
Abstract
Tooth agenesis and orofacial clefts represent the most common developmental anomalies and their co-occurrence is often reported in patients as well in animal models. The aim of the present systematic review is to thoroughly investigate the current literature (PubMed, EMBASE) to identify the genes and genomic loci contributing to syndromic or non-syndromic co-occurrence of tooth agenesis and orofacial clefts, to gain insight into the molecular mechanisms underlying their dual involvement in the development of teeth and facial primordia. Altogether, 84 articles including phenotype and genotype description provided 9 genomic loci and 26 gene candidates underlying the co-occurrence of the two congenital defects: MSX1, PAX9, IRF6, TP63, KMT2D, KDM6A, SATB2, TBX22, TGFα, TGFβ3, TGFβR1, TGFβR2, FGF8, FGFR1, KISS1R, WNT3, WNT5A, CDH1, CHD7, AXIN2, TWIST1, BCOR, OFD1, PTCH1, PITX2, and PVRL1. The molecular pathways, cellular functions, tissue-specific expression and disease association were investigated using publicly accessible databases (EntrezGene, UniProt, OMIM). The Gene Ontology terms of the biological processes mediated by the candidate genes were used to cluster them using the GOTermMapper (Lewis-Sigler Institute, Princeton University), speculating on six super-clusters: (a) anatomical development, (b) cell division, growth and motility, (c) cell metabolism and catabolism, (d) cell transport, (e) cell structure organization and (f) organ/system-specific processes. This review aims to increase the knowledge on the mechanisms underlying the co-occurrence of tooth agenesis and orofacial clefts, to pave the way for improving targeted (prenatal) molecular diagnosis and finally to reflect on therapeutic or ultimately preventive strategies for these disabling conditions in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Developmental tooth abnormalities, including mild and more severe forms of tooth agenesis (TA), have often been reported in patients affected with orofacial clefts (OFCs) (Ranta 1986; Aspinall et al. 2014). We recently observed that the same genes whose mutations were shown to cause TA, such as MSX1 and PAX9 (Seo et al. 2013), often also contain SNPs as genetic risk factors for OFCs.
Both, TA and OFCs represent two of the most common developmental orofacial birth defects. While hypodontia—the agenesis of 1–5 teeth (excluding agenesis of third molars)—is highly prevalent (more than 5 % in some populations), severe TA—oligodontia with agenesis of 6 teeth or more (excluding agenesis of third molars)—has been estimated to affect 1 individual in 1000 worldwide (Rakhshan and Rakhshan 2015; Polder et al. 2004). For OFCs, the overall prevalence has been estimated as 1 in 700–1000 live births (Mossey and Catilla 2003). These statistics, however, do not convey the considerable variation across studies depending on the severity of the phenotype; the study design, the cohort ethnicity and the geographical location also affect the prevalence (Khalaf et al. 2014; Murthy and Bhaskar 2009; Vastardis et al. 1996). Both conditions lead to significant life-long complications that require extensive multidisciplinary treatments, and represent severe psychosocial and economic burdens for their families and for society (Mossey et al. 2009).
Based on the number of missing teeth, TA is conventionally divided into three forms: hypodontia, oligodontia and anodontia (Klein et al. 2013). Hypodontia (HD) is used for one to five missing teeth, whereas oligodontia (OD) is used for six or more missing teeth (Fig. 1). Anodontia (AD) is the most severe condition with complete lack of tooth development in the deciduous and permanent dentition (Fig. 1). As the third molars are missing in up to 20 % of the populations worldwide, making it a very common finding, these teeth are excluded from the classification (Vastardis et al. 1996; Graber 1978). Based on the severity and the anatomical regions involved, OFCs are also classified into different phenotypic categories ranging from microforms to rare complete overt facial clefts, i.e., oblique facial cleft, where the gap may extend to the nose, the cheeks, the eyes, the ears till the forehead (Fig. 2). The three main OFC phenotypes are represented by cleft lip (CL), cleft palate (CP) and cleft lip and palate (CLP), which can be uni- or bilateral (Fig. 2).

Forms of tooth agenesis. Panel of tooth agenesis (TA) forms in the permanent dentition, listed according to the number of absent teeth. Frontal intraoral pictures and orthopantograms (OPTs) of two adult patients affected with hypodontia, a without cleft and b with cleft (repaired cleft lip involving the alveolar ridge, marked by dashed blue circle), respectively. Frontal intraoral pictures and OPTs of two adult patients affected by oligodontia, c without cleft and d with cleft (repaired cleft lip and palate involving the alveolar ridge, marked by dashed blue circle), respectively. e Internal intraoral pictures (maxillary dental arch, left; mandibular dental arch, right) and OPT of an adult patient affected by complete anodontia, without orofacial clefts (copyright: Wang et al. 2013). X-axis: presence or absence of orofacial cleft in combination with TA. Y-axis: number of absent teeth (hypodontia, 1–5 missing teeth; oligodontia 6–31 missing teeth; anodontia, 32 missing teeth)

Forms of orofacial clefts. Panel of orofacial cleft forms, listed according to the severity based on the cleft extension and orofacial regions affected. Cleft lip types (frontal views): microform (a) (copyright: Cleft lip—A comprehensive review. Shkoukani et al., Front Pediatr. 2013); unilateral incomplete cleft lip (b); bilateral incomplete cleft lip (c); unilateral complete cleft lip (d); bilateral complete cleft lip (e). Cleft palate types (occlusal views): bifid uvula (f); cleft of the soft palate (g); cleft of hard and soft palate (h). Unilateral cleft lip and palate (i): frontal view of the patient in childhood and occlusal view of the same patient in adulthood, where the cleft palate has been repaired (surgical scars marked with blue arrows). Bilateral cleft lip and palate (j): frontal view of the patient in childhood, with protruding vermilion, and occlusal view of the same patient in adulthood, where the cleft palate is still partially open. Unilateral facial cleft extending from the oral region till the eye (K) (copyright: Garg and Goyal 2009). X-axis: type of orofacial cleft. Y-axis: severity based on the cleft extension (intraoral region, perioral region, whole face)
In CL, the nasal and lip primordia fail to fuse resulting in a gap of the upper lip and the disruption of the orbicularis oris muscle, with a variable degree of severity ranging from microforms, i.e., forme fruste CL, to complete unilateral or bilateral clefting. CP is characterized by either a submucosal or an overt cleft in the anterior hard palate or posterior soft palate, with variable disorientation of palatal muscles, arising from the fusion failure of lateral palatal shelves. The mildest form of soft CP involves only the uvula, while in the most severe cases the cleft extends through soft and secondary hard palate. CLP is a combination of the previously described phenotypes, usually divided into two classes: incomplete CLP (a.k.a. cleft lip and alveolus) when the upper lip, alveolar ridge and part of the hard palate (primary palate) are affected, or complete CLP, when the cleft develops along the entire mouth length from the nostrils to the uvula. Despite their common features, CLP, CP and CL emerge from the disruption of distinct morphogenetic processes at different stages of embryological development (Shkoukani et al. 2013).
Both TA and OFCs can occur as isolated conditions without any other recognizable anomaly (non-syndromic forms) or associated with structural abnormalities of other anatomical regions (syndromic forms) (Cobourne 2004; Klein et al. 2013). Over 80 syndromes include TA among their typical features, especially HD, while over 275 syndromes include at least one of the different subtypes of OFCs (Klein et al. 2013; Leslie and Marazita 2013). Interestingly, syndromic forms of TA and OFCs may arise within the same syndromes: this is the case for van der Woude syndrome (VWS) (OMIM# 119300), which includes OFCs with dental anomalies and lip fistulas (Kondo et al. 2002).
In a recent comprehensive study based on the largest international cohort of individuals with OFC investigated so far (Howe et al. 2015), it has been shown that a wide spectrum of dental anomalies, characterized by alteration in tooth number, size, shape, timing of formation and eruption, is more frequently detected in individuals with OFC than in the population without these birth defects, although this evidence is restricted to the upper jaw. The prevalence of TA in and outside the cleft area, as well as its location in the upper versus lower jaw, has been reported to be significantly higher in patients with OFC compared to individuals without a cleft (Shapira et al. 1999; Aspinall et al. 2014). TA has been described to occur approximately three times more frequently on the cleft than on the non-cleft side (Ranta 1972), and its severity increases with the OFC phenotype severity (Ranta 1986). The cause of the co-occurrence of these dental abnormalities and OFCs has also been debated. According to Howe et al. (2015), the dental features may result from local mechanical circumstances at the time of the cleft formation or from conditions of blood supply during early postnatal surgical interventions.
In their geometric morphometric study in a Neo/Null and Neo/Wt mouse model, Green et al. (2015) show that the facial/nasal prominences can fail to fuse due to their misalignment as a result of decreased mesenchymal growth. Failure of tooth germ development can also be caused by mutations in genes which regulate mesenchymal cell proliferation (like MSX1), fitting the common genetic origin hypothesis (Eerens et al. 2001). Such gene variants could therefore—besides causing TA—also increase the risk for OFC development, if a proper alignment of the midfacial prominences is not achieved in time. Moreover, the absence of developing tooth germ structures (like thickened dental laminas in the growing palatal processes) could itself also underlie the subtle volumetric shape changes contributing to the failure of optimal geometric alignment of the approaching orofacial prominences. In the Online Mendelian Inheritance in Man (OMIM) database an overall large genetic heterogeneity for selective TA (STHAG) is described, but so far only STHAG type 1 (OMIM# 106600) includes the annotation ‘with or without orofacial cleft’, which draws back to a heterozygous mutation affecting MSX1 (Table 1, Supplementary Table 4) (van den Boogaard et al. 2000). Combined TA and OFC phenotypes in humans have, however, been also shown to result from rare variants of IRF6 and TP63, both in syndromic and non-syndromic cases (Celli et al. 1999; McGrath et al. 2001; Brunner et al. 2002a, b; Kondo et al. 2002).
The present study aims to systematically review the literature to provide a comprehensive panel of genes and loci reported to be associated to the co-occurrence of TA and OFCs in patients (syndromic and non-syndromic cases), including supporting evidence in animal models when available. This will not only increase the knowledge on the genetic risk factors and mechanisms underlying the co-occurrence of TA and OFCs, but will also pave the way to improve (prenatal) targeted diagnosis.
Materials and methods
The literature search was systematically performed using two publicly available literature databases, PubMed (http://www.ncbi.nlm.nih.gov/pubmed) and EMBASE (https://ovidsp.tx.ovid.com/sp-3.17.0a/ovidweb.cgi), in August 2015. In each database, three separate searches were performed based on search terms belonging to three broad topics—genetics, orofacial clefts and tooth agenesis (Supplementary Table 1)—to avoid the risk of overlooking interesting articles. The individual searches were carried out using free text search combined with subject headings (Supplementary Table 1). In each database, the articles resulting from the individual searches were then overlapped to highlight only those containing terms from the three fields of interest in their abstract and title. Next, the final lists of overlapping articles from PubMed and EMBASE were both exported into EndNote X7 (Thomson Reuters, http://endnote.com), where the duplicates were removed and the article texts were retrieved.
In the first selection phase, the non-English language studies were excluded as well as the conference and meeting reports. Subsequently, the remaining articles were entirely screened and hence selected according to the inclusion criteria. In principle, the articles were included when describing evidence of genes or genetic loci—in human or in animal models—whose disruption may cause orofacial clefts (OFCs), specifically CL, CP or CL/P, and tooth agenesis (TA), including AD, OD or HD (especially located outside the cleft area), with or without other phenotypes. The evidence that leads to the inclusion of articles was based on phenotyping using clinical examination, X-rays, or histology in case of animal experiments, and on genotyping such as polymerase chain reaction and genome-wide association studies. The lack of molecular diagnosis, the absence of OFC or TA or the unclear phenotype description was reason enough to exclude an article. The authors M.P. and F.C. of this review first carried out the content-based selection of the articles individually while the disagreements about the study eligibility were solved by discussion and further careful check of the published data. In case both first authors found uncertainty in classifying an article, the authors of that article were contacted to ask for further clarifications before deciding on its inclusion or exclusion.
The molecular pathways, cellular functions, tissue-specific expression and disease association of the candidate genes collected from the included articles were investigated using publicly accessible databases, such as EntrezGene (www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene), UniProt (www.uniprot.org/) and OMIM (http://www.omim.org/), highlighting the aspects that further support the hypothesis of association between the genes and the co-occurrence of OFCs and TA. In addition, the Gene Ontology terms indicating the biological processes mediated by these candidate genes were used to cluster them using the GO tool names GOTermMapper (Lewis-Sigler Institute for Integrative Genomics, Princeton University, http://go.princeton.edu/cgi-bin/GOTermMapper) based on the map2slim script, part of the GO Perl package (Boyle et al. 2004; Harris et al. 2004). This tool maps the granular GO annotations for each gene to a set of broad, high-level GO parent terms (GO-slim terms), allowing to bin the genes into general categories, which can eventually be summarized in even broader super-clusters.
Apart from genes, genomic loci were also collected: for each locus, the genomic coordinates were defined using UCSC Genome Browser (https://genome.ucsc.edu/index.html) and the encompassed genes (RefSeq genes) were retrieved with Table Browser, setting GRCh38/hg38 as the human genome assembly.
Results
Inclusion and exclusion of articles in our study and dataflow chart
Our systematic search of the literature initially yielded 347 unique articles, of which 263 had to be excluded due to incompliance with the inclusion criteria (as to language, origin, availability or content) (Fig. 3; Supplementary Table 2). Based on phenotype details provided by the authors of five articles, three of them were included and two were excluded (Fig. 3). Hence, 84 articles of which fifteen reviews, three GeneReviews and one editorial, in addition to research articles and research letters, were finally included (Supplementary Table 3). Five selected articles describing studies that do not confirm the association between specific genes and the combination of OFCs and TA were also included and were classified as negative evidence.

Search flowchart. The literature search was performed using PubMed, which provided 166 articles, and EMBASE, which provided 281 articles, combining to a total of 447 articles. After the removal of duplicates (100), the selection process was carried out in two steps. In the first selection, the references where screened based on the document specifics: non-English articles (20), conference reports (10) and not available articles (10) were removed. The second selection of the remaining 307 articles was based on the contents, considering the molecular diagnosis and the combined phenotypes (TA and OFCs) present in patients and animal models, excluding 221 articles. For five articles the authors were contacted, and three of them were subsequently included. The final number of selected articles was 84, including research articles, case reports, research letters and reviews
From these 84 references, we identified 26 genes and 9 genomic loci presenting different types of evidences, ranging from borderline to significant associations even confirmed in animal models in some cases. The 26 candidate genes are described according to the evidence available in the current literature.
Msx1 and pax9
MSX1 and its main protein–protein interactor PAX9 are both transcription factors, members of the homeoprotein families which are co-expressed during craniofacial development and in different stages of tooth morphogenesis (Ogawa et al. 2005, 2006; Nakatomi et al. 2010). MSX1 encodes a member of the muscle segment homeobox gene family, which acts as a transcriptional repressor during embryogenesis via the core transcription complex and other homeoproteins. MSX1 has been proven through mouse models and molecular and biochemical analyses on human tissues to play a main role in limb-pattern formation, tumor growth inhibition and craniofacial development, particularly in odontogenesis (EntrezGene; Davidson 1995; Lallemand et al. 2005; Park et al. 2005; Ogawa et al. 2006).
The MSX1 signaling loop also involves other essential homeobox genes, such as BMP genes, hence mediating the reciprocal epithelial–mesenchymal tissue interaction and regulating the development of both the craniofacial skeleton and the teeth (Zhang et al. 2002; De Coster et al. 2007). Although our systematic literature search did not identify any study proving evidence of association between BMP genes and the co-occurrence of the features discussed, it would be intriguing to further investigate this pathway, since the BMP gene family includes proposed OFC-causing genes (Ogawa et al. 2006; Lin et al. 2008; Suzuki et al. 2009; He et al. 2010; Suazo et al. 2010; Sahoo et al. 2011; Williams et al. 2012; Zawiślak et al. 2014; Liu et al. 2005) as well as genes involved in early tooth development, which disruption may result in tooth agenesis (Tompkins 2006; De Coster et al. 2007).
MSX1 mutations are associated with the non-syndromic co-occurrence of CP and TA, especially HD, in humans (Table 1; Supplementary Table 4) (Carey and Viskochil 2002; Lidral and Reising 2002; Slayton et al. 2003; Vieira 2003; Wong and Hagg 2004; Modesto et al. 2006; Wilkie 2009; Kouskoura et al. 2011; Liang et al. 2012; Leslie and Marazita 2013). Similarly, Msx1-deficient mice exhibit severe craniofacial abnormalities, including clefting of the secondary palate and lack of teeth (Table 3) (Satokata and Maas 1994; Kavitha et al. 2010; Nakatomi et al. 2010).
Nowadays, a variable combination of selective TA with OFC (STHAG1) (OMIM# 106600) has been characterized in three affected members of a Dutch family whose genotyping revealed a heterozygous MSX1 stop mutation inherited across generations (Table 1; Supplementary Table 4) (van den Boogaard et al. 2000). Later, a similar combined phenotype has been described as co-segregating with a different MSX1 missense mutation in a Chinese family (Table 1; Supplementary Table 4) (Liang et al. 2012), supporting the hypothesis of the dual role of this gene in the etiology of TA and OFCs.
Even though MSX1 mutations are known to cause non-syndromic OFCs and TA, Nieminen et al. (2003) described the case of a patient with Wolf–Hirschhorn syndrome (WHS) (OMIM# 194190) due to a complete deletion of the MSX1 gene (Table 2; Supplementary Table 4), which is located in the deleted region in chromosome 4p, whose craniofacial features included CP as well as TA (Paradowska-Stolarz 2014).
Mutations of PAX9, the main protein–protein interactor of MSX1, have also been described as potentially causative for combined OFCs and TA. Specifically, PAX9 is a member of the paired box family of transcription factors, which plays critical roles in embryogenesis, mainly skeletogenesis, tooth formation, palatogenesis and neural tube development (EntrezGene; Balling et al. 1996; Peters et al. 1998a; Hamachi et al. 2003; Hu et al. 2014; Monsoro-Burq 2015). Genetic disturbances of MSX1 and PAX9 are associated with TA, located both inside and outside the cleft area (Seo et al. 2013). In mouse, Pax9 and Msx1 are co-expressed during craniofacial development, and in double-mutant mice for these two genes, incompletely penetrant CL and absence of lower incisors have been reported (Table 3) (Nakatomi et al. 2010), suggesting that reduction of PAX9 and MSX1 gene dosage in humans may increase the risk for combined OFC and TA. However, this hypothesis was not confirmed in the study of Tallon-Walton et al. (2010).
Focusing on PAX9 only, the first evidence of PAX9 association with TA and OFCs arose from a Pax9 −/− knockout mouse model described by Peters et al. (1998b), and was later confirmed in human by the study from Das et al. (2003) who reported a novel PAX9 missense mutation and an exonic insertion in families with autosomal dominant TA where some of the members also showed CL/P (Table 3; Supplementary Table 4) (Kist et al. 2007; Kavitha et al. 2010).
Irf6
The IRF6 gene encodes a member of the interferon regulatory transcription factor family; more specifically, the only member that is not related to immunological and inflammatory functions, but with morphogenesis, especially oral ectoderm and periderm formation, lip formation and spatio-temporal regulation of palatal shelf migration, adhesion and fusion (Richardson et al. 2009; Kousa and Schutte 2015). IRF6 mutations are recognized as primary genetic causes of isolated and syndromic OFCs (Kondo et al. 2002; Zucchero et al. 2004; Blanton et al. 2005; Ingraham et al. 2006; Park et al. 2007; Beaty et al. 2010; Ludwig et al. 2012).
The most common OFC syndrome is the van der Woude syndrome (VWS) (OMIM# 119300), which represents 2 % of all syndromic CL/P. In 68 % of the cases, this syndrome is caused by IRF6 mutations or deletions (Sander et al. 1995; Schutte et al. 1999; Kondo et al. 2002; de Lima et al. 2009). The dominant traits with variable expressivity and low penetrance are OFCs, HD and lip pits usually present in combination (Schinzel and Klausler 1986; Wienker et al. 1987). A number of studies describing IRF6 missense, frameshift or stop mutations causing VWS in patients showing the co-occurrence of CL/P and/or CP and TA have been found in our literature search, resulting in a list of more than 33 cases, some of them belonging to VWS families (Table 2, Supplementary Table 4) (Vieira 2003; Wang et al. 2003; Ghassibé et al. 2004; Item et al. 2004; Wong and Hagg 2004; Ye et al. 2005; Peyrard-Janvid et al. 2005; Minones-Suarez et al. 2012; Klein et al. 2013; Peyrard-Janvid et al. 2014). As further confirmation, another case report presented two patients with VWS belonging to the same family with the typical features of this syndrome, including both CL/P and HD. However, in this specific case, the gene appears fully missing as encompassed by a large deletion inherited in the affected members of this family (Wong et al. 1999) (Table 4; Supplementary Table 6). Since this deletion, del(1)(q32), encompasses 198 genes in total, the contribution of other genes located within the deleted region cannot be excluded (Supplementary Table 6).
In contrast, a study by Ali et al. (2009) failed to report the association between IRF6 markers and this syndrome in a cohort of Indian VWS families, supporting the evidence that other genes may contribute to the etiology of this syndrome, such as GHRL3.
Apart from the VWS, different mutations in the same gene lead to another syndrome associated with OFCs, named popliteal pterygium syndrome (PPS) (OMIM# 119500) (Kondo et al. 2002), which shares some clinical features of VWS with the addition of webbed skin of the legs, genital malformations and oral synechiae. From our literature search, a PPS family was found based on the combination of OFC and TA in one affected member due to an inherited IRF6 mutation (Table 2; Supplementary Table 4) (Peyrard-Janvid et al. 2005, 2014).
Furthermore, the contribution of IRF6 variation to non-syndromic OFCs has been sturdily proven. Originally, a GWAS study identified the IRF6 region as a susceptibility locus for non-syndromic OFCs (Beaty et al. 2010), which has been later confirmed by several further studies in human and in mouse models. The role of IRF6 in non-syndromic OFCs in combination with TA located outside the cleft area was thoroughly investigated by Letra et al. (2012) in a cohort of 134 Brazilian patients affected by both these conditions, thus identifying a borderline-associated IRF6 marker (rs658860) in the sub-group of subjects showing CP and TA (Table 1; Supplementary Table 4). As further evidence, a statistically significant association was found between co-occurring OFCs and TA and an SNP in the AP-2α binding site of the IRF6 promoter in a large study based on 93 Latvian patients with isolated OFCs (Table 1) (Krasone et al. 2014). On the contrary, Pegelow et al. (2008) did not find any significant association between different IRF6 SNPs and non-syndromic CL/P in 17 Swedish OFC families that included 13 members affected with OFC, further supporting the hypothesis of a minor contribution of other genes to the pathogenesis of these conditions.
Tp63
The IRF6 gene is one of the main targets of another transcription factor, p63 (tumor protein 63). Disruption of a p63-binding site upstream to IRF6 due to a small insertion has been seen to cause VWS in a family where the IRF6 gene was not mutated (Fakhouri et al. 2014), proving that the syndrome may be caused by an upstream disruption which does not directly affect the causative gene sequence.
TP63 encodes for a member of the p53 family of transcription factors, named p63, for which unlike p53, a role in tumorigenesis has not been defined so far, while its role in proliferation, development and commitment to stratified epithelial tissues has been extensively characterized in humans as well as in animal models (EntrezGene; UniProt; Yang et al. 1998). Tp63 −/− knockout mice show typical developmental defects in epithelium-related structures including skin, hair, limbs, palate and mammary glands (Mills et al. 1999; Yang et al. 1999). In humans, the disruption of TP63 regulation leads to abnormalities of the skin, the limb and the orofacial structure, resulting from the impaired transcription of its targets which include not only IRF6 but also other cleft-associated genes, such as TFAP2α and RIPK4 (McDade et al. 2012; Mitchell et al. 2012). Mutations in the TP63 gene itself have been associated with multiple syndromes, called p63 syndromes: ectrodactyly-ectodermal dysplasia-clefting (EEC) (OMIM# 129900), split-hand/foot malformation type 4 (SHFM4) (OMIM# 605289), ankyloblepharon-ectodermal dysplasia-cleft syndrome (AEC) (OMIM# 106260), acro-dermato-ungual-lacrimal-tooth syndrome (ADULT) (OMIM# 103285), limb-mammary syndrome (LMS) (OMIM# 603543) and Rapp–Hodgkin syndrome (RHS) (OMIM# 129400). Of these, the EEC syndrome most frequently shows co-occurrence of OFCs and TA (Itin and Fistarol 2004; Kouskoura et al. 2011; Tadini et al. 2013). In our systematic search, TP63 mutations have been seen to likely contribute to the syndromic co-occurrence of TA and OFCs, in relation to different p63 syndromes.
In 2010, two studies described novel TP63 mutations in six patients with EEC exhibiting OFCs and HD (Table 2; Supplementary Table 4) (Clements et al. 2010; Yin et al. 2010). One year later, an editorial by Sripathomsawat et al. (2011) reviewed two Thai patients with EEC and six previously published Dutch families focusing mainly on the oral and dental features, with particular attention on OFCs and TA.
Cabanillas et al. (2011) characterized one patient showing a combination of B cell leukemia and ectodermal dysplasia including CP and TA, theoretically caused by a pathogenic maternally inherited heterozygous germline mutation of the TP63 gene (Table 2; Supplementary Table 4). The review by Tadini et al. (2013) focused on TP63-related diseases, describing CL/P and TA or anodontia (AD) as a typical feature of RHS while CP with or without bifid uvula and TA as a hallmark of LMS syndrome. The core clinical features of the LMS were defined upon the investigation of a large Dutch family, in which affected individuals were characterized by severe limb and gland anomalies, CP and TA (van Bokhoven et al. 1999). The genetic defect was mapped to the subtelomeric region of chromosome 3q, which led to the identification of causative TP63 mutations in EEC syndrome, and subsequently related conditions including LMS.
Another syndrome-causing TP63 mutation was defined by McGrath et al. (2001) who reported on an AEC family with phenotypes including CLP and TA due to a TP63 missense mutation, later confirmed in a case report by Clements et al. (2012) describing an AEC patient with a CLP and TA (Table 2; Supplementary Table 4). Intriguingly, Clements et al. (2010) proposed that RHS and AEC represent a variable spectrum of the same genetic disorder, investigating four cases of which two showed bilateral CLP and TA due to two missense mutations of TP63 gene (Table 2; Supplementary Table 4). Interestingly, a case report described a patient with ADULT syndrome-like phenotype associated with CP and TA, who was found to be heterozygous for a de novo mutation in TP63 (Table 2; Supplementary Table 4) (Prontera et al. 2011). The peculiar aspect of this case is represented by the unusual combination of features: ADULT differs from EEC and LMS mainly by the absence of CL/P, but in this case CP was also present, thus the authors suggested to combine the three phenotypic spectra into a unique syndrome called ELA (Prontera et al. 2011). Patients with mixed phenotypic variations seen in EEC, AEC and RHS were previously described by Steele et al. (2005), one of these showed CLP and TA in addition to other anomalies, resulting from another TP63 SNP (Table 2; Supplementary Table 4) (Steele et al. 2005). The new and variable phenotypic features noted in these patients emphasize the wide spectrum of diseases caused by mutations in TP63.
The TGF pathway
The transforming growth factors (TGFs) represent a large family of proteins whose members regulate a remarkable range of biologic processes by acting on the transcription of genes controlling cell proliferation, differentiation, death, adhesion, migration and positioning. This superfamily is further divided into two classes, TGFα and TGFβ, which are not structurally nor genetically related but both modulating similar cell responses through different receptor mechanisms (TGF preferentially with EGF receptor, EGFR, while TGFβ via TGFβ receptors, TGFβRs) (Brachmann et al. 1989; Wong et al. 1989; Wrana et al. 1994; Heldin et al. 2009; Macias et al. 2015).
One of the most well-characterized members of the TGFβ subfamily is TGFβ3, a secreted protein that plays an essential role in embryogenesis by modulating mesenchymal cell proliferation, differentiation, migration and extracellular matrix production, via transmembrane TGFβRs which then transduce the signal from the cell surface to the cytoplasm mainly via SMAD proteins (EntrezGene; Wrana et al. 1994; Derynck and Zhang 2003; Massagué et al. 2005; Derynck et al. 2014; Macias et al. 2015). Diseases associated with TGFβ3 mutations include Loeys–Dietz syndrome-5 (LDS5) (a.k.a. Rienhoff syndrome) (OMIM# 615582) and arrhythmogenic right ventricular dysplasia (OMIM# 107970).
In the literature, OFCs with TA outside the cleft region was found to be positively associated with TGFβ3 variants, compared with non-OFC controls (Slayton et al. 2003). This evidence has been confirmed also in animal models, where mutant mice for TGFβ3 have been described as affected by HD and CP (Table 3) (Vieira 2003).
TGFβ3 represents one of the main ligands of two serine/threonine protein kinase receptors, TGFβR1 and TGFβR2, which have also been investigated in relation to syndromic OFCs (Loeys et al. 2005). Moreover, these genes have been associated with Marfan syndrome (OMIM# 154700), Loeys–Dietz syndrome (LDS) (OMIM# 609192; OMIM# 610168), features of which include CP (Loeys et al. 2005), and Kallmann syndrome (KAL, a.k.a. hypogonadotropic hypogonadism with anosmia) (OMIM# 147950). Interestingly, a study based on 14 patients with KAL whose phenotypic spectrum includes CP and tooth anomalies, found causative non-exonic mutations in TGFβR1 and TGFβR2 (Table 2; Supplementary Table 4). Although it is not specified whether TA is included in the analyzed dental abnormalities, this evidence remains interesting since patients with KAL share phenotypes with patients suffering from LDS type 2, suggesting a possible minor role for the TGFβR-mediated pathway in KAL (Bottani et al. 2006).
Unlike TGFβ3, TGFα encodes a ligand for EGFR that works synergistically with the TGFβ pathway to regulate cell proliferation, differentiation and embryonic development (Brachmann et al. 1989; Wong et al. 1989). A variety of positive and negative results have been reported concerning the association between OFC and TGFα, which is highly expressed in the medial edge epithelium of the palatal shelves at the time of palatal fusion (EntrezGene; Letra et al. 2012). Variants in TGFα have also been described as a possible risk factor for OFCs in case of maternal exposure to cigarette smoke, alcohol consumption or improper retinoic acid intake (Ardinger et al. 1989; Chenevix-Trench et al. 1992; Feng et al. 1994; Shaw et al. 1996a, b; Pezzetti et al. 1998; Jugessur et al. 2003; Zeiger et al. 2005; Letra et al. 2012). In addition, previous evidences have suggested that a possible interaction between IRF6 and TGFα may contribute to TA (Vieira et al. 2007).
In our literature search, a case–control study based on the genotyping of 406 Brazilian Caucasian patients with non-syndromic OFC (106 affected by TA) found a significant association between IRF6 as well as TGFα markers and the combination of OFCs and TA (Table 1; Supplementary Table 4) (Letra et al. 2012), representing a further clue of a possible role of TGFα in the dual pathogenesis of these orofacial defects.
Satb2
Originally identified as KIAA1034, SATB2 encodes a transcription regulator and chromatin remodeling factor, belonging to the homeobox proteins (SATB Homeobox 2). Its expression starts in the embryo and is later conserved in adult tissues, such as the spinal cord, the kidneys, and the central nervous system (UniProtKB; Zhao et al. 2014). This homeobox protein acts in concert with the BMP signaling pathway to modulate skeletogenesis by triggering several critical transcription factors like RUNX2, the master and the earliest osteogenic transcription factor (Zhao et al. 2014). A number of studies confirmed that SATB2 is strongly expressed in the developing craniofacial regions during mammalian embryogenesis, where it regulates osteoblast differentiation and craniofacial patterning determination (Britanova et al. 2006; Dobreva et al. 2006; Zhao et al. 2014). Consequently, mutations of this gene lead to increased apoptosis in the craniofacial mesenchyme and to impaired expression patterns of three genes, PAX9, ALX4 and MSX1, implicated in the regulation of craniofacial development in humans and mice, resulting in facial clefts (Dobreva et al. 2006; Zhao et al. 2014). In a large number of studies, the contribution of SATB2 variants to OFCs in human has been confirmed, especially CP, both in non-syndromic OFC (OMIM# 119530) as well as in syndromes such as Glass syndrome (OMIM# 612313), and Pierre Robin sequence with or without ankyloglossia and cleft-associated intellectual disability (OMIM# 261800) (FitzPatrick et al. 2003; Beaty et al. 2006; Britanova et al. 2006; Leoyklang et al. 2007; Rosenfeld et al. 2009; Urquhart et al. 2009; Rainger et al. 2014). In addition, recent evidence suggests a possible link between SATB2 and dental anomalies including TA (Rosenfeld et al. 2009; Kaiser et al. 2015). Regarding the co-occurrence of these pathogenic conditions, a case report describes a male patient with multiple associated phenotypes, including CP and TA, who carries a small intragenic duplication in the SATB2 gene affecting three coding exons (Table 2; Supplementary Table 4) (Lieden et al. 2014). In addition, the heterozygous loss-of-function mutations of SATB2 have been seen to result in micrognathia and CP both in mice and humans. In a recent study, two patients both affected by CP and TA were described with translocations, the breakpoints in which were mapped to SATB2 and PLCL1, t(2;11)(q33.1;p13) and t(1;2)(p34;q33), further supporting the hypothesis of a causative role of SATB2 in a common etiologic mechanism shared between OFCs and TA (Table 1; Supplementary Table 4) (Rainger et al. 2014).
Tbx22
A highly conserved gene family involved in the embryonic patterning from Drosophila to vertebrates is the T-box family, whose members are derived from events of gene duplication and cluster dispersion (Packham and Brook 2003).The key role played by TBX proteins during many aspects of embryonic development has been demonstrated by the generation of targeted T-box gene deletions in zebrafish and mouse (Bollag et al. 1994; Agulnik et al. 1996; Packham and Brook 2003). These models confirm that TBX factors are responsible for the decision of paraxial mesoderm to follow a mesodermal or neuronal pathway (Chapman and Papaioannou 1998). Due to its essential role in human palatogenesis, mutations in one of the TGF members, TBX22, have been reported in patients with OFCs and TA as well as in OFC-associated syndromes, such as inherited X-linked cleft palate with ankyloglossia (OMIM# 303400) and in Abruzzo–Erickson syndrome (OMIM# 302905) (Braybrook et al. 2001, 2002; Herr et al. 2003; Marçano et al. 2004; Suphapeetiporn et al. 2007; Kim et al. 2009; Pauws et al. 2009, 2013; Acevedo et al. 2010; Kantaputra et al. 2011; Kaewkhampa et al. 2012; Gurramkonda et al. 2015). The speculation about its contribution to OFCs and TA originated from two sources. First, one individual was found to present both OFC and TA likely due to a TBX22 missense mutation in a study based on a large cohort of patients with ankyloglossia and patients with sporadic isolated OFC (Table 2; Supplementary Table 4) (Kantaputra et al. 2011). Second, a case report describing a male patient with complete unilateral CLP and TA identified a hemizygous missense mutation in TBX22 (Table 1; Supplementary Table 4) (Kaewkhampa et al. 2012).
Chd7 and fgfr1/fgf8
On chromosome 8, two specific loci, 8p11.23 and 8q12.2, have been associated with the etiology of Kallmann syndrome (a.k.a. hypogonadotropic hypogonadism type 2 with anosmia) (OMIM# 147950) whose minor phenotypic manifestations include OFC and TA (Layman 2013). These two loci encompass two genes, proposed as causative genes of KAL: FGFR1 and CHD7, respectively (Beate et al. 2012; Layman 2013).
Located in 8q12.2, CHD7 gene encodes a DNA-binding protein that acts as a positive transcriptional regulator by binding to enhancer elements in the nucleoplasm, and its disruption leading to Kallmann syndrome or CHARGE syndrome (OMIM# 214800).
The other locus, 8p11.23, contains other genes, including FGFR1 (a.k.a. KAL2) and FGF8, both considered as main players in Kallmann syndrome. Mutations in FGFR1 are also described as causative for other syndromes, some of them including OFCs and dental anomalies (Kim et al. 2005; Riley et al. 2007; Stoler et al. 2009; Simonis et al. 2013), like a gain-of-function FGFR1 mutation associated with Kallmann syndrome and loss-of-function mutations in craniosynostosis presenting OFCs (Dodé et al. 2003). FGFR1 is a member of the fibroblast growth factor receptor (FGFR) family, a group of tyrosine kinase receptors belonging to the FGF pathway, which regulates a wide range of cell responses, such as angiogenesis, cell migration, and embryonic development, including skeletal formation (EntrezGene; Muenke and Schell 1995). This FGF signaling pathway contains also the ligands of these receptors, such as FGF8. Interestingly, FGFR1 as well as FGFR2 are well-characterized OFC-associated genes, but have been only recently investigated for possible involvement in TA (Huang et al. 2015; Hosokawa et al. 2009).
Rare sequence variants (defined as genetic variants with a minor allele frequency lower than 1 % in control populations) in FGFR1 (10 %) and CHD7 (6 %) are the most common autosomal causes of Kallmann syndrome, whereas another causative gene, KAL1, has been estimated to have a prevalence of 5–10 % in affected males (X-linked recessive) (Layman 2013). Costa-Barbosa et al. (2013) performed a detailed phenotypic comparison in a large group of 151 KAL subjects harboring known rare sequence variants, in eight genes belonging to six molecular pathways, which included CHD7 and FGFR1/FGF8. The co-occurrence of TA and OFC was observed in only two patients with rare sequence variants affecting CHD7 (Table 2; Supplementary Table 4), and although interesting as a clue suggesting the existence of a connection between the gene and the phenotypes of interest, the low number of cases was not sufficient to emerge as a statistically significant phenotype predictor. In contrast, among patients with CL/P, 54 in total, a significant association resulted in the sub-group of patients with CL/P showing TA (39 %) and mutations in the FGF8/FGFR1 (Table 2; Supplementary Table 4). Albuisson et al. (2005) studied a cohort of 98 patients with Kallmann syndrome, seven of whom contained mutations in FGFR1 related to OFCs and TA: of these, no one has been reported with the combined phenotypes; however, two patients with different FGFR1 mutations (p.D129A and p.V273 M) showed CP while another patient (c.1093_1094delAG) showed TA. Although no patients showed the combination of the phenotypes in this cohort, the study still raises interesting hypothesis since the same gene is affected and apparently related to both TA and OFCs even if in different subjects. Altogether, in our search we identified seven FGFR1 mutations that have been proposed as causative in seven patients with Kallmann syndrome, exhibiting CL/P and TA among other main phenotypes (Supplementary Table 4) (Zenaty et al. 2006; Xu et al. 2007, 2015; Bailleul-Forestier et al. 2010; Tommiska et al. 2014), representing relevant insights into a possible common FGFR1-related mechanism that may contribute to the dual etiology of OFCs and TA.
The WNT signaling pathway
The wingless-type MMTV integration site family (Wnt family) consists of structurally related genes encoding secreted signaling proteins implicated in several developmental processes, such as cell fate regulation and patterning during embryogenesis (EntrezGene; Dale 1998; Yin and Bian 2015). Together with the TGFβ signaling pathway, the canonical Wnt/β-catenin pathway provides most genes related to the network active during the initiation phase of palatogenesis and odontogenesis (Smalley and Dale 1999; Bae et al. 2015; Yin and Bian 2015). At the same time, Wnt signaling has been confirmed as implicated in oncogenesis at a later stage of life by a large number of studies since the late 1990s (e.g., Dale 1998; Morin 1999; Smalley and Dale 1999). In the last decade, mutations affecting the WNT10A member of this family have emerged as frequent causes of syndromic as well as non-syndromic TA (van den Boogaard et al. 2012; Arte et al. 2013; He et al. 2013; Abdalla et al. 2014; Alves-Ferreira et al. 2014; Kantaputra et al. 2014; Mues et al. 2014; Song et al. 2014; Vink et al. 2014). In addition, a WNT10A polymorphism is described to be associated with a significantly increased risk for OFC in a Chinese cohort (Feng et al. 2014; Beaty et al. 2006). However, unlike WNT3 and WNT5, no studies currently published have investigated WNT10A gene in relation to these combined orofacial phenotypes.
Mutations in WNT3 are well-known causes of syndromic tetra-amelia with CLP (OMIM# 273395), but the disruption of this gene has recently also been described as involved in non-syndromic OFC with TA (Table 2; Supplementary Table 4) (Yao et al. 2011; Mostowska et al. 2012). Interestingly, Menezes et al. (2010) identified a significant association between a marker located close to WNT3 gene in the group of patients affected by bilateral CL/P and agenesis of the lateral incisors. Specifically, this point mutation (rs142167, personal communication) is located in the intronic sequence of NSF, a gene flanking WNT3 and encoding a transporter involved in the vesicle-mediated trafficking within the Golgi cisternae (UniProt). However, since the effect of this mutation via NSF or through the close WNT3 gene is still not clear, further investigations are needed. In the same gene family, WNT5A has been reported by Person et al. (2010) as the causative gene of autosomal Robinow syndrome (ADRS) (OMIM# 180700) and in a recent update Roifman et al. (2015) described TA as a typical feature and CLP as a less common phenotype. Furthermore, mutations in other canonical WNT signaling-related genes have been shown to cause either TA with or without OFCs or other associated disorders, such as AXIN2, playing an important role in the regulation of β-catenin stability in the cytoplasm, and LRP6 functioning as a transmembrane co-receptor of Frizzled proteins (EntrezGene; Sarkar and Sharpe 1999; Bodine and Komm 2006). For LRP6, its role in lip formation and odontogenesis has been studied in mice and in patients (Song et al. 2009; Massink et al. 2015; Ockeloen et al. 2016) while the role of AXIN2 is not yet fully defined although its involvement in embryogenesis and oncogenesis is clear. Intriguingly, a pathogenic AXIN2 mutation has been described as causative for both TA and cancer development in a Finnish family where the TA phenotype segregated with colorectal cancer predisposition (Lammi et al. 2004). In a case–control study including 500 patients with non-syndromic OFC and 500 unrelated controls, an AXIN2 polymorphism (Table 1, Supplementary Table 4) showed association (rs7591, p = 0.01) with the co-occurrence of unilateral right CL/P with TA, stimulating the interest in this gene that may be involved in both pathogenic processes (Letra et al. 2009).
Cdh1
CDH1 (cadherin 1) belongs to the cadherin superfamily of transmembrane adhesion proteins, which play important roles in craniofacial morphogenesis (Taneyhill 2008), specifically during the formation of facial cartilages and bones as well as during dental development (Verstraeten et al. 2010), either by controlling cell–cell adhesion or interacting with Wnt intracellular signaling (Di Benedetto et al. 2015; Schambony et al. 2004; Bienz 2005; Brembeck et al. 2006). To date, mutations affecting this gene have been described in families presenting a combination of gastric cancer and CL/P (Letra et al. 2009; Frebourg et al. 2006; Vogelaar et al. 2013). In a wide case–control study, 500 Brazilian patients with OFC and 500 unrelated controls were analyzed to investigate the role of CDH1 and AXIN2 markers in OFC etiology. Interestingly, the sub-group of patients with OFC showing also TA, considered as cleft sub-phenotype in this study, revealed an association of one CDH1 marker (rs11642413, p = 0.008) and one AXIN2 marker (rs7591, p = 0.01) with unilateral right CL/P (Table 1; Supplementary Table 4) (Letra et al. 2009).
Other candidate genes rarely associated with co-occurrence of orofacial clefting and tooth agenesis
Kmt2d and kdm6a
KMT2D (a.k.a. MLL2), which encodes an SET-domain-containing protein of lysine-specific histone methyltransferases responsible for trimethylation of histone H3 at lysine 4 (H3K4me3), and KDM6A, a histone H3 lysine 27 (H3K27)-specific demethylase, have been recognized as the main causative genes of Kabuki syndrome (a.k.a. Niikawa–Kuroki syndrome) (OMIM# 147920, OMIM# 300867, respectively). These two enzymes modulate the gene expression by epigenetic modifications, playing a critical role in craniofacial, heart and brain development (Van Laarhoven et al. 2015). KMT2D-related Kabuki syndrome (type 1) (OMIM# 147920) is inherited in an autosomal dominant manner and KMT2D mutations are present in 34–76 % of patients with KS, while KDM6A-related KS (type 2) (OMIM# 300867) is less frequent and inherited in an X-linked manner (Adam et al. 1993; Van Laarhoven et al. 2015). This syndrome has peculiar craniofacial phenotypes, including as minor CL/P features, hypodontia and lower lip pits in some cases, which can lead to a misdiagnosis of VWS (Matsumoto and Niikawa 2003; David-Paloyo et al. 2014).
Patients with a KMT2D mutation are more likely to have the distinctive Kabuki facial phenotype, which may reflect the fact that a portion of those without a KMT2D mutation may have been misdiagnosed. However, in the literature, molecular analyses confirmed the presence of a KMT2D mutation in only one patient with KS exhibiting the co-occurrence of CP and HD (Table 2; Supplementary Table 4) (David-Paloyo et al. 2014).
Ofd1
The X-linked gene OFD1 has been recognized as a causative gene of the oral–facial–digital syndrome type 1 (OFD1) (OMIM# 311200) (Klein et al. 2013). This gene encodes a centrosomal protein implicated in embryonic development by regulating the canonical Wnt signaling pathways and the sonic hedgehog (Shh) signal during the early embryonic specification of the left–right axis in mammals (EntrezGene; Macca and Franco 2009). In OFD1 syndrome, CP is present in more than 50 % of the affected patients, and also another minor OFC subtype, the cleft alveolus, is commonly reported in patients with OFD. In addition, the lower lateral incisors are missing in 50 % of the individuals, which is also associated with fibrous bands in the region (Klein et al. 2013). In their NCBI GeneReview, Toriello and Franco (1993) indicate that in OFD1 mainly median clefts or (pseudo)clefts of the upper lip are present. In a case series found in our search, two OFD1 siblings sharing the same mutation were described; only one of them had TA and a cleft alveolar ridge (Table 2; Supplementary Table 4) (Shimojima et al. 2013).
Bcor
The BCL6 corepressor gene, BCOR, encodes a protein that inhibits gene expression by sequence-specific DNA-binding proteins such as BCL6 and MLLT3 when recruited to their promoter regions (UniProt). In addition, this gene is known to interact with AP-2α, a known OFC gene (Milunsky et al. 2008; Rahimov et al. 2008), and WNT10A, a known TA gene. Syndromes associated with variants in BCOR include oculofaciocardiodental syndrome (OFCD) (a.k.a. syndromic microphthalmia type 2, OMIM# 300166), which is known to be associated with both OFC and TA. In a review by Kantaputra (2014), the OFCD syndrome has been described to be associated with several dental and orofacial anomalies including HD and craniofacial features including CP. As confirmation, Feberwee et al. (2014) indeed found two patients affected with OFCD carrying BCOR point mutations, one affected by CP and mild HD while the other by CP and OD, although a concrete evidence that these two phenotypes are ‘typical’ features of OFCD syndrome is lacking (Table 2; Supplementary Table 4).
Twist1
Within the basic helix–loop–helix (bHLH) transcription factor family, which plays an essential role in cell lineage determination and differentiation, TWIST1 (twist family BHLH transcription factor 1) was found by Busche et al. (2011) as the only gene affected by a microdeletion of 7p21 in three patients (Table 2; Supplementary Table 4). Although a wide range of phenotypes was present in these subjects, such as features resembling typical traits of blepharophimosis–ptosis–epicanthus inversus syndrome (BPES) (OMIM# 110100) and Saethre–Chotzen syndrome (OMIM# 101400), CP and TA were present in one of these patients.
Pitx2
A transcriptional regulator, member of the PITX homeobox family, is encoded by PITX2 (paired-like homeodomain 2) and is involved in the morphogenesis of the eyes, the teeth and abdominal organs (EntrezGene). Mutations in this gene are associated with Axenfeld–Rieger syndrome type 1 (RIEG1) (OMIM# 180500), iridogoniodysgenesis syndrome type 2 (IRID2) (OMIM# 137600), and sporadic cases of Peters anomaly (OMIM# 604229).
Main characteristics of Axenfeld-Rieger syndrome type 1 (RIEG1) include severe TA that is associated with midfacial hypoplasia and CP (Kavitha et al. 2010). Although evidences in humans currently lack, Pitx2 knockout mice typically exhibit TA, CP and abnormal development of the maxilla and mandible (Table 3) (Kouskoura et al. 2011), supporting the hypothesis of a conserved relation between this gene and orofacial defects in human.
Ptch1
In our literature search, patched 1 (PTCH1) gene encoding a member of the patched family which functions as a receptor for Indian hedgehog (IHH), desert hedgehog (DHH) and mainly for sonic hedgehog (SHH) was also identified. Shh represents a key inductive signal for a variety of patterning events that take place in the early embryo, and consequently PTCH1 is also involved in embryonic development. Interestingly, mutations in SHH cause holoprosencephaly (OMIM# 142945), whose wide phenotypic spectrum also includes CL/P and the presence of a single median upper central incisor, which may be considered as a mild form of TA (Roessler et al. 1996; Orioli et al. 2002).
A study based on a transgenic mouse model expressing Shh, ligand of Ptch1, in basal epithelium under the control of a specific Keratin-14 promoter showed that an increased activity of Shh in this tissue prevents apoptosis, palatal shelf fusion and tooth development at the bud stage (Table 3) (Cobourne et al. 2009). PTCH1 is one of the causative genes for nevoid basal cell carcinoma syndrome (a.k.a. basal cell nevus syndrome, OMIM# 109400), which includes OFC and TA as secondary features of its core characteristics, including also jaw cysts, basal cell tumors and skeletal abnormalities (Cobourne et al. 2009; Lam et al. 2013).
Pvrl1
Mutations in PVRL1, encoding an adhesion protein contributing to the adherent and tight junction formation in epithelial and endothelial cells, are known to cause CL/P-ectodermal dysplasia syndrome (CLPED, a.k.a. Zlotogora syndrome) (OMIM# 225060) as well as non-syndromic CL/P (EntrezGene; Suzuki et al. 2000; Sözen et al. 2001; Turhani et al. 2005; Avila et al. 2006; Scapoli et al. 2006; Sözen et al. 2009). So far, combined CL/P and HD has only been diagnosed in one CLPED patient who exhibited a homozygous nonsense mutation in the PVRL1 gene (Supplementary Table 4) (Yoshida et al. 2015).
Kiss1r
KISS1R gene encodes for a galanin-like G protein-coupled receptor that plays a role in endocrine function regulation and puberty onset by binding its ligand, metastin, and triggering a signaling via phospholipase C and G(q) proteins (EntrezGene; UniProtKB). This gene is known as the causative gene of hypogonadotropic hypogonadism type 8 with or without anosmia (OMIM# 614837) (Acierno et al. 2003; de Roux et al. 2003; Brioude et al. 2013). Interestingly, mutations of KISS1R have recently been linked to Kallmann syndrome: specifically, Xu et al. (2015) reported on a Kallmann patient exhibiting CL and TA (Table 2; Supplementary Table 4). Although a single evidence is not enough to draw any conclusion, the relation of the KISS1R mutation with Kallmann syndrome including the co-occurrence of OFC and TA is worth to be further investigated.
GO term analysis and gene clustering
To find the hypothesized common etiological genetic factors explaining the co-occurrence of TA and OFC, we further analyzed the data as follows using a Gene Ontology (GO) term mapping tool. The GO terms related to the biological processes mediated by the 26 candidate genes were mapped to 51 broad categories, which were subsequently combined to generate six super-clusters (Supplementary Table 5): (a) anatomical development, (b) cell division, growth and motility, (c) cell metabolism and catabolism, (d) cell transport, (e) cell structure organizations and (f) organ/system-specific processes.
Anatomical development, the first cluster, includes a total of 23 genes related with embryogenesis, morphogenesis, anatomical structure formation and growth (in alphabetical order): AXIN2, BCOR, CDH1, CHD7, FGF8, FGFR1, IRF6, KDM6A, KMT2D, MSX1, OFD1, PAX9, PITX2, PTCH1, PVRL1, SATB2, TGFβ3, TGFβR1, TGFβR2, TP63, TWIST1, WNT3, and WNT5A. Cell division, growth and motility, the second cluster, largely overlaps with the first cluster, encompassing 23 genes involved in different processes that range from cell division and proliferation, over differentiation, to cell motility and adhesion. Excluding PAX9 and BCOR, the other 21 genes of the first cluster are present also in the second, which in addition includes TGFα and KISS1R. Similarly, 23 genes are included in the third cluster, for cell metabolism and catabolism: AXIN2, BCOR, CDH1, CHD7, FGF8, FGFR1, IRF6, KDM6A, KISS1R, KMT2D, MSX1, PAX9, PITX2, PTCH1, SATB2, TBX22, TGFα, TGFβ3, TGFβR1, TGFβR2, TP63, TWIST1, and WNT5A. Other biological processes highly represented in our set of candidate genes are the cell transport and signal transduction, comprising 21 genes which correspond to the second cluster excluding SATB2 and OFD1. In addition, 17 candidate genes are also implicated in the cellular structure organization, specifically membrane formation and cytoskeleton assembly: AXIN2, BCOR, CDH1, CHD7, KMT2D, KDM6A, OFD1, PTCH1, PVRL1, SATB2, TGFα, TGFβ3, TGFβR1, TP63, TWIST1, and WNT5A. The last cluster includes 17 genes contributing to organ/system-specific processes, such as the immune system, the neurological system and the circulatory system processes: CDH1, CHD7, FGF8, FGFR1, IRF6, KMT2D, MSX1, PITX2, PVRL1, SATB2, TGFβ3, TGFβR1, TGFβR2, TP63, TWIST1, WNT3, and WNT5A.
Genomic loci likely associated to co-occurrence of orofacial clefting and tooth agenesis
In our literature search, some genomic loci, either deleted or containing mutations, were reported in patients with the co-occurrence of TA and OFCs. Schinzel and Schmid (1980) reported a patient with a deletion of 1q21–q25 [del(1)(q21–q25)] exhibiting OFC and TA (Table 4). This large deletion encompassed 702 genes, including protein-coding, non-coding genes, miRNAs and long non-coding RNAs (Supplementary Table 6).
A new ectodermal dysplasia-like syndrome, named del(2q32) syndrome, has been proposed in a case report of a patient with a 26 Mb interstitial deletion involving the region 2q31.2–q33.2, who showed multiple phenotypes including CP and TA, as well as severe intellectual disability and ectodermal anomalies (Rifai et al. 2010) (Table 4, Supplementary Table 6). Another 3.7 Mb deletion affecting the locus 4p16.3 was described by Maas et al. (2008) in a patient with Wolf–Hirschhorn syndrome (OMIM# 194190) exhibiting CP and TA along with intellectual disability, microcephaly at birth and other abnormalities (Table 4; Supplementary Table 6). Three studies reported on patients with oropalatal Bettex–Graf dysplasia, combining CP and HD or OD with a fragile site located in the region 16q22 (Table 4; Supplementary Table 6) (Bettex et al. 1998; Janiszewska-Olszowska et al. 2013; McKenzie et al. 2002).
Another locus associated with a known cleft syndrome, DiGeorge syndrome (OMIM# 188400), is located on chromosome 22 containing the main causative gene, TBX1. In four young patients with DiGeorge syndrome, the deletion of the locus 22q11 was diagnosed along with a CP and TA (Table 4; Supplementary Table 6) (Heliövaara et al. 2011).
A family-based study published in 2012 (Yildirim et al. 2012) confirmed the significant association between an intragenic SNP (rs987525) affecting a long non-coding RNA gene, LINC00976, located in the 8q24.21 region (Table 4; Supplementary Tables 4 and 6), and the co-occurrence of OFCs and TA. This evidence is particularly interesting since rs987525 has been demonstrated to be a susceptibility marker for non-syndromic CL/P in human as well as in animal models although the molecular mechanisms explaining the involvement of LINC00976 in OFC development remain unknown (Birnbaum et al. 2009; Mangold et al. 2011; Uslu et al. 2014). On the contrary, no studies have been published so far to validate a possible association between the 8q24 locus and TA, thus our hypothesis of a possible association represents a first input to stimulate molecular and functional studies in vitro and in vivo to shed light on potentially novel pathogenic mechanisms for TA and OFCs.
Discussion
The aim of this systematic review is to thoroughly and systematically investigate the available literature to collect a panel of genes and loci likely contributing to the co-occurrence of TA and OFC in humans, possibly confirmed in animal models, and to speculate on the possible key pathways involved in physiological tooth development and in facial primordia migration, proliferation and fusion.
Although the co-occurrence of dental anomalies, specifically TA, and OFCs is frequently seen clinically, a comprehensive molecular and genetic exploration of the possible key genes for the common pathogenesis has not been performed so far. This is mainly explained by the fact that the mildest forms of TA are often neglected or overlooked compared to the more severe OFCs.
Overall, from this systematic literature search we identified 84 articles fulfilling our inclusion criteria. Based on them, 26 genes and 9 genomic loci emerged as related to the oral defects of interest (Tables 1, 2, 3; Supplementary Tables 4 and 6). Among the 26 genes, the majority belongs to known OFC- or TA-related pathways (Tables 1, 2, 3; Supplementary Table 4). Some of these genes encode transcription factors differentially involved in the regulation of embryonic developmental events, working synergistically in some cases: MSX1 and PAX9, CHD7, TWIST1, TP63 and IRF6, and the homeodomain proteins SATB2, TBX22 and PITX2. Other candidates encode for effectors of signaling pathways that lead to the modulation of cell differentiation, migration or adhesion. This is the case for CDH1 and PVRL1, the SHH receptor PTCH1, for TGFα, TGFβ3 and its receptors TGFβR1/2, for FGF8 and FGFR1, and for AXIN2, WNT3 and its regulator OFD1. In addition, we found two enzymes, KMT2D and KDM6, that specifically modify chromatin structure, thus regulating transcription by epigenetic modifications. Moreover, the DNA-binding repressor protein, BCOR, was also identified, acting by inhibiting gene expression when recruited to specific promoter regions.
To speculate on the possible clustering of these 26 genes (Tables 1, 2, 4; Supplementary Table 4), the GO terms indicating the biological processes which involve these genes have been collected and subsequently mapped to broader GO-slim categories. Based on these generic GO-slim categories, we proposed a gene super-clustering, including six partially overlapping super-clusters: (a) anatomical development, (b) cell division, growth and motility, (c) cell metabolism and catabolism, (d) cell transport and signal transduction, (e) cell structure organizations and (f) organ/system-specific processes (Supplementary Table 5). As we expected, 23 of the 26 candidate genes described in this review are implicated in embryogenesis, morphogenesis, anatomical patterning and maturation, as well as neural crest formation, further supporting the hypothesis about their involvement in tooth, lip and palate formation. In addition, 23 of the 26 candidate genes contribute to general cellular metabolism and catabolism as well as general cell processes, such as differentiation, proliferation and migration, while 21 are related to cell transport and signal transduction. Out of 26 candidate genes, 17 are involved in cell structure organization, like membrane and cytoskeleton formation. While, another group of 17 candidate genes has been associated with organ and system-specific processes, such as those which take place specifically in the nervous system and in the circulatory system. To discriminate which of the presented candidate genes have molecular functions that indeed underlie the failure of facial primordia migration and fusion, and the disruption of tooth development, and those whose functions could not explain their dual role in the pathogenesis of these conditions, further molecular analyses based on genotyping and population screening in concert with animal model studies and bioinformatics tools are necessary.
The fact that OFCs and TA are congenital birth defects starting to develop in the orofacial region of the 6–12-week human embryo largely explains why the GO term-based super-clustering (first super-cluster) is focusing on genes known to drive embryogenesis and oral morphogenesis. The clustering based on the GO terms in relation to biological processes have been used to identify common genes and pathways involved in TA and OFCs, and to predict possible new interactions between candidates in the same biological processes. However, since the clustering is a bioinformatics prediction, it will be of interest to validate the resulting clusters and to underpin the related hypotheses with functional studies.
Some of the presented genes, like TP63 and IRF6, are well-known and widely studied genes, especially in relation to OFCs, and in our GO term analysis they are present in all the clusters, suggesting that those genes are governing broad molecular networks. The TP63 gene plays a critical role in epithelial differentiation and in our gene set it represents the gene most likely contributing to the pathogenesis of both TA and OFCs as it takes part in the development and maintenance of stratified epithelial tissues, mediating the interactions between the mesenchyme and the epithelium. Mutations in TP63 also underlie several dysmorphology syndromes including clefts or cleft features.
The IRF6 gene is a target of TP63 that activates IRF6 transcription through the IRF6 enhancer element (Dixon et al. 2011). IRF6 is related to the formation of connective tissue (for example in the palate), the palatal rugae and underlying the dental epithelium (Blackburn et al. 2012; Chu et al. 2016). IRF6 can be considered as the second gene most likely playing a role in the combined OFC–TA phenotype since it is the causative gene of van der Woude syndrome, whose typical features are OFCs, TA and lip pits.
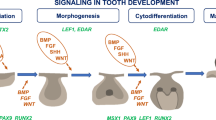
Similarly, MSX1 and PAX9 have also been associated with the co-occurrence of OFCs and TA, as implicated in the development of cephalic structures and dental development both in humans and animal models. Interestingly, some of the candidate genes belong to five major gene families, WNTs, FGFs, BMPs, TGFs and PAXs, protein families that are essential in different phases of neural crest development, the structure from which originate the facial primordia, the palatal shelves, the alveolar ridge and the teeth. During the initial neural crest cell specification phase, the concerted activity of the WNT, FGF and BMP pathways induces the expression of neural plate border (or neural crest) genes, which turn on the expression of a distinct set of transcription factors including MSX1 and PAXs, and neural crest cells genes such as TWIST1 and Myc, representing the neural crest specification module factors that determine the neural or non-neural fate of the neural crest cells (Simões-Costa and Bronner 2015).
In addition, a new level of gene expression regulation has been recently added to the cis-transcriptional regulation programs, i.e., the epigenetic regulation based on chromatin remodeling. For the neural crest specification, the removal of repressive methylation marks is necessary as well as the addition of acetylation marks to relax the chromatin structure making it more accessible to DNA-binding transcription factors. Interestingly, two of the presented candidate genes, KMT2D and KDM6A (a methyltransferase and a demethylase, respectively), regulate the chromatin methylation status and they are both causes of Kabuki syndrome, whose wide spectrum of features includes also OFCs and TA.
As the neural crest is one of the most conserved structures in vertebrates, the overlap of genes active during neural crest formation, odontogenesis, palatogenesis and facial primordia development may reflect the importance of the teeth and the palate, also in terms of human evolution and survival.
Although it would have been interesting to check for maternal genetic effects in these combined OFC–TA phenotypes, it was striking that in most articles relevant for our literature search the genotype of the mother was not provided. As our study almost exclusively focuses on inherited genetic effects, based on the genotypes of the described cases, we could not draw any conclusion on a possible implication of maternal genetic effects, which in other studies are acknowledged to affect the susceptibility of the embryo, and increasing the risk to develop a cleft. We, therefore, estimate that from the current stage of knowledge in the literature concerning the co-occurrence of OFC and TA, we are not able to provide any clues on the maternal genetic effects on the incidence or prevalence of these phenotypes.
As our review is a survey of scientific literature to identify genes involved in cases with co-occurrence of OFCs and TA, epidemiological analyses are beyond the specific aim of our study. We are aware indeed that some of the identified genes could be involved in OFC cases with TA by chance but, based on the sole data that is available in the literature, it seems impossible to track them down at this stage. However, the results of this study provide an intriguing list of candidate genes which could eventually be tested in prospective studies to sort out those that were not specifically associated with OFC–TA co-occurrence.
Nevertheless, with our review we aim to boost the research in this direction: we would like to draw the attention of clinicians and researchers working in TA and OFC field, on the investigation of the co-occurrence of these two defects, rather than focusing on the one or the other single oral defect. If the new combinatorial perspective will be adopted and developed in the following years, it would be easier in the future to get a concrete overview of the risk of TA–OFC co-occurrence in the population.
It would also be interesting to get a sense of how much of risk for each defect is attributable to recognized genetic loci, but our review is neither a meta-analysis nor an epidemiologic study. Presently, the number of patients reported in the literature is far too low for most of the genes, so that a sturdy statistical analysis is not possible. Therefore, we decided to report the number of patients found for each locus/gene (Tables 1, 2, 4) to give the reader a sense of how rarely the TA–OFC co-occurrence develops in case of gene/locus mutations. This way we avoided to perform a statistical analysis that would never have been unbiased and reliable in the present situation.
It is too early for translating the review findings into therapeutic strategies for congenital birth defects like orofacial clefts with (or without) tooth agenesis, as these birth defects are different from other genetic diseases like retinitis pigmentosa (Bassuk et al. 2016) and Duchenne’s muscular dystrophy (Wojtal et al. 2016) for which postnatal treatments with genetic editing in patient-derived stem cells are currently under development. However, the identified OFC–TA genes could eventually be included in postnatal diagnostic gene panels. Advantages of postnatal genetic testing not only includes more precise (sub)phenotyping of the patient, but also targeted physical examination of the patients’ parents as well as members of his/her broader family. The latter can also be approached for genotyping and for deep phenotyping, including OFC and TA (sub)phenotypes. In fact, in children born with apparently isolated OFC, an NGS-based screening of a panel of genes including both syndromic and TA-OFC genes could be useful for the diagnosis of cleft syndromes and tooth agenesis, and lead to earlier onset of therapies. However, the diagnosis of a case with OFC (with or without TA) but without other severe malformations does not influence future pregnancy planning as these conditions are treatable. In next pregnancies, testing OFC–TA genes could eventually be carried out on amniocytes to anticipate diagnosis. For these cases, however, termination of pregnancy should never be an option offered by clinical geneticists.
In addition, the exact role of these genes, loci and pathways in orofacial development should first be further elucidated with functional in vitro and in vivo studies to increase our understanding of the molecular mechanisms that lead to orofacial clefts in case of disruption. Tooth development, palatal shelf migration and lip formation, although depending on different tissues, timing and dynamics, are based on similar processes including cell migration and fusion. It is not fully clear if the same genes drive these processes in different tissues, but the co-occurrence of TA and OFCs due to the disruption of specific genes may support this hypothesis.
Intriguingly, not only the disrupted gene but even the location of the mutations within the gene can lead to diverse phenotypes. A recently published review (Liang et al. 2016) shows how the location of mutations in the MSX1 homeodomain always causes TA with or without other phenotypes while mutations outside the homeodomain are mostly associated with non-syndromic OFCs. Following this hypothesis, it would be interesting for further functional studies to expand the molecular investigation to different protein domains in relation to different spectra of phenotypes, thus improving the diagnostic potential of these gene panels and the knowledge of molecular pathogenic mechanisms affecting the orofacial region.
References
Abdalla EM, Mostowska A, Jagodzinski PP, Dwidar K, Ismail SR (2014) A novel WNT10A mutation causes non-syndromic hypodontia in an Egyptian family. Arch Oral Biol 59:722–728. doi:10.1016/j.archoralbio.2014.04.004
Acevedo AC, da Fonseca JA, Grinham J, Doudney K, Gomes RR, de Paula LM, Stanier P (2010) Autosomal-dominant ankyloglossia and tooth number anomalies. J Dent Res 89:128–132. doi:10.1177/0022034509356401
Acierno JS Jr, Shagoury JK, Bo-Abbas Y, Crowley WF Jr, Seminara SB (2003) A locus for autosomal recessive idiopathic hypogonadotropic hypogonadism on chromosome 19p13.3. J Clin Endocrinol Metabol 88:2947–2950. doi:10.1210/jc.2003-030423
Adam MP, Hudgins L, Hannibal M (1993) Kabuki syndrome. In: Pagon RA et al. (eds) GeneReviews(R). University of Washington, Seattle University of Washington, Seattle. All rights reserved, Seattle (WA)
Agulnik SI et al (1996) Evolution of mouse T-box genes by tandem duplication and cluster dispersion. Genetics 144:249–254
Albuisson J et al (2005) Kallmann syndrome: 14 novel mutations in KAL1 and FGFR1 (KAL2). Hum Mutat 25:98–99. doi:10.1002/humu.9298
Ali A, Singh SK, Raman R (2009) Coding region of IRF6 gene may not be causal for Van der Woude syndrome in cases from India. Cleft Palate-Craniofac J 46:541–544. doi:10.1597/08-202.1
Alves-Ferreira M, Pinho T, Sousa A, Sequeiros J, Lemos C, Alonso I (2014) Identification of genetic risk factors for maxillary lateral incisor agenesis. J Dent Res 93:452–458. doi:10.1177/0022034514523986
Ardinger HH, Buetow KH, Bell GI, Bardach J, VanDemark DR, Murray JC (1989) Association of genetic variation of the transforming growth factor-alpha gene with cleft lip and palate. Am J Hum Genet 45:348–353
Arte S, Parmanen S, Pirinen S, Alaluusua S, Nieminen P (2013) Candidate gene analysis of tooth agenesis identifies novel mutations in six genes and suggests significant role for WNT and EDA signaling and allele combinations. PLoS One 8:e73705. doi:10.1371/journal.pone.0073705
Aspinall A, Raj S, Jugessur A, Marazita M, Savarirayan R, Kilpatrick N (2014) Expanding the cleft phenotype: the dental characteristics of unaffected parents of Australian children with non-syndromic cleft lip and palate. International journal of paediatric dentistry/the British Paedodontic Society [and] the International Association of Dentistry for. Children 24:286–292. doi:10.1111/ipd.12072
Avila JR et al (2006) PVRL1 variants contribute to non-syndromic cleft lip and palate in multiple populations. Am J Med Genet Part A 140:2562–2570. doi:10.1002/ajmg.a.31367
Bae CH, Kim TH, Ko SO, Lee JC, Yang X, Cho ES (2015) Wntless regulates dentin apposition and root elongation in the mandibular molar. J Dent Res 94:439–445. doi:10.1177/0022034514567198
Bailleul-Forestier I, Gros C, Zenaty D, Bennaceur S, Leger J, de Roux N (2010) Dental agenesis in Kallmann syndrome individuals with FGFR1 mutations. Int J Paediatr Dent 20:305–312. doi:10.1111/j.1365-263X.2010.01056.x
Balling R, Helwig U, Nadeau J, Neubuser A, Schmahl W, Imai K (1996) Pax genes and skeletal development. Ann N Y Acad Sci 785:27–33
Bassuk AG, Zheng A, Li Y, Tsang SH, Mahajan VB (2016) Precision medicine: genetic repair of retinitis pigmentosa in patient-derived stem cells. Sci Rep 6:19969. doi:10.1038/srep19969
Beate K, Joseph N, de Nicolas R, Wolfram K (2012) Genetics of isolated hypogonadotropic hypogonadism: role of GnRH receptor and other genes. Int J Endocrinol 2012:147893. doi:10.1155/2012/147893
Beaty TH et al (2006) Analysis of candidate genes on chromosome 2 in oral cleft case-parent trios from three populations. Hum Genet 120:501–518. doi:10.1007/s00439-006-0235-9
Beaty TH et al (2010) A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet 42:525–529. doi:10.1038/ng.580
Bettex M, Graf B, Winkler B, Gerber-Huber S (1998) Oro-palatal dysplasia Bettex-Graf—a new syndrome. Eur J Pediatr Surg 8:4–8
Bienz M (2005) beta-Catenin: a pivot between cell adhesion and Wnt signaling. Curr Biol 15:R64–R67. doi:10.1016/j.cub.2004.12.058
Birnbaum S et al (2009) Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet 41:473–477. doi:10.1038/ng.333
Blackburn J et al (2012) The role of Irf6 in tooth epithelial invagination. Dev Biol 365:61–70. doi:10.1016/j.ydbio.2012.02.009
Blanton SH, Cortez A, Stal S, Mulliken JB, Finnell RH, Hecht JT (2005) Variation in IRF6 contributes to nonsyndromic cleft lip and palate. Am J Med Genet Part A 137:259–262. doi:10.1002/ajmg.a.30887
Bodine PV, Komm BS (2006) Wnt signaling and osteoblastogenesis. Rev Endocr Metabol Disord 7:33–39. doi:10.1007/s11154-006-9002-4
Bollag RJ, Siegfried Z, Cebra-Thomas JA, Garvey N, Davison EM, Silver LM (1994) An ancient family of embryonically expressed mouse genes sharing a conserved protein motif with the T locus. Nat Genet 7:383–389. doi:10.1038/ng0794-383
Bottani A, Pardo B, Bouchardy I, Schoumans J, Toutain A, Conrad B (2006) No major contribution of the TGFBR1- and TGFBR2-mediated pathway to Kabuki syndrome. Am J Med Genet Part A 140:903–905. doi:10.1002/ajmg.a.31168
Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G (2004) GO: TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20:3710–3715. doi:10.1093/bioinformatics/bth456
Brachmann R, Lindquist PB, Nagashima M, Kohr W, Lipari T, Napier M, Derynck R (1989) Transmembrane TGF-alpha precursors activate EGF/TGF-alpha receptors. Cell 56:691–700
Braybrook C et al (2001) The T-box transcription factor gene TBX22 is mutated in X-linked cleft palate and ankyloglossia. Nat Genet 29:179–183. doi:10.1038/ng730
Braybrook C et al (2002) Craniofacial expression of human and murine TBX22 correlates with the cleft palate and ankyloglossia phenotype observed in CPX patients. Hum Mol Genet 11:2793–2804
Brembeck FH, Rosario M, Birchmeier W (2006) Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev 16:51–59. doi:10.1016/j.gde.2005.12.007
Brioude F et al (2013) Two families with normosmic congenital hypogonadotropic hypogonadism and biallelic mutations in KISS1R (KISS1 receptor): clinical evaluation and molecular characterization of a novel mutation. PLoS One 8:e53896. doi:10.1371/journal.pone.0053896
Britanova O, Depew MJ, Schwark M, Thomas BL, Miletich I, Sharpe P, Tarabykin V (2006) Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am J Hum Genet 79:668–678. doi:10.1086/508214
Brunner HG, Hamel BC, Bokhoven Hv H (2002a) P63 gene mutations and human developmental syndromes. Am J Med Genet 112:284–290. doi:10.1002/ajmg.10778
Brunner HG, Hamel BC, Van Bokhoven H (2002b) The p63 gene in EEC and other syndromes. J Med Genet 39:377–381
Busche A, Graul-Neumann LM, Zweier C, Rauch A, Klopocki E, Horn D (2011) Microdeletions of chromosome 7p21, including TWIST1, associated with significant microcephaly, facial dysmorphism, and short stature. Eur J Med Genet 54:256–261. doi:10.1016/j.ejmg.2011.02.002
Cabanillas M, Torrelo A, Monteagudo B, Suarez-Amor O, Ramirez-Santos A, Gonzalez-Vilas D, de las Heras C (2011) A novel heterozygous point mutation in the p63 gene in a patient with ectodermal dysplasia associated with B-cell leukemia. Pediatr Dermatol 28:707–710. doi:10.1111/j.1525-1470.2011.01474.x
Carey JC, Viskochil DH (2002) Status of the human malformation map: 2002. Am J Med Genet 115:205–220. doi:10.1002/ajmg.10987
Celli J et al (1999) Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 99:143–153
Chapman DL, Papaioannou VE (1998) Three neural tubes in mouse embryos with mutations in the T-box gene Tbx6. Nature 391:695–697. doi:10.1038/35624
Chenevix-Trench G, Jones K, Green AC, Duffy DL, Martin NG (1992) Cleft lip with or without cleft palate: associations with transforming growth factor alpha and retinoic acid receptor loci. Am J Hum Genet 51:1377–1385
Chu EY et al (2016) Full spectrum of postnatal tooth phenotypes in a novel Irf6 cleft lip model. J Dent Res. doi:10.1177/0022034516656787
Clements SE, Techanukul T, Coman D, Mellerio JE, McGrath JA (2010) Molecular basis of EEC (ectrodactyly, ectodermal dysplasia, clefting) syndrome: five new mutations in the DNA-binding domain of the TP63 gene and genotype-phenotype correlation. Br J Dermatol 162:201–207. doi:10.1111/j.1365-2133.2009.09496.x
Clements SE et al (2012) Mutations in AEC syndrome skin reveal a role for p63 in basement membrane adhesion, skin barrier integrity and hair follicle biology. Br J Dermatol 167:134–144. doi:10.1111/j.1365-2133.2012.10888.x
Cobourne MT (2004) The complex genetics of cleft lip and palate. Eur J Orthod 26:7–16
Cobourne MT, Xavier GM, Depew M, Hagan L, Sealby J, Webster Z, Sharpe PT (2009) Sonic hedgehog signalling inhibits palatogenesis and arrests tooth development in a mouse model of the nevoid basal cell carcinoma syndrome. Dev Biol 331:38–49. doi:10.1016/j.ydbio.2009.04.021
Costa-Barbosa FA et al (2013) Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J Clin Endocrinol Metabol 98:E943–E953. doi:10.1210/jc.2012-4116
Dale TC (1998) Signal transduction by the Wnt family of ligands. Biochem J 329(Pt 2):209–223
Das P, Hai M, Elcock C, Leal SM, Brown DT, Brook AH, Patel PI (2003) Novel missense mutations and a 288-bp exonic insertion in PAX9 in families with autosomal dominant hypodontia. Am J Med Genet Part A 118:35–42. doi:10.1002/ajmg.a.10011
David-Paloyo FP, Yang X, Lin JL, Wong FH, Wu-Chou YH, Lo LJ (2014) Lower lip pits: van der Woude or Kabuki syndrome? Cleft Palate-Craniofac J 51:729–734. doi:10.1597/12-258
Davidson D (1995) The function and evolution of Msx genes: pointers and paradoxes. Trends Genet 11:405–411
De Coster PJ, Mortier G, Marks LA, Martens LC (2007) Cranial suture biology and dental development: genetic and clinical perspectives. J oral Pathol Med 36:447–455. doi:10.1111/j.1600-0714.2007.00553.x
de Lima RL et al (2009) Prevalence and nonrandom distribution of exonic mutations in interferon regulatory factor 6 in 307 families with Van der Woude syndrome and 37 families with popliteal pterygium syndrome. Genet Med 11:241–247. doi:10.1097/GIM.0b013e318197a49a
de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E (2003) Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA 100:10972–10976. doi:10.1073/pnas.1834399100
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signaling. Nature 425:577–584. doi:10.1038/nature02006
Derynck R, Muthusamy BP, Saeteurn KY (2014) Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr Opin Cell Biol 31:56–66. doi:10.1016/j.ceb.2014.09.001
Di Benedetto A et al (2015) Osteogenic differentiation of mesenchymal stem cells from dental bud: role of integrins and cadherins. Stem Cell Res 15:618–628. doi:10.1016/j.scr.2015.09.011
Dixon MJ, Marazita ML, Beaty TH, Murray JC (2011) Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet 12:167–178. doi:10.1038/nrg2933
Dobreva G et al (2006) SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 125:971–986. doi:10.1016/j.cell.2006.05.012
Dodé C et al (2003) Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 33:463–465. doi:10.1038/ng1122
Eerens K, Vlietinck R, Heidbuchel K, Van Olmen A, Derom C, Willems G, Carels C (2001) Hypodontia and tooth formation in groups of children with cleft, siblings without cleft, and nonrelated controls. Cleft Palate-Craniofac J 38:374–378. doi:10.1597/1545-1569(2001)038<0374:hatfig>2.0.co;2
Fakhouri WD et al (2014) An etiologic regulatory mutation in IRF6 with loss- and gain-of-function effects. Hum Mol Genet 23:2711–2720. doi:10.1093/hmg/ddt664
Feberwee HE et al (2014) Novel BCOR mutations in patients with oculofaciocardiodental (OFCD) syndrome. Clin Genet 85:194–197. doi:10.1111/cge.12125
Feng H et al (1994) Evidence, from family studies, for linkage disequilibrium between TGFA and a gene for nonsyndromic cleft lip with or without cleft palate. Am J Hum Genet 55:932–936
Feng C, Duan W, Zhang D, Zhang E, Xu Z, Lu L (2014) C392T polymorphism of the Wnt10a gene in non-syndromic oral cleft in a northeastern Chinese population. Br J Oral Maxillofac Surg 52:751–755. doi:10.1016/j.bjoms.2014.06.001
FitzPatrick DR et al (2003) Identification of SATB2 as the cleft palate gene on 2q32–q33. Hum Mol Genet 12:2491–2501. doi:10.1093/hmg/ddg248
Frebourg T et al (2006) Cleft lip/palate and CDH1/E-cadherin mutations in families with hereditary diffuse gastric cancer. J Med Genet 43:138–142. doi:10.1136/jmg.2005.031385
Garg A, Goyal S (2009) Tessier number 5 cleft. Indian Pediatr 46:907
Ghassibé M, Revencu N, Bayet B, Gillerot Y, Vanwijck R, Verellen-Dumoulin C, Vikkula M (2004) Six families with van der Woude and/or popliteal pterygium syndrome: all with a mutation in the IRF6 gene. J Med Genet 41:e15
Graber LW (1978) Congenital absence of teeth: a review with emphasis on inheritance patterns. J Am Dent Assoc 96:266–275
Green RM et al (2015) Tfap2a-dependent changes in mouse facial morphology result in clefting that can be ameliorated by a reduction in Fgf8 gene dosage. Dis Models Mech 8:31–43. doi:10.1242/dmm.017616
Gurramkonda VB, Hussain SA, Murthy J, Lakkakula BV (2015) Two promoter polymorphisms in TBX22 are associated with the risk of NSCLP in Indian women. Clin Dysmorphol 24:140–143. doi:10.1097/mcd.0000000000000088
Hamachi T, Sasaki Y, Hidaka K, Nakata M (2003) Association between palatal morphogenesis and Pax9 expression pattern in CL/P embryos with clefting during palatal development. Arch Oral Biol 48:581–587
Harris MA et al (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32:D258–D261. doi:10.1093/nar/gkh036
He F et al (2010) Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Dev Biol 347:109–121. doi:10.1016/j.ydbio.2010.08.014
He H, Han D, Feng H, Qu H, Song S, Bai B, Zhang Z (2013) Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PLoS One 8:e80393. doi:10.1371/journal.pone.0080393
Heldin CH, Landstrom M, Moustakas A (2009) Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol 21:166–176. doi:10.1016/j.ceb.2009.01.021
Heliövaara A, Rantanen I, Arte S (2011) Dental development and tooth agenesis in children with velocardiofacial syndrome. Int J Paediatr Dent 21:446–450. doi:10.1111/j.1365-263X.2011.01148.x
Herr A, Meunier D, Muller I, Rump A, Fundele R, Ropers HH, Nuber UA (2003) Expression of mouse Tbx22 supports its role in palatogenesis and glossogenesis. Dev Dyn 226:579–586. doi:10.1002/dvdy.10260
Hosokawa R, Deng X, Takamori K, Xu X, Urata M, Bringas P Jr, Chai Y (2009) Epithelial-specific requirement of FGFR2 signaling during tooth and palate development. J Exp Zool Part B, Mol Dev Evol 312:343–350. doi:10.1002/jez.b.21274
Howe BJ et al (2015) Spectrum of dental phenotypes in nonsyndromic orofacial clefting. J Dent Res 94:905–912. doi:10.1177/0022034515588281
Hu X, Lin C, Shen B, Ruan N, Guan Z, Chen Y, Zhang Y (2014) Conserved odontogenic potential in embryonic dental tissues. J Dent Res 93:490–495. doi:10.1177/0022034514523988
Huang F, Hu X, Fang C, Liu H, Lin C, Zhang Y, Hu X (2015) Expression profile of critical genes involved in FGF signaling pathway in the developing human primary dentition. Histochem Cell Biol 144:457–469. doi:10.1007/s00418-015-1358-7
Ingraham CR et al (2006) Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat Genet 38:1335–1340. doi:10.1038/ng1903
Item CB et al (2004) Gene symbol: IRF6. Disease: Van der Woude syndrome. Hum Genet 115:175
Itin PH, Fistarol SK (2004) Ectodermal dysplasias. Am J Med Genet Part C, Semin Med Genet 131:45–51. doi:10.1002/ajmg.c.30033
Janiszewska-Olszowska J, Gawrych E, Dydyk A, Studniak E, Biadun-Poplawska A, Zajaczek S (2013) Oro-palatal dysplasia Bettex-Graf–clinical findings, genetic background, treatment. J Cranio-Maxillo-Fac Surg 41:e29–e32. doi:10.1016/j.jcms.2012.06.002
Jugessur A et al (2003) Cleft palate, transforming growth factor alpha gene variants, and maternal exposures: assessing gene-environment interactions in case-parent triads. Genet Epidemiol 25:367–374. doi:10.1002/gepi.10268
Kaewkhampa A, Jotikasthira D, Malaivijitnond S, Kantaputra P (2012) TBX22 mutation associated with cleft lip/palate, hypodontia, and limb anomaly. Cleft Palate-Craniofac J 49:240–244. doi:10.1597/10-208
Kaiser AS, Maas B, Wolff A, Sutter C, Janssen JW, Hinderhofer K, Moog U (2015) Characterization of the first intragenic SATB2 duplication in a girl with intellectual disability, nearly absent speech and suspected hypodontia. Eur J Hum Genet 23:704–707. doi:10.1038/ejhg.2014.163
Kantaputra PN (2014) BCOR mutations and unstoppable root growth: a commentary on oculofaciocardiodental syndrome: novel BCOR mutations and expression in dental cells. J Hum Genet 59:297–299. doi:10.1038/jhg.2014.31
Kantaputra PN et al (2011) Cleft lip with cleft palate, ankyloglossia, and hypodontia are associated with TBX22 mutations. J Dent Res 90:450–455. doi:10.1177/0022034510391052
Kantaputra P, Kaewgahya M, Jotikasthira D, Kantaputra W (2014) Tricho-odonto-onycho-dermal dysplasia and WNT10A mutations. Am J Med Genet Part A 164:1041–1048. doi:10.1002/ajmg.a.36388
Kavitha B, Priyadharshini V, Sivapathasundharam B, Saraswathi TR (2010) Role of genes in oro-dental diseases. Indian J Dent Res 21:270–274. doi:10.4103/0970-9290.66646
Khalaf K, Miskelly J, Voge E, Macfarlane TV (2014) Prevalence of hypodontia and associated factors: a systematic review and meta-analysis. J Orthod 41:299–316. doi:10.1179/1465313314y.0000000116
Kim HG et al (2005) Hypogonadotropic hypogonadism and cleft lip and palate caused by a balanced translocation producing haploinsufficiency for FGFR1. J Med Genet 42:666–672. doi:10.1136/jmg.2004.026989
Kim SM, Lee JH, Jabaiti S, Lee SK, Choi JY (2009) Tbx22 expressions during palatal development in fetuses with glucocorticoid-/alcohol-induced C57BL/6 N cleft palates. J Craniofac Surg 20:1316–1326. doi:10.1097/SCS.0b013e3181ae6686
Kist R, Greally E, Peters H (2007) Derivation of a mouse model for conditional inactivation of Pax9. Genesis 45:460–464. doi:10.1002/dvg.20295
Klein OD, Oberoi S, Huysseune A, Hovorakova M, Peterka M, Peterkova R (2013) Developmental disorders of the dentition: an update. Am J Med Genet Part C, Semin Med Genet 163:318–332. doi:10.1002/ajmg.c.31382
Kondo S et al (2002) Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet 32:285–289. doi:10.1038/ng985
Kousa YA, Schutte BC (2015) Toward an orofacial gene regulatory network. Dev Dyn. doi:10.1002/dvdy.24341
Kouskoura T et al (2011) The genetic basis of craniofacial and dental abnormalities. Schweizer Monatsschrift fur Zahnmedizin 121:636–646
Krasone K, Lace B, Akota I, Care R, Deeley K, Kuchler EC, Vieira AR (2014) IRF6 AP-2a binding site promoter polymorphism is associated with oral clefts in Latvia. Stomatologija 16:132–136
Lallemand Y, Nicola MA, Ramos C, Bach A, Cloment CS, Robert B (2005) Analysis of Msx1; Msx2 double mutants reveals multiple roles for Msx genes in limb development. Development 132:3003–3014. doi:10.1242/dev.01877
Lam C, Ou JC, Billingsley EM (2013) “PTCH”-ing it together: a basal cell nevus syndrome review. Dermatol Surg 39:1557–1572. doi:10.1111/dsu.12241
Lammi L et al (2004) Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 74:1043–1050. doi:10.1086/386293
Layman LC (2013) Clinical genetic testing for Kallmann syndrome. J Clin Endocrinol Metabol 98:1860–1862. doi:10.1210/jc.2013-1624
Leoyklang P, Suphapeetiporn K, Siriwan P, Desudchit T, Chaowanapanja P, Gahl WA, Shotelersuk V (2007) Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum Mutat 28:732–738. doi:10.1002/humu.20515
Leslie EJ, Marazita ML (2013) Genetics of cleft lip and cleft palate. Am J Med Genet Part C, Seminars Med Genet 163:246–258. doi:10.1002/ajmg.c.31381
Letra A, Menezes R, Granjeiro JM, Vieira AR (2009) AXIN2 and CDH1 polymorphisms, tooth agenesis, and oral clefts Birth defects research Part A. Clin Mol Teratol 85:169–173. doi:10.1002/bdra.20489
Letra A et al (2012) Interaction between IRF6 and TGFA genes contribute to the risk of nonsyndromic cleft lip/palate. PLoS One 7:e45441. doi:10.1371/journal.pone.0045441
Liang J, Zhu L, Meng L, Chen D, Bian Z (2012) Novel nonsense mutation in MSX1 causes tooth agenesis with cleft lip in a Chinese family. Eur J Oral Sci 120:278–282. doi:10.1111/j.1600-0722.2012.00965.x
Liang J, Von den Hoff J, Lange J, Ren Y, Bian Z, Carels CE (2016) MSX1 mutations and associated disease phenotypes: genotype-phenotype relations. Eur J Hum Genet. doi:10.1038/ejhg.2016.78
Lidral AC, Reising BC (2002) The role of MSX1 in human tooth agenesis. J Dent Res 81:274–278
Lieden A, Kvarnung M, Nilssson D, Sahlin E, Lundberg ES (2014) Intragenic duplication–a novel causative mechanism for SATB2-associated syndrome. Am J Med Genet Part A 164:3083–3087. doi:10.1002/ajmg.a.36769
Lin JY et al (2008) Association of bone morphogenetic protein 4 gene polymorphisms with nonsyndromic cleft lip with or without cleft palate in Chinese children. DNA Cell Biol 27:601–605. doi:10.1089/dna.2008.0777
Liu W, Sun X, Braut A, Mishina Y, Behringer RR, Mina M, Martin JF (2005) Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 132:1453–1461. doi:10.1242/dev.01676
Loeys BL et al (2005) A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 37:275–281. doi:10.1038/ng1511
Lu MF, Pressman C, Dyer R, Johnson RL, Martin JF (1999) Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature 401:276–278. doi:10.1038/45797
Ludwig KU et al (2012) Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat Genet 44:968–971. doi:10.1038/ng.2360
Maas NM et al (2008) Genotype-phenotype correlation in 21 patients with Wolf-Hirschhorn syndrome using high resolution array comparative genome hybridisation (CGH). J Med Genet 45:71–80. doi:10.1136/jmg.2007.052910
Macca M, Franco B (2009) The molecular basis of oral-facial-digital syndrome, type 1. Am J Med Genet Part C, Semin Med Genet 151:318–325. doi:10.1002/ajmg.c.30224
Macias MJ, Martin-Malpartida P, Massague J (2015) Structural determinants of Smad function in TGF-beta signaling. Trends Biochem Sci 40:296–308. doi:10.1016/j.tibs.2015.03.012
Mangold E, Ludwig KU, Nothen MM (2011) Breakthroughs in the genetics of orofacial clefting. Trends Mol Med 17:725–733. doi:10.1016/j.molmed.2011.07.007
Marçano AC et al (2004) TBX22 mutations are a frequent cause of cleft palate. J Med Genet 41:68–74
Massagué J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19:2783–2810. doi:10.1101/gad.1350705
Massink MP et al (2015) Loss-of-function mutations in the WNT co-receptor LRP6 cause autosomal-dominant oligodontia. Am J Hum Genet 97:621–626. doi:10.1016/j.ajhg.2015.08.014
Matsumoto N, Niikawa N (2003) Kabuki make-up syndrome: a review. Am J Med Genet Part C, Semin Med Genet 117:57–65. doi:10.1002/ajmg.c.10020
McDade SS et al (2012) Genome-wide analysis of p63 binding sites identifies AP-2 factors as co-regulators of epidermal differentiation. Nucleic Acids Res 40:7190–7206. doi:10.1093/nar/gks389
McGrath JA et al (2001) Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet 10:221–229
McKenzie F, Turner A, Withers S, Dalzell P, McGlynn M, Kirk EP (2002) Dominant inheritance of cleft palate, microstomia and micrognathia–possible linkage to the fragile site at 16q22 (FRA16B). Clin Dysmorphol 11:237–241
Menezes R et al (2010) Studies with Wnt genes and nonsyndromic cleft lip and palate Birth defects research Part A. Clin Mol Teratol 88:995–1000. doi:10.1002/bdra.20720
Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A (1999) p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398:708–713. doi:10.1038/19531
Milunsky JM et al (2008) TFAP2A mutations result in branchio-oculo-facial syndrome. Am J Hum Genet 82:1171–1177. doi:10.1016/j.ajhg.2008.03.005
Minones-Suarez L, Mas-Vidal A, Fernandez-Toral J, Llano-Rivas I, Gonzalez-Garcia M (2012) A novel mutation in the IRF6 gene associated with facial asymmetry in a family affected with Van der Woude syndrome. Pediatr Dermatol 29:768–770. doi:10.1111/j.1525-1470.2011.01575.x
Mitchell K et al (2012) Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am J Hum Genet 90:69–75. doi:10.1016/j.ajhg.2011.11.013
Modesto A, Moreno LM, Krahn K, King S, Lidral AC (2006) MSX1 and orofacial clefting with and without tooth agenesis. J Dent Res 85:542–546
Monsoro-Burq AH (2015) PAX transcription factors in neural crest development. Semin Cell Dev Biol 44:87–96. doi:10.1016/j.semcdb.2015.09.015
Morin PJ (1999) beta-catenin signaling and cancer. BioEssays 21:1021–1030. doi:10.1002/(sici)1521-1878(199912)22:1<1021:aid-bies6>3.0.co;2-p
Mossey PA, Catilla E (2003) Global registry and database on craniofacial anomalies: report of a WHO Registry Meeting on Craniofacial Anomalies. Paper presented at the Geneva World Health Organization, http://www.who.int/genomics/anomalies/en/CFA-RegistryMeeting-2001.pdf
Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC (2009) Cleft Lip and palate. Lancet 374:1773–1785. doi:10.1016/s0140-6736(09)60695-4
Mostowska A, Hozyasz KK, Biedziak B, Wojcicki P, Lianeri M, Jagodzinski PP (2012) Genotype and haplotype analysis of WNT genes in non-syndromic cleft lip with or without cleft palate. Eur J Oral Sci 120:1–8. doi:10.1111/j.1600-0722.2011.00938.x
Muenke M, Schell U (1995) Fibroblast-growth-factor receptor mutations in human skeletal disorders Trends in genetics. TIG 11:308–313
Mues G, Bonds J, Xiang L, Vieira AR, Seymen F, Klein O, D’Souza RN (2014) The WNT10A gene in ectodermal dysplasias and selective tooth agenesis. Am J Med Genet Part A 164:2455–2460. doi:10.1002/ajmg.a.36520
Murthy J, Bhaskar L (2009) Current concepts in genetics of nonsyndromic clefts. Indian J Plast Surg 42:68–81. doi:10.4103/0970-0358.53004
Nakatomi M et al (2010) Genetic interactions between Pax9 and Msx1 regulate lip development and several stages of tooth morphogenesis. Dev Biol 340:438–449. doi:10.1016/j.ydbio.2010.01.031
Nieminen P, Kotilainen J, Aalto Y, Knuutila S, Pirinen S, Thesleff I (2003) MSX1 gene is deleted in Wolf-Hirschhorn syndrome patients with oligodontia. J Dent Res 82:1013–1017
Ockeloen CW et al (2016) Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet Med. doi:10.1038/gim.2016.10
Ogawa T, Kapadia H, Wang B, D’Souza RN (2005) Studies on Pax9-Msx1 protein interactions. Arch Oral Biol 50:141–145. doi:10.1016/j.archoralbio.2004.09.011
Ogawa T, Kapadia H, Feng JQ, Raghow R, Peters H, D’Souza RN (2006) Functional consequences of interactions between Pax9 and Msx1 genes in normal and abnormal tooth development. J Biol Chem 281:18363–18369. doi:10.1074/jbc.M601543200
Orioli IM, Vieira AR, Castilla EE, Ming JE, Muenke M (2002) Mutational analysis of the Sonic Hedgehog gene in 220 newborns with oral clefts in a South American (ECLAMC) population. Am J Med Genet 108:12–15
Packham EA, Brook JD (2003) T-box genes in human disorders Human molecular genetics 12 Spec No 1:R37–44
Paradowska-Stolarz AM (2014) Wolf-Hirschhorn syndrome (WHS)—literature review on the features of the syndrome. Adv Clin Exp Med 23:485–489
Park K et al (2005) Homeobox Msx1 interacts with p53 tumor suppressor and inhibits tumor growth by inducing apoptosis. Cancer Res 65:749–757
Park JW et al (2007) Association between IRF6 and nonsyndromic cleft lip with or without cleft palate in four populations. Genet Med 9:219–227. doi:10.1097/GIM.0b013e3180423cca
Pauws E et al (2009) Tbx22null mice have a submucous cleft palate due to reduced palatal bone formation and also display ankyloglossia and choanal atresia phenotypes. Hum Mol Genet 18:4171–4179. doi:10.1093/hmg/ddp368
Pauws E et al (2013) X-linked CHARGE-like Abruzzo-Erickson syndrome and classic cleft palate with ankyloglossia result from TBX22 splicing mutations. Clin Genet 83:352–358. doi:10.1111/j.1399-0004.2012.01930.x
Pegelow M et al (2008) Familial non-syndromic cleft lip and palate–analysis of the IRF6 gene and clinical phenotypes. Eur J Orthod 30:169–175. doi:10.1093/ejo/cjm097
Person AD et al (2010) WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev Dynam 239:327–337. doi:10.1002/dvdy.22156
Peters H, Neubuser A, Balling R (1998a) Pax genes and organogenesis: Pax9 meets tooth development. Eur J Oral Sci 106(Suppl 1):38–43
Peters H, Neubuser A, Kratochwil K, Balling R (1998b) Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev 12:2735–2747
Peyrard-Janvid M et al (2005) Novel and de novo mutations of the IRF6 gene detected in patients with Van der Woude or popliteal pterygium syndrome. Eur J Hum Genet 13:1261–1267. doi:10.1038/sj.ejhg.5201493
Peyrard-Janvid M et al (2014) Dominant mutations in GRHL3 cause Van der Woude Syndrome and disrupt oral periderm development. Am J Hum Genet 94:23–32. doi:10.1016/j.ajhg.2013.11.009
Pezzetti F, Scapoli L, Martinelli M, Carinci F, Bodo M, Carinci P, Tognon M (1998) A locus in 2p13-p14 (OFC2), in addition to that mapped in 6p23, is involved in nonsyndromic familial orofacial cleft malformation. Genomics 50:299–305. doi:10.1006/geno.1998.5273
Pitteloud N et al (2006) Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA 103:6281–6286. doi:10.1073/pnas.0600962103
Polder BJ, Van’t Hof MA, Van der Linden FP, Kuijpers-Jagtman AM (2004) A meta-analysis of the prevalence of dental agenesis of permanent teeth. Commun Dent Oral Epidemiol 32:217–226. doi:10.1111/j.1600-0528.2004.00158.x
Prontera P, Garelli E, Isidori I, Mencarelli A, Carando A, Silengo MC, Donti E (2011) Cleft palate and ADULT phenotype in a patient with a novel TP63 mutation suggests lumping of EEC/LM/ADULT syndromes into a unique entity: ELA syndrome. Am J Med Genet Part A 155:2746–2749. doi:10.1002/ajmg.a.34270
Rahimov F et al (2008) Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet 40:1341–1347. doi:10.1038/ng.242
Rainger JK et al (2014) Disruption of SATB2 or its long-range cis-regulation by SOX9 causes a syndromic form of Pierre Robin sequence. Hum Mol Genet 23:2569–2579. doi:10.1093/hmg/ddt647
Rakhshan V, Rakhshan H (2015) Meta-analysis of congenitally missing teeth in the permanent dentition: Prevalence, variations across ethnicities, regions and time. Int Orthod 13:261–273. doi:10.1016/j.ortho.2015.06.008
Ranta R (1972) A comparative study of tooth formation in the permanent dentition of Finnish children with cleft lip and palate. orthopantomogr Study Proc Finn Dent Soc Suomen Hammaslaakariseuran toimituksia 68:58–66
Ranta R (1986) A review of tooth formation in children with cleft lip/palate. Am J Orthod Dentofac Orthop 90:11–18
Richardson RJ, Dixon J, Jiang R, Dixon MJ (2009) Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum Mol Genet 18:2632–2642. doi:10.1093/hmg/ddp201
Rifai L et al (2010) Ectodermal dysplasia-like syndrome with mental retardation due to contiguous gene deletion: further clinical and molecular delineation of del(2q32) syndrome. Am J Med Genet Part A 152:111–117. doi:10.1002/ajmg.a.33164
Riley BM et al (2007) A genome-wide linkage scan for cleft lip and cleft palate identifies a novel locus on 8p11–23. Am J Med Genet Part A 143:846–852. doi:10.1002/ajmg.a.31673
Roessler E et al (1996) Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet 14:357–360. doi:10.1038/ng1196-357
Roifman M, Brunner H, Lohr J, Mazzeu J, Chitayat D (2015) Autosomal dominant Robinow syndrome. In: Pagon RA et al (eds) GeneReviews®. University of Washington, Seattle University of Washington, Seattle, pp 1993–2016
Rosenfeld JA et al (2009) Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS One 4:e6568. doi:10.1371/journal.pone.0006568
Sahoo T et al (2011) Microdeletion 20p12.3 involving BMP2 contributes to syndromic forms of cleft palate. Am J Med Genet Part A 155:1646–1653. doi:10.1002/ajmg.a.34063
Sander A et al (1995) Microsatellite-based fine mapping of the Van der Woude syndrome locus to an interval of 4.1 cM between D1S245 and D1S414. Am J Hum Genet 56:310–318
Sarkar L, Sharpe PT (1999) Expression of Wnt signalling pathway genes during tooth development. Mech Dev 85:197–200
Satokata I, Maas R (1994) Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet 6:348–356. doi:10.1038/ng0494-348
Scapoli L et al (2006) Study of the PVRL1 gene in Italian nonsyndromic cleft lip patients with or without cleft palate. Ann Hum Genet 70:410–413. doi:10.1111/j.1529-8817.2005.00237.x
Schambony A, Kunz M, Gradl D (2004) Cross-regulation of Wnt signaling and cell adhesion. Differ Res Biol Divers 72:307–318. doi:10.1111/j.1432-0436.2004.07207002.x
Schinzel A, Klausler M (1986) The Van der Woude syndrome (dominantly inherited lip pits and clefts). J Med Genet 23:291–294
Schinzel A, Schmid W (1980) Interstitial deletion of the long arm of chromosome 1, del(1)(q21 leads to q25) in a profoundly retarded 8-year-old girl with multiple anomalies. Clin Genet 18:305–313
Schutte BC et al (1999) Microdeletions at chromosome bands 1q32-q41 as a cause of Van der Woude syndrome. Am J Med Genet 84:145–150
Seo YJ, Park JW, Kim YH, Baek SH (2013) Associations between the risk of tooth agenesis and single-nucleotide polymorphisms of MSX1 and PAX9 genes in nonsyndromic cleft patients The. Angle Orthod 83:1036–1042. doi:10.2319/020513-104.1
Shapira Y, Lubit E, Kuftinec MM (1999) Congenitally missing second premolars in cleft lip and cleft palate children. Am J Orthod Dentofac Orthop 115:396–400
Shaw GM, Wasserman CR, Block G, Lammer EJ (1996a) High maternal vitamin A intake and risk of anomalies of structures with a cranial neural crest cell contribution. Lancet 347:899–900
Shaw GM, Wasserman CR, Lammer EJ, O’Malley CD, Murray JC, Basart AM, Tolarova MM (1996b) Orofacial clefts, parental cigarette smoking, and transforming growth factor-alpha gene variants. Am J Hum Genet 58:551–561
Shimojima K, Shimada S, Sugawara M, Yoshikawa N, Niijima S, Urao M, Yamamoto T (2013) Challenges in genetic counseling because of intra-familial phenotypic variation of oral-facial-digital syndrome type 1. Congenit Anom 53:155–159. doi:10.1111/j.1741-4520.2012.00384.x
Shkoukani MA, Chen M, Vong A (2013) Cleft lip—a comprehensive review. Front Pediatr 1:53. doi:10.3389/fped.2013.00053
Simões-Costa M, Bronner ME (2015) Establishing neural crest identity: a gene regulatory recipe. Development 142:242–257. doi:10.1242/dev.105445
Simonis N et al (2013) FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. J Med Genet 50:585–592. doi:10.1136/jmedgenet-2013-101603
Slayton RL, Williams L, Murray JC, Wheeler JJ, Lidral AC, Nishimura CJ (2003) Genetic association studies of cleft lip and/or palate with hypodontia outside the cleft region. Cleft Palate-Craniofac J 40:274–279. doi:10.1597/1545-1569(2003)040<0274:gasocl>2.0.co;2
Smalley MJ, Dale TC (1999) Wnt signalling in mammalian development and cancer. Cancer Metastasis Rev 18:215–230
Song L et al (2009) Lrp6-mediated canonical Wnt signaling is required for lip formation and fusion. Development 136:3161–3171. doi:10.1242/dev.037440
Song S, Zhao R, He H, Zhang J, Feng H, Lin L (2014) WNT10A variants are associated with non-syndromic tooth agenesis in the general population. Hum Genet 133:117–124. doi:10.1007/s00439-013-1360-x
Sözen MA, Suzuki K, Tolarova MM, Bustos T, Fernandez Iglesias JE, Spritz RA (2001) Mutation of PVRL1 is associated with sporadic, non-syndromic cleft lip/palate in northern Venezuela. Nat Genet 29:141–142. doi:10.1038/ng740
Sözen MA, Hecht JT, Spritz RA (2009) Mutation analysis of the PVRL1 gene in caucasians with nonsyndromic cleft lip/palate. Genet Test Mol Biomark 13:617–621. doi:10.1089/gtmb.2009.0052
Sripathomsawat W, Tanpaiboon P, Heering J, Dötsch V, Hennekam RC, Kantaputra P (2011) Phenotypic analysis of Arg227 mutations of TP63 with emphasis on dental phenotype and micturition difficulties in EEC syndrome. Am J Med Genet A 155A(1):228–232. doi:10.1002/ajmg.a.33768
Steele JA, Hansen H, Arn P, Kwong PC (2005) Spectrum of phenotypic manifestations from a single point mutation of the p63 gene, including new cutaneous and immunologic findings. Pediatr Dermatol 22:415–419. doi:10.1111/j.1525-1470.2005.00105.x
Stoler JM, Rosen H, Desai U, Mulliken JB, Meara JG, Rogers GF (2009) Cleft palate in Pfeiffer syndrome. J Craniofac Surg 20:1375–1377. doi:10.1097/SCS.0b013e3181ae42e4
Suazo J, Santos JL, Jara L, Blanco R (2010) Association between bone morphogenetic protein 4 gene polymorphisms with nonsyndromic cleft lip with or without cleft palate in a chilean population. DNA Cell Biol 29:59–64. doi:10.1089/dna.2009.0944
Suphapeetiporn K, Tongkobpetch S, Siriwan P, Shotelersuk V (2007) TBX22 mutations are a frequent cause of non-syndromic cleft palate in the Thai population. Clin Genet 72:478–483. doi:10.1111/j.1399-0004.2007.00891.x
Suzuki K, Hu D, Bustos T, Zlotogora J, Richieri-Costa A, Helms JA, Spritz RA (2000) Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat Genet 25:427–430. doi:10.1038/78119
Suzuki S et al (2009) Mutations in BMP4 are associated with subepithelial, microform, and overt cleft lip. Am J Hum Genet 84:406–411. doi:10.1016/j.ajhg.2009.02.002
Tadini G, Santagada F, Brena M, Pezzani L, Nannini P (2013) Ectodermal dysplasias: the p63 tail. Giornale italiano di dermatologia e venereologia 148:53–58
Tallon-Walton V, Nieminen P, Arte S, Ustrell-Torrent JM, Carvalho-Lobato P, Manzanares-Cespedes MC (2010) Oral findings in Midline Syndrome: a case report and literature review. Med oral, patologia oral y cirugia bucal 15:e579–e582
Taneyhill LA (2008) To adhere or not to adhere: the role of Cadherins in neural crest development. Cell Adhes Migr 2:223–230
Tommiska J, Kansakoski J, Christiansen P, Jorgensen N, Lawaetz JG, Juul A, Raivio T (2014) Genetics of congenital hypogonadotropic hypogonadism in Denmark. Eur J Med Genet 57:345–348. doi:10.1016/j.ejmg.2014.04.002
Tompkins K (2006) Molecular mechanisms of cytodifferentiation in mammalian tooth development. Connect Tissue Res 47:111–118. doi:10.1080/03008200600727756
Toriello HV, Franco B (1993) Oral-Facial-Digital Syndrome Type I. In: Pagon RA et al. (eds) GeneReviews(R). University of Washington, Seattle University of Washington, Seattle. All rights reserved, Seattle (WA)
Turhani D, Item CB, Watzinger E, Sinko K, Watzinger F, Lauer G, Ewers R (2005) Mutation analysis of CLPTM 1 and PVRL 1 genes in patients with non-syndromic clefts of lip, alveolus and palate. J Cranio-Maxillo-Fac Surg 33:301–306. doi:10.1016/j.jcms.2005.04.004
Urquhart J, Black GC, Clayton-Smith J (2009) 4.5 Mb microdeletion in chromosome band 2q33.1 associated with learning disability and cleft palate. Eur J Med Genet 52:454–457. doi:10.1016/j.ejmg.2009.06.003
Uslu VV, Petretich M, Ruf S, Langenfeld K, Fonseca NA, Marioni JC, Spitz F (2014) Long-range enhancers regulating Myc expression are required for normal facial morphogenesis. Nat Genet 46:753–758. doi:10.1038/ng.2971
van Bokhoven H et al (1999) Limb mammary syndrome: a new genetic disorder with mammary hypoplasia, ectrodactyly, and other Hand/Foot anomalies maps to human chromosome 3q27. Am J Hum Genet 64:538–546. doi:10.1086/302246
van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK (2000) MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet 24:342–343. doi:10.1038/74155
van den Boogaard MJ et al (2012) Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet 49:327–331. doi:10.1136/jmedgenet-2012-100750
Van Laarhoven PM et al (2015) Kabuki syndrome genes KMT2D and KDM6A: functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum Mol Genet 24:4443–4453. doi:10.1093/hmg/ddv180
Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE (1996) A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet 13:417–421. doi:10.1038/ng0896-417
Verstraeten B, Sanders E, van Hengel J, Huysseune A (2010) Zebrafish teeth as a model for repetitive epithelial morphogenesis: dynamics of E-cadherin expression. BMC Dev Biol 10:58. doi:10.1186/1471-213x-10-58
Vieira AR (2003) Oral clefts and syndromic forms of tooth agenesis as models for genetics of isolated tooth agenesis. J Dent Res 82:162–165
Vieira AR, Modesto A, Meira R, Barbosa AR, Lidral AC, Murray JC (2007) Interferon regulatory factor 6 (IRF6) and fibroblast growth factor receptor 1 (FGFR1) contribute to human tooth agenesis. Am J Med Genet Part A 143:538–545. doi:10.1002/ajmg.a.31620
Vink CP et al (2014) Variability in dentofacial phenotypes in four families with WNT10A mutations. Eur H Hum Genet 22:1063–1070. doi:10.1038/ejhg.2013.300
Vogelaar IP et al (2013) Identification of germline mutations in the cancer predisposing gene CDH1 in patients with orofacial clefts. Hum Mol Genet 22:919–926. doi:10.1093/hmg/dds497
Wang Y, Zhao H, Zhang X, Feng H (2003) Novel identification of a four-base-pair deletion mutation in PITX2 in a Rieger syndrome family. J Dent Res 82:1008–1012
Wang J et al (2013) PAX9 polymorphism and susceptibility to sporadic non-syndromic severe anodontia: a case-control study in southwest China. J Appl Oral Sci 21:256–264. doi:10.1590/1679-775720130079
Wienker TF, Hudek G, Bissbort S, Mayerova A, Mauff G, Bender K (1987) Linkage studies in a pedigree with Van der Woude syndrome. J Med Genet 24:160–162
Wilkie AO (2009) Pitfalls in the phylogenomic evaluation of human disease-causing mutations. J Biol 8:26. doi:10.1186/jbiol127
Williams ES, Uhas KA, Bunke BP, Garber KB, Martin CL (2012) Cleft palate in a multigenerational family with a microdeletion of 20p12.3 involving BMP2. Am J Med Genet Part A 158:2616–2620. doi:10.1002/ajmg.a.35594
Wojtal D et al (2016) Spell checking nature: versatility of CRISPR/Cas9 for developing treatments for inherited disorders. Am J Hum Genet 98:90–101. doi:10.1016/j.ajhg.2015.11.012
Wong FK, Hagg U (2004) An update on the aetiology of orofacial clefts. Hong Kong Med J 10:331–336
Wong ST et al (1989) The TGF-alpha precursor expressed on the cell surface binds to the EGF receptor on adjacent cells, leading to signal transduction. Cell 56:495–506
Wong FK et al (1999) Clinical and genetic studies of Van der Woude syndrome in Sweden. Acta Odontol Scand 57:72–76
Wrana JL, Attisano L, Wieser R, Ventura F, Massague J (1994) Mechanism of activation of the TGF-beta receptor. Nature 370:341–347. doi:10.1038/370341a0
Xu N, Qin Y, Reindollar RH, Tho SP, McDonough PG, Layman LC (2007) A mutation in the fibroblast growth factor receptor 1 gene causes fully penetrant normosmic isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metabol 92:1155–1158. doi:10.1210/jc.2006-1183
Xu H et al (2015) Novel FGFR1 and KISS1R mutations in Chinese kallmann syndrome males with Cleft Lip/Palate. BioMed Res Int 2015:649698. doi:10.1155/2015/649698
Yang A et al (1998) p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 2:305–316
Yang A et al (1999) p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398:714–718. doi:10.1038/19539
Yao T, Yang L, Li PQ, Wu H, Xie HB, Shen X, Xie XD (2011) Association of Wnt3A gene variants with non-syndromic cleft lip with or without cleft palate in Chinese population. Arch Oral Biol 56:73–78. doi:10.1016/j.archoralbio.2010.09.002
Ye XQ, Jin HX, Shi LS, Fan MW, Song GT, Fan HL, Bian Z (2005) Identification of novel mutations of IRF6 gene in Chinese families with Van der Woude syndrome. Int J Mol Med 16:851–856
Yildirim M, Seymen F, Deeley K, Cooper ME, Vieira AR (2012) Defining predictors of cleft lip and palate risk. J Dent Res 91:556–561. doi:10.1177/0022034512444928
Yin W, Bian Z (2015) The Gene Network Underlying Hypodontia. J Dent Res 94:878–885. doi:10.1177/0022034515583999
Yin W, Ye X, Shi L, Wang QK, Jin H, Wang P, Bian Z (2010) TP63 gene mutations in Chinese P63 syndrome patients. J Dent Res 89:813–817. doi:10.1177/0022034510366804
Yoshida K, Hayashi R, Fujita H, Kubota M, Kondo M, Shimomura Y, Niizeki H (2015) Novel homozygous mutation, c.400C > T (p.Arg134*), in the PVRL1 gene underlies cleft lip/palate-ectodermal dysplasia syndrome in an Asian patient The. J Dermatol 42:715–719. doi:10.1111/1346-8138.12882
Zawiślak A, Wozniak K, Jakubowska A, Lubinski J, Kawala B, Znamirowska-Bajowska A (2014) Polymorphic variants in VAX1 gene (rs7078160) and BMP4 gene (rs762642) and the risk of non-syndromic orofacial clefts in the Polish population. Dev Period Med 18:16–22
Zeiger JS, Beaty TH, Liang KY (2005) Oral clefts, maternal smoking, and TGFA: a meta-analysis of gene-environment interaction. Cleft Palate-Craniofac J 42:58–63. doi:10.1597/02-128.1
Zenaty D, Bretones P, Lambe C, Guemas I, David M, Leger J, de Roux N (2006) Paediatric phenotype of Kallmann syndrome due to mutations of fibroblast growth factor receptor 1 (FGFR1). Mol Cell Endocrinol 254–255:78–83. doi:10.1016/j.mce.2006.04.006
Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y (2002) Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 129:4135–4146
Zhao X, Qu Z, Tickner J, Xu J, Dai K, Zhang X (2014) The role of SATB2 in skeletogenesis and human disease. Cytokine Growth Factor Rev 25:35–44. doi:10.1016/j.cytogfr.2013.12.010
Zucchero TM et al (2004) Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Eng J Med 351:769–780. doi:10.1056/NEJMoa032909
Acknowledgments
The authors acknowledge the support received from the European Science Foundation within the EUROCleftNet Programme (FC, ESF exchange visit Grants, No. 4918) and the Foundation Friends for OFC Research (CC, MP). Furthermore, we would like to thank Dr. Celeste van Heumen and Mrs. Dirkje Ter Meer (Radboud UMC of Nijmegen, the Netherlands) for providing the radiographs of the patient affected with anodontia of the permanent dentition, included in our figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
Additional information
M. Phan and F. Conte contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Literature searches: histories and search terms. (DOCX 24 kb)
Supplementary Table 2
All articles resulting from the literature search. (XLSX 46 kb)
Supplementary Table 3
Articles included based on the contents. (XLSX 19 kb)
Supplementary Table 4
Human mutations described as affecting the candidate genes reported in the included articles. (XLSX 28 kb)
Supplementary Table 5
Slim GO term analysis and gene clustering. (XLSX 23 kb)
Supplementary Table 6
Genomic loci identified from the included articles and their encompassed genes. (XLSX 39 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Phan, M., Conte, F., Khandelwal, K.D. et al. Tooth agenesis and orofacial clefting: genetic brothers in arms?. Hum Genet 135, 1299–1327 (2016). https://doi.org/10.1007/s00439-016-1733-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1733-z