
Abstract
Background
Osteogenesis imperfecta (OI) is a heterogeneous connective tissue disorder characterized by an increased tendency for fractures throughout life. Autosomal dominant (AD) mutations in COL1A1 and COL1A2 are causative in approximately 85% of cases. In recent years, recessive variants in genes involved in collagen processing have been found. Hypodontia (< 6 missing permanent teeth) and oligodontia (≥ 6 missing permanent teeth) have previously been reported in individuals with OI. The aim of the present cross-sectional study was to investigate whether children and adolescents with OI and oligodontia and hypodontia also present with variants in other genes with potential effects on tooth development. The cohort comprised 10 individuals (7.7–19.9 years of age) with known COL1A1/A2 variants who we clinically and radiographically examined and further genetically evaluated by whole-genome sequencing. All study participants were treated at the Astrid Lindgren Children’s Hospital at Karolinska University Hospital, Stockholm (Sweden’s national multidisciplinary pediatric OI team). We evaluated a panel of genes that were associated with nonsyndromic and syndromic hypodontia or oligodontia as well as that had been found to be involved in tooth development in animal models.
Results
We detected a homozygous nonsense variant in CREB3L1, p.Tyr428*, c.1284C > A in one boy previously diagnosed with OI type III. COL1A1 and COL1A2 were the only two genes among 9 individuals which carried a pathogenic mutation. We found rare variants with unknown significance in several other genes related to tooth development.
Conclusions
Our findings suggest that mutations in COL1A1, COL1A2, and CREB3L1 may cause hypodontia and oligodontia in OI. The findings cannot exclude additive effects from other modifying or interacting genes that may contribute to the severity of the expressed phenotype. Larger cohorts and further functional studies are needed.
Similar content being viewed by others
Background
Osteogenesis imperfecta (OI; MIM 166200, 166210, 259420, and 166220) is a heterogeneous connective tissue disorder characterized by an increased tendency for fractures throughout life. Other symptoms include growth deficiency, joint laxity, prolonged bleeding tendency, bruises, premature hearing loss, blue sclerae, and dentinogenesis imperfecta (DGI). The reported prevalence of OI is approximately 7 in 100,000 individuals [1]. The disease has traditionally been classified into four main types based on primary clinical findings and pattern of inheritance [2]. Severity ranges from a slightly increased fracture tendency to perinatal lethality. A new OI nomenclature comprising five main types has been proposed [3]; however, the Sillence classification is still the most frequently used.
Autosomal dominant (AD) mutations in the COL1A1 and COL1A2 genes are causative in approximately 85% of cases [4]. In addition, mutations in IFITM5 have been found with an autosomal dominant pattern of inheritance [5]. OI-causing variants often present with unique features, but the interfamilial phenotypic variability can be extensive, despite the same causing mutation [4]. In recent years, recessive variants in genes involved in collagen processing have been found [6]. Among these, Symoens et al. [7] identified a homozygous deletion in cyclic AMP responsive element-binding protein 3-like I (CREB3L1), which in one Turkish family caused a severe form of OI. CREB3L1 encodes a transcription factor which is also known as OASIS (Old Astrocyte Specifically Induced Substance).
Tooth agenesis denotes congenital absence of one or more permanent teeth. Hypodontia is defined by agenesis of fewer than six teeth, while oligodontia is congenital absence of six or more permanent teeth, excluding third molars. Tooth agenesis can occur either as an isolated trait (nonsyndromic) or as part of a syndromic trait (syndromic), which reflects the genetic and phenotypic heterogeneity of the condition [8]. The prevalence of hypodontia in the general population ranges from 6 to 8% in Nordic studies [9,10,11] with oligodontia occurring in 1–2‰ [11,12,13,14]. Mutations in several genes have been associated with nonsyndromic tooth agenesis, among others the PAX9, MSX1, AXIN2, EDA, EDARADD, and EDAR genes [15,16,17,18,19]. Tooth agenesis has previously been reported in patients with OI [20, 21]. Malmgren et al. [22] found hypodontia in 11%, and oligodontia in 6% of children and adolescents with varying severity of OI. Seventy-five percent of the individuals presenting with oligodontia harbored mutations in COL1A1 and COL1A2 that were predicted to cause qualitatively changed protein.
Tooth development is a process under strict genetic control [23,24,25]. The complex process is regulated by a number of genes including transcription factors, growth factors, signaling molecules and other intra- and extracellular molecules [26].
Phenotypic variability of individuals with mutations in COL1A1/A2 brings the question whether these individuals have other pathogenic mutations that exclusively cause tooth agenesis. In this study, we investigated 10 OI patients with and without tooth agenesis harboring a COL1A1/A2 variant in order to identify whether these individuals present with pathogenic mutations in other coding genes that have an important role in tooth development.
Subjects and methods
This cross-sectional study included 10 children and adolescents (aged 7.7–19.9 years) with OI who had participated in a previous study [22]: 7 presenting with oligodontia; 2 presenting with hypodontia; and a participant from this previous study who had OI type IV but who was newly diagnosed with oligodontia. We evaluated the cohort further genetically. 3 participants were classified as OI type I, 4 as OI type III, and 3 as OI type IV (Table 1).
All study participants had been treated at the Astrid Lindgren Children’s Hospital at Karolinska University Hospital, Stockholm (Sweden’s national multidisciplinary pediatric OI team). All children and adolescents underwent a detailed clinical and radiographic evaluation of panoramic radiographs performed by two experienced pediatric dentists (KA and BM) regarding total number of permanent tooth germs and teeth. All included individuals were aged > 7 years to enable evaluation of all permanent teeth. In cases of dental agenesis, a family history was taken. The clinical examination included recording signs of DGI.
Venous blood samples were collected from all individuals. Furthermore, seven family members of patient no.1 accepted to provide their saliva samples for segregation analysis. Genomic DNA was isolated at the Clinical Genetics Unit of Karolinska University Hospital.
Written informed consent for participation and sample collection was obtained from all recruited individuals and/or their legal guardians in accordance with the Declaration of Helsinki. The Swedish regional ethics committee in Stockholm approved the study protocol (Daybook no. 157/99, 2014/254–31, and 2012/2106–31/4).
Whole-genome sequencing and bioinformatics analysis
Libraries were prepared for sequencing on Illumina HiSeqX (Illumina Inc., San Diego, CA, USA) from the genomic DNA using the Illumina TruSeq DNA PCR-Free kit with a mean insert size of 400 bp. On average, this resulted in approximately 480 million mapped unique sequences with a mean coverage of > 37, i.e., 30x coverage for 80% of reference sequences. We used an in-house pipeline developed by the Science for Life Laboratory, Stockholm, Sweden to map reads to the human reference genome (hg19) and to call variants. Data were aligned to the reference genome using bwa (v0.7.12) [27].
We deduplicated, recalibrated, and indel realigned raw alignments using GATK (v3.3–0-geee94ec) [28]. The quality control information was gathered using Qualimap (v2.2) [29] and single-nucleotide variants (SNVs) and indels were called using the HaplotypeCaller in GATK. Supplementary Table 1 presents general statistics of WGS data to show sequencing quality. We further processed the variants with GenotypeGVCFs, VariantRecalibrator, ApplyRecalibration, VariantFiltration and SelectVariants tools in GATK (v3.7), which were then functionally annotated using Variant Effect Predictor (VEP; version 89) [30] and loaded into a database using GEMINI (v0.20.0) [31]. We followed two approaches. We excluded variants with minor allele frequency of 0.1% and higher in 1000 Genomes Project (1000G), 6500 NHLBI-GO Exome Sequencing Project (ESP), Swedish Genome Project (SweGen) [32] and Exome and Genome Aggregation Consortium (ExAC and gnomAD). Variants located in repetitive sequence regions were excluded. Only non-synonymous variants, frameshift indels, and putative splice site variants were considered for further analysis. We used Combined Annotation Dependent Depletion (CADD) to score the pathogenicity of SNVs [33]. Evolutionary conservation of variants was evaluated with Genomic Evolutionary Rate Profiling (GERP) [34]. The variants were explored in the database using built-in tools in GEMINI, and variants were visualized on Integrated Genome Viewer (IGV) [35].
Structural variants were analyzed using the FindSV pipeline (https://github.com/J35P312/FindSV) which merges calls from CNVnator V0.3.2 [36] and TIDDIT [37]. The structural variants were annotated by VEP and filtered based on the quality flag of the variant. The filtered and annotated variants were then sorted based on a local structural variant frequency database consisting of variants from 1000 healthy individuals from the SweGen project. The reads at breakpoints were visualized in the IGV. The structural variants that fall into intergenic regions as well as intronic deletions and duplications were excluded.
Based on literature reviews, in a systematic manner, we created a panel of genes that are associated with nonsyndromic and syndromic hypodontia or oligodontia as well as of genes involved in tooth development in animal models [8, 38, 39] (Supplementary Table 2). In our downstream analysis of the whole-genome sequencing (WGS) data, we also used this gene panel to investigate both single nucleotide and structural variants in genes that have roles in tooth agenesis and tooth development. Here, we applied a minor allele frequency higher than 1% for the population databases to filter out common single nucleotide variants. In the structural variant analysis, we focused only on breakpoints located in the exons of coding genes that were listed in our gene list (Supplementary Table 2).
PCR and sanger sequencing
We performed PCR and Sanger sequencing to validate variants of interest and for segregation analysis using standard PCR and Sanger sequencing protocols. Primers are available upon request.
Results
Clinical findings and confirmation of COL1A1/A2 variants
Mean age at the most recent evaluation of dental phenotype in the 10 individuals was 12.8 ± 3.7 years (range 7.7–19.9 years). Missing premolars were the most frequent finding, 92% (61/66). The individuals included into this study presented with a heterozygous variant in COL1A1, 70% (7/10) and COL1A2 (3/10). The majority of COL1A1 and COL1A2 variants were de novo mutations (Table 1). Whole genome sequencing confirmed pathogenic COL1A1/A2 mutations in 9 of the 10 individuals. One boy (patient no. 6) previously diagnosed with OI type III had been diagnosed with a splice variant in COL1A1 (c.3208-6C > T). This variant was assessed as likely benign in this investigation since it is a common single-nucleotide polymorphism (SNP) (gnomAD v2.1 MAF: 0.00068) and we concluded that it was not responsible for the patient’s OI phenotype. However, we identified a homozygous nonsense variant in CREB3L1, p.Tyr428*, c.1284C > A (NM_052854.3) (Fig. 1a). At the time of analysis, this variant was not found in any publicly available population database but is recently published [40]. It creates a stop codon, resulting in a premature termination of protein at position 428 instead of 519. The CADD score was very high (36.0) and Tyr428 is evolutionarily conserved across species (Fig. 1b). The mutation was located on the C-terminal of OASIS which is localized in the rough endoplasmic reticulum. Based on the literature, it was assessed as the most likely disease-causing variant, and we discarded the COL1A1 variant.

The CREB3L1 variant visualized on an Integrated Genome Viewer (IGV) and schematic representation of the domain structures of human OASIS. (a) The homozygous variant at chr11(hg19): 46341840 C > A in whole genome sequencing data on IGV in patient no 6. The variant is located in CREB3L1. It creates a stop codon at position 428 and causes early termination of the protein (p.Tyr428*, c.1284C > A). (b) Schematic representation of the domain structures of human OASIS. Different domains are colored in different shades of grey. Mutations identified in this study (p.Tyr428*) and identified by Keller et al. (p.Lys312del) have been shown on the protein. The mutation is located directly after the S1P recognition motif (RSLL), highlighted in grey. Multiple sequence alignment shows evolutionary conservation of Tyr428 across species
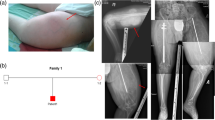
The boy harboring the CREB3L1 variant (patient no. 6) had no family history of OI or tooth agenesis. None of the four siblings were affected by OI or tooth agenesis. At the time of the oral examination, the proband presented with a mixed dentition. Investigation revealed six missing permanent tooth germs, all premolars, and a severe malocclusion including mandibular overjet, unilateral open bite, and bimaxillary crowding (Fig. 2). The boy exhibited neither clinical nor radiographic signs of DGI in the deciduous or permanent dentitions (Table 1). Further details regarding the non-dental findings in the proband have recently been described in detail [40].

Clinical and radiographic findings in a 12-year-old boy identified with a CREB3L1 variant. (a) A severe malocclusion with crowding and lateral open bite on the right side and cross-bite on the left. No clinical signs of dentinogenesis imperfecta (DGI). (b) Panoramic radiograph showing absence of six permanent tooth germs in the premolar regions. The radiograph is difficult to interpret due the extensive crowding. No signs of DGI
Genomic analyses
Rare variants in genes related to tooth development
Variants in genes known to be relevant in tooth development were found in all individuals (Table 2). Excluding COL1A1 and COL1A2, we identified 23 heterozygous variants in 21 genes. The majority (n = 21) were SNVs resulting in missense variants in highly conserved positions (Table 2). Of the identified missense variants, 17% (4/23) could be filtered out as mutations in these genes cause other conditions that were not phenotypically expressed by our probands (BMPER-diaphanospondylodysostosis with AR inheritance; GLI3-mainly developmental disorders with AD inheritance pattern; ERBB3-lethal congenital contracture syndrome 2, and PTCH1-Gorlin syndrome). These variants were thus deemed unlikely to be the cause of the patients’ symptoms.
After filtering according to our protocol, two missense variants in NOTCH2 were considered interesting from a biological perspective: one in patient no. 5 (p.(Gly1354Ala), c.4061G > C), and one in patient no. 6 (p.(Asn753Asp), c.2257A > G). According to GERP predictions, both variants occur at highly conserved positions. One frameshift variant was found in KDM5B, and a premature stop in BRPF3 was assessed as a multi-nucleotide polymorphism (MNP). Both genes are important for regulation of epigenetic events.
Variants in known genes associated with tooth agenesis and tooth development
We focused on genes (Supplementary Table 2) that were previously reported to cause hypodontia/oligodontia in humans and play a role in tooth development in mouse models [8, 38, 39]. Here, we applied a lenient filter, set the minor allele frequency (MAF) threshold to 1%, and kept rare variants. We identified 26 variants in 26 genes related to tooth development based on the literature. All individuals were carriers of rare variants in genes of interest. Of the variants, 11 were assessed as probably/possibly damaging by PolyPhen-2 prediction and were identified in 70% (7/10) individuals (Tables 3 and 4).
One boy (patient no.1) with OI type IV presented with two missense variants: in AXIN2 (rs200883019; p.Thr511Met, c.1532C > T) and in PTCH1 (p.Asp986Tyr, c.2956G > T). Both of these genes are known to be expressed during tooth development. Segregation analysis showed that the mother, the maternal grandfather, and the maternal aunt were carriers of the AXIN2 variant and presented with hypodontia.
We could not find any rare, likely pathogenic structural variants that disrupt genes involved in tooth development. Furthermore, no significant differences in distribution of variants between individuals with oligodontia and hypodontia could be seen. Except for the rare pathogenic variants in COL1A1, COL1A2, and CREB3L1, we were unable to identify any other pathogenic variant in a shared gene in the cohort that could explain the phenotype of oligodontia.
Discussion
In this study, we used WGS in our search to identify potential gene candidates that would explain the OI phenotype with presence of oligodontia. We hypothesized that individuals presenting with these features harbored variants in modifying genes related to COL1A1/A2 and tooth development. We found rare variants with unknown significance in several other genes related to tooth development.
A splice variant in COL1A1, c.3208-6C > T, a variant that was not predicted to markedly affect mRNA splicing, had previously been identified using Sanger sequencing in one of our investigated individuals (patient no. 6), a boy diagnosed with OI type III [1, 22, 41]. Using WGS, we detected a rare homozygous stop gain variant in CREB3L1 (p.Tyr428*, c.1284C > A). CREB3L1 encodes a transcription factor which is also known as OASIS (Old Astrocyte Specifically Induced Substance), an ER-stress inducer that has been found to be essential to correct expression of collagen type I by affecting transcription of COL1A1 and secretion of matrix proteins [42]. It has been shown that ER-stress causes the N-terminal part of OASIS to be transported to the nucleus where it binds to the osteoblast-specific unfolded protein response element (UPRE) regulatory region in the promoter of COL1A1. This induces a high level of tissue-specific procollagen type I expression [42].
To our knowledge, this is the first time the dental and maxillofacial phenotype of an individual with a CREB3L1 variant has been described in detail. Our patient presented with oligodontia and severe malocclusion including a mandibular overjet, unilateral open bite, and bimaxillary crowding. However, no DGI could be detected clinically or radiographically. We previously reported that presence of clinical and radiographic signs of DGI is associated with mutations in COL1A1 and COL1A2 that cause a qualitatively changed protein [41]. Based on our findings, we suggest that homozygous loss-of-function mutations of CREB3L1 may contribute to arrest of tooth development in specific dental developmental areas but is not enough to cause severe signs of DGI. The expression of collagen type I is seen early in tooth development. In the rat, expression has been detected in the dental epithelium and mesenchyme during the bud stage and during the cap and bell stages [43]. The secretion of predentin is initiated during the late bell stage of tooth development. This may indicate that cells are more sensitive to CREB3L1 during earlier stages of tooth development, where the altered secretion of collagen type I and its interaction with modifier genes may contribute to arrest of tooth development and cause dental agenesis. Lindahl et al. found markedly low levels of CREB3L1 mRNA in primary human osteoblast-like cell (hOB) (16%) and fibroblasts (21%), but collagen I levels were only reduced I hOBs (5–10%) indicating tissue-specific sensitivity [40]. OI-causing loss-of-function mutations in CREB3L1 have been described in two consanguineous families in two previous studies [7, 44]. These studies reported a 3-bp in-frame deletion (p.Lys312del, c.934_936delAAG) in exon 7 [44] and a homozygous whole gene deletion [7]. In the study by Keller et al. [44], heterozygous carriers of the variants were mildly affected by OI and homozygosity caused prenatal/perinatal lethal OI similar to that seen in OI type II. Except for reduced skull mineralization, no other craniofacial characteristics of probands presenting with homozygous variants in CREB3L1 have been described. However, a heterozygous carrier was found to have blue sclerae, but normal teeth [7]. Since complete loss of CREB3L1 causes prenatal/perinatal lethality as in both of the previous cases, we believe that, since the nonsense mutation is relatively close to the 3′ end of CREB3L1, the transcript might escape from nonsense-mediated decay and translate into a partially functioning protein with a missing C-terminal end.
Several signaling pathways are of specific importance in tooth development and expressed during all stages of tooth development; these are the TGFβ, Shh, FGF, Wnt, and Eda (ectodysplasin) pathways. Thus, the search for presence of rare variants involved in these pathways was of specific interest in this study. Another potential candidate was NOTCH2, a gene involved in the Notch signaling pathway. Two missense variants in NOTCH2 were interesting from a biological perspective: one in patient no. 5 (p.(Gly1354Ala), c.4061G > C), and one in patient no. 6 (p.(Asn753Asp), c.2257A > G). Both presented with high GERP and CADD scores. NOTCH2 is a highly conserved mammalian homologue of the Drosophila Notch gene, which encodes a transmembrane protein important for various cell fate decisions including differentiation, proliferation, and apoptosis during development. It has been shown to be expressed during several stages of tooth development in the mouse [45, 46]. Odontoblasts initiate dentinogenesis during the bell stage. Notch2 expression was not observed in odontoblasts at this stage of development [46]. None of our patients with a rare variant in NOTCH2 exhibited any clinical or radiographic signs of DGI.
One missense variant was found in GLI3 (p.Ala924Thr, c.2770G > A) in a girl (patient no. 2) with OI type IV. We also detected a PTCH1 variant (p.Asp986Tyr, c.2956G > T) in a boy (patient no. 1) with OI type IV. Both genes are involved in the Shh pathway. Shh signaling is essential in tooth development and can be detected in the oral epithelium from embryonic day 11.0 (E.11) in mice [47]. However, we assessed the PTCH1 variant to be a less likely candidate as a mutation in this gene would cause Gorlin syndrome (MIM 601309) with features including multiple nevoid basal-cell epitheliomas, jaw cysts, and bifid ribs [48]. Furthermore, the GLI3 variant could be discarded as no developmental disturbance was present. The boy (patient no. 1) also presented with a variant in AXIN2 (p.Thr511Met, c.1532C > T). The proband’s mother, grandfather, and one aunt, all carriers of the variant, were unaffected by OI but presented with hypodontia. Interestingly, none of them could be diagnosed with oligodontia. This finding indicates that the more severe phenotype seen in our proband may be due to additive effects of the variants in COL1A1 and AXIN2 and their modifying or interacting genes.
However, it is worth noting that one maternal uncle with a wild type allele of AXIN2, reported that he was missing one or two permanent teeth. This finding points to the oligogenic/polygenic characteristics of tooth agenesis and may be explained by Falconer’s polygenic threshold model. The hypothesis is based on the assumption that liability to tooth agenesis is multifactorial and follows a normal distribution (hypodontia is diagnosed in 6–8% of individuals in the general population). Hypodontia presents when a particular threshold is exceeded and is shifted in close relatives of an affected individual leading to an increased susceptibility within the family. Whole-genome sequencing of all individuals would have increased the chance for identification of more loci contributing to the phenotype. AXIN2 encodes the axis inhibition protein 2, a protein that facilitates β-catenin degradation by forming a destruction complex in the canonical Wnt signaling pathway. A nonsense mutation in AXIN2 (p.Arg656*) was previously found to cause oligodontia and a predisposition to colorectal cancer in a four-generation Finnish family [49]. Another variant (p.Trp663*, c.1989G > A) was associated with oligodontia, ectodermal dysplasia, and neoplastic syndrome [50]. Several additional mutations, mostly missense variants, have also been associated with nonsyndromic tooth agenesis [15, 51,52,53]. The AXIN2 variant in this study, having a medium impact severity, was common in the Finnish population (gnomAD v2.1 Finnish: 1.3%). MAF was also common in Northern European populations: 0.03% in the gnomAD, suggesting that it is a common polymorphism in Nordic populations.
Limitations of this study include the small sample size. Nevertheless, the included number of individuals is high within a cohort of subjects with OI and oligodontia. Interpreting functional impact of missense changes in genes involved in tooth development is challenging and functional assays are necessary to understand how these changes can potentially affect the amount, structure and function of the expressed protein and cause a dental phenotype that is variable between individuals with OI. There is also a need for segregation analysis in the investigated individuals to further decrease the list of potential gene candidates. In our bioinformatics evaluation, we focused on protein-coding sequences. We did not evaluate rare variants in non-coding genome. We found a missense variant in AXIN2, but detected no pathogenic variants in MSX1, PAX9, EDA, EDAR, or EDARADD, genes that previously have been found in nonsyndromic oligodontia. Nevertheless, we found common and rare variants in these genes, most of them located outside of coding regions.
Based on the findings in this study, OI is associated with an increased risk of hypodontia as well as oligodontia. It is important to diagnose agenesis at an early age in order to optimize treatment planning and treatment. We suggest a panoramic radiograph at 7 years of age in children and adolescents with OI. We detected oligodontia in all types of OI, indicating that severe agenesis may also be found in individuals with OI of mild severity. Furthermore, our findings indicate that the possibility of undiagnosed OI should be considered in individuals presenting with oligodontia and hypodontia. Further evaluation regarding presence of medical and dental variables associated with OI may be indicated in such evaluation, and have previously been described by us in detail [54]. If suspicion of OI remains, referral to a pediatrician is indicated.
Conclusions
In summary, we found several rare missense variants in all investigated probands. No significant differences in distribution of variants between individuals with oligodontia and hypodontia could be seen. Aside from the pathogenic variants in COL1A1, COL1A2 and CREB3L1, we were unable to identify any other mutual variant related to collagen type I that could explain the phenotype with osteogenesis imperfecta and oligodontia. Based on our findings, we suggest that the cause of the expressed phenotype is the collagen I related mutation, but that additive effects from rare variants in several other known genes participating in dental development may be important. The findings of this study provide a snapshot of and give further insight into the genetic heterogeneity of hypo/oligodontia in individuals with OI. We stress the importance of further studies, including more individuals with OI and oligodontia and OI without oligodontia to evaluate potential gene candidates further. Finally, we suggest CREB3L1 as a candidate gene to be included in the genetic investigation of individuals presenting with oligodontia.
Availability of data and materials
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AD:
-
Autosomal dominant
- CADD:
-
Combined annotation dependent depletion
- DGI:
-
Dentinogenesis imperfecta
- ER:
-
Endoplasmic reticulum
- GERP:
-
Genomic evolutionary rate profiling
- MAF:
-
Minor allele frequency
- MNP:
-
Multi-nucleotide polymorphism
- OASIS:
-
Old astrocyte specifically induced substance
- OI:
-
Osteogenesis imperfecta
- SNV:
-
Single-nucleotide variants
- WGS:
-
Whole-genome sequencing
References
Lindahl K, Astrom E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren O, Kindmark A. Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Hum Genet. 2015;23(8):1042–50.
Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16(2):101–16.
Van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 2014;164a(6):1470–81.
Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Korkko J, Prockop DJ, De Paepe A, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28(3):209–21.
Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, et al. A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012;91(2):349–57.
Marini JC, Forlino A, Bachinger HP, Bishop NJ, Byers PH, Paepe A, Fassier F, Fratzl-Zelman N, Kozloff KM, Krakow D, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052.
Symoens S, Malfait F, D'Hondt S, Callewaert B, Dheedene A, Steyaert W, Bachinger HP, De Paepe A, Kayserili H, Coucke PJ. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J Rare Dis. 2013;8:154.
Ye X, Attaie AB. Genetic basis of Nonsyndromic and Syndromic tooth agenesis. J Pediatr Genet. 2016;5(4):198–208.
Aasheim B, Ogaard B. Hypodontia in 9-year-old Norwegians related to need of orthodontic treatment. Scand J Dent Res. 1993;101(5):257–60.
Backman B, Wahlin YB. Variations in number and morphology of permanent teeth in 7-year-old Swedish children. Int J Paediatr Dent. 2001;11(1):11–7.
Haavikko K. Hypodontia of permanent teeth. An orthopantomographic study. Suom Hammaslaak Toim. 1971;67(4):219–25.
Grahnén H. Hypodontia in the permanent dentition - a clinical and genetical investigation. Odontol Revy. 1956;7:1–100.
Rolling S, Poulsen S. Agenesis of permanent teeth in 8138 Danish schoolchildren: prevalence and intra-oral distribution according to gender. Int J Paediatr Dent. 2009;19(3):172–5.
Schalk-van der Weide Y, Beemer FA, Faber JA, Bosman F. Symptomatology of patients with oligodontia. J Oral Rehabil. 1994;21(3):247–61.
Bergendal B, Klar J, Stecksen-Blicks C, Norderyd J, Dahl N. Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am J Med Gen A. 2011;155a(7):1616–22.
Han D, Gong Y, Wu H, Zhang X, Yan M, Wang X, Qu H, Feng H, Song S. Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur J Med Gen. 2008;51(6):536–46.
Hansen L, Kreiborg S, Jarlov H, Niebuhr E, Eiberg H. A novel nonsense mutation in PAX9 is associated with marked variability in number of missing teeth. Eur J Oral Sci. 2007;115(4):330–3.
Lidral AC, Reising BC. The role of MSX1 in human tooth agenesis. J Dent Res. 2002;81(4):274–8.
Liang J, Song G, Li Q, Bian Z. Novel missense mutations in PAX9 causing oligodontia. Arch Oral Biol. 2012;57(6):784–9.
Lukinmaa PL, Ranta H, Ranta K, Kaitila I, Hietanen J. Dental findings in osteogenesis imperfecta: II. Dysplastic and other developmental defects. J Craniofac Genet Dev Biol. 1987;7(2):127–35.
O'Connell AC, Marini JC. Evaluation of oral problems in an osteogenesis imperfecta population. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87(2):189–96.
Malmgren B, Andersson K, Lindahl K, Kindmark A, Grigelioniene G, Zachariadis V, Dahllof G, Astrom E. Tooth agenesis in osteogenesis imperfecta related to mutations in the collagen type I genes. Oral Dis. 2017;23(1):42–9.
Nieminen P. Genetic basis of tooth agenesis. J Exp Zool B Mol Dev Evol. 2009;312b(4):320–42.
Thesleff I. The genetic basis of tooth development and dental defects. Am J Med Genet A. 2006;140(23):2530–5.
Vastardis H. The genetics of human tooth agenesis: new discoveries for understanding dental anomalies. Am J Orthod Dentofacial Orthop. 2000;117(6):650–6.
Uchibe K, Shimizu H, Yokoyama S, Kuboki T, Asahara H. Identification of novel transcription-regulating genes expressed during murine molar development. Dev Dyn. 2012;241(7):1217–26.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2009;25(14):1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Okonechnikov K, Conesa A, Garcia-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics (Oxford, England). 2016;32(2):292–4.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics (Oxford, England). 2010;26(16):2069–70.
Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. 2013;9(7):e1003153.
Ameur A, Dahlberg J, Olason P, Vezzi F, Karlsson R, Martin M, Viklund J, Kahari AK, Lundin P, Che H, et al. SweGen: a whole-genome data resource of genetic variability in a cross-section of the Swedish population. Eur J Hum Gen. 2017;25(11):1253–60.
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15(7):901–13.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–84.
Eisfeldt J, Vezzi F, Olason P, Nilsson D, Lindstrand A. TIDDIT, an efficient and comprehensive structural variant caller for massive parallel sequencing data. F1000Research. 2017;6:664.
Balic A, Thesleff I. Tissue interactions regulating tooth development and renewal. Curr Top Dev Biol. 2015;115:157–86.
Phan M, Conte F, Khandelwal KD, Ockeloen CW, Bartzela T, Kleefstra T, van Bokhoven H, Rubini M, Zhou H, Carels CE. Tooth agenesis and orofacial clefting: genetic brothers in arms? Hum Genet. 2016;135(12):1299–327.
Lindahl K, Astrom E, Dragomir A, Symoens S, Coucke P, Larsson S, Paschalis E, Roschger P, Gamsjaeger S, Klaushofer K, et al. Homozygosity for CREB3L1 premature stop codon in first case of recessive osteogenesis imperfecta associated with OASIS-deficiency to survive infancy. Bone. 2018;114:268–77.
Andersson K, Dahllof G, Lindahl K, Kindmark A, Grigelioniene G, Astrom E, Malmgren B. Mutations in COL1A1 and COL1A2 and dental aberrations in children and adolescents with osteogenesis imperfecta - a retrospective cohort study. PLoS One. 2017;12(5):e0176466.
Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, Sekiya H, Tsumagari K, Ochiai K, Yoshinaga K, et al. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol. 2009;11(10):1205–11.
Webb PP, Moxham BJ, Ralphs JR, Benjamin M. Immunolocalisation of collagens in the developing rat molar tooth. Eur J Oral Sci. 1998;106(Suppl 1):147–55.
Keller RB, Tran TT, Pyott SM, Pepin MG, Savarirayan R, McGillivray G, Nickerson DA, Bamshad MJ, Byers PH. Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively. Genet Med. 2018;20(4):411–19.
Mitsiadis TA, Lardelli M, Lendahl U, Thesleff I. Expression of notch 1, 2 and 3 is regulated by epithelial-mesenchymal interactions and retinoic acid in the developing mouse tooth and associated with determination of ameloblast cell fate. J Cell Biol. 1995;130(2):407–18.
Mitsiadis TA, Romeas A, Lendahl U, Sharpe PT, Farges JC. Notch2 protein distribution in human teeth under normal and pathological conditions. Exp Cell Res. 2003;282(2):101–9.
Bitgood MJ, McMahon AP. Hedgehog and bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev Biol. 1995;172(1):126–38.
Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med. 1960;262:908–12.
Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74(5):1043–50.
Marvin ML, Mazzoni SM, Herron CM, Edwards S, Gruber SB, Petty EM. AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am J Med Gen A. 2011;155a(4):898–902.
Liu H, Ding T, Zhan Y, Feng H. A novel AXIN2 missense mutation is associated with non-Syndromic Oligodontia. PLoS One. 2015;10(9):e0138221.
Wong S, Liu H, Bai B, Chang H, Zhao H, Wang Y, Han D, Feng H. Novel missense mutations in the AXIN2 gene associated with non-syndromic oligodontia. Arch Oral Biol. 2014;59(3):349–53.
Yue H, Liang J, Yang K, Hua B, Bian Z. Functional analysis of a novel missense mutation in AXIN2 associated with non-syndromic tooth agenesis. Eur J Oral Sci. 2016;124(3):228–33.
Andersson K, Malmgren B, Astrom E, Dahllof G. Dentinogenesis imperfecta type II in Swedish children and adolescents. Orphanet journal of rare diseases. 2018;13(1):145.
Acknowledgments
We thank our patients and their families for participation in this study. The authors acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure, NGI, and Uppmax for providing assistance in massive parallel sequencing and computational infrastructure. The study was supported by grants from the Stockholm County Council (ALF) and Norrbacka-Eugeniastiftelsen.
Funding
The study was supported by grants from the Stockholm County Council (ALF) and Norrbacka-Eugeniastiftelsen. The funding body had no role in design of the study, collection, analysis, interpretation of data or manuscript writing. Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
Authors’ roles: KA, BM, EÅ, AN, FT and GD contributed to conception and design. KA, BM and EÅ contributed to acquisition of data. KA and FT analyzed and interpreted the data. KA and FT drafted the manuscript. All authors critically revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent for participation and sample collection was obtained from all recruited individuals and/or their legal guardians in accordance with the Declaration of Helsinki. The Swedish regional ethics committee in Stockholm approved the study protocol (Daybook no. 157/99, 2014/254–31, and 2012/2106–31/4).
Consent for publication
Consent for publication of individual photographs and radiographs was obtained.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Summary statistics of whole-genome sequencing data.
Additional file 2: Table S2.
Genes involved in tooth development that were evaluated.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Andersson, K., Malmgren, B., Åström, E. et al. Mutations in COL1A1/A2 and CREB3L1 are associated with oligodontia in osteogenesis imperfecta. Orphanet J Rare Dis 15, 80 (2020). https://doi.org/10.1186/s13023-020-01361-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01361-4