-01837-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-021-01837-x