
Abstract
Background
Pediatric cataract is a clinically and genetically heterogeneous disease which is a significant cause of lifelong visual impairment and treatable blindness. Our study aims to investigate the genotype spectrum in a group of Chinese patients with pediatric cataract.
Methods
We enrolled 39 families with pediatric cataract from October 2015 to April 2016. DNA samples of the probands were analyzed by target next-generation sequencing. Variants were validated using Sanger sequencing in the probands and available family members.
Results
In our cohort of 39 cases with different types of pediatric cataract, 23 cases were found to harbor putative pathogenic variants in 15 genes: CRYAA, CRYBA1, CRYBA4, CRYBB1, CRYGC, CRYGD, MIP, GCNT2, IARS2, NHS, BCOR, BFSP2, FYCO1, MAF, and PAX6. The mutation detection rates in the familial and sporadic cases were 75 and 47.8%, respectively. Of the 23 causative variants, over half were novel.
Conclusions
This is a rare report of systematic mutation screening analysis of pediatric cataract in a comparably large cohort of Chinese patients. Our observations enrich the mutation spectrum of pediatric cataract. Next-generation sequencing provides significant diagnostic information for pediatric cataract cases, especially when considering sporadic and subtle syndromal cases.
Similar content being viewed by others

Background
Pediatric cataract is often referred to as congenital or infantile cataract, characterized by any opacity of the lens presenting at birth or in the first year of life. With a global prevalence of 3–6 in 10,000 live births and accounting for 10% of childhood blindness worldwide, pediatric cataract is one of the most common causes of visual impairment and blindness in children [1,2,3]. Pediatric cataract either occurs as a systemic (syndromic) disease or as an isolated (non-syndromic) disease with or without other ocular malformations such as microcornea, microphthalmia, or anterior segment dysgenesis [4, 5]. While autosomal dominant inheritance is most common, autosomal recessive and X-linked inheritance have also been reported, indicating some degree of genetic heterogeneity in pediatric cataract. However, only 8–25% of cases have cataract-linked, inherited mutations [6]. Approximately 81.2% of pediatric cataract cases do not have a family history, suggesting that a significant proportion of cases are sporadic, but many of these cases lack a known underlying genetic cause [7].
Mutations in over 318 genes associated with cataracts had been reported prior to 29 January 2018 (http://cat-map.wustl.edu/), including genes coding for crystallins, intermediate filament proteins, cytoskeleton proteins, gap junction proteins, lens membrane proteins, and lens-associated transcription factors [8]. In this study, we characterize the clinical manifestations and identify pathogenic variants in a cohort of 39 pediatric cataract cases with a variety of inheritance patterns, including a high proportion of sporadic cases in non-consanguineous families. Determining the precise genetic causes of pediatric cataract has significant clinical relevance for defining clinical diagnoses, implementing early treatment strategies, and guiding genetic counseling.
Methods
Participants
Thirty-nine probands with bilateral pediatric cataract were investigated as part of this study, including 22 total cataracts, three perinuclear cataracts, two nuclear cataracts, one posterior polar cataract, and 11 undetermined types. All patients with histories of intrauterine infection, drug exposure, metabolic disorders, or malnutrition were excluded. A positive family history was observed in 41.02% (16/39), and non-syndromic cataract was the most common presentation (34/39). A pedigree analysis of the 16 familial cases suggested that 13 were caused by an autosomal dominant mode of inheritance and the remaining three were likely caused by autosomal recessive variants with no consanguinity. While 38 probands were diagnosed within the first year of life, the remaining proband was diagnosed at 5 years old. 15 probands also had nystagmus, six had microphthalmia and/or microcornea, and five had extra-ocular features.
Panel design, library preparation and next-generation sequencing
A panel of amplicons, targeting the coding exons and 25 bp flanking intronic sequences of 80 cataract-associated genes, was designed by combining data from the Online Mendelian Inheritance in Man (https://omim.org/) and an independent search of PubMed literature (https://www.ncbi.nlm.nih.gov/pubmed). The gene list is shown in Additional file 1: Table S1. The panel included 1811 amplicons with lengths ranging from 125 to 375 base pairs, covering 98.16% of the bases in the target regions.
Libraries were constructed using the Ion AmpliSeq Library Kit v2.0, and DNA fragments from individual samples were ligated with barcoded sequencing adaptors using the Ion Xpress Barcode Adapter 1–16 Kit according to the manufacturer’s instructions. Bar-coded libraries were selectively amplified by emulsion PCR, and ion sphere particles with qualified DNA were isolated and sequenced on Ion 318 Chips using the vendor-provided sequencing kit on the Ion Personal Genome Machine Sequencer (Life Technologies, Carlsbad, CA). Variants were initially called using Ion Torrent Variant Caller version 4.0 software and subsequently visualized using the Integrative Genomics Viewer to facilitate the detection of false variant calls. Confirmed variants were annotated using ANNOVAR (http://wannovar.wglab.org/), and respective minor allele frequencies were assesed in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP), 1000genomes (http://www.1000genomes.org/), Exome Variant Server (http://evs.gs.washington.edu/EVS/) and Exome Aggregation Consortium (ExAC) databases (http://exac.broadinstitute.org/). Heterozygous variants with minor allele frequencies > 0.01 were filtered out. Variants were validated using Sanger sequencing in the probands and available family members, and then analyzed for possible pathogenic significance according to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines [9].
Haplotype analysis and allele specific PCR
Six short tandem repeat (STR) microsatellite markers flanking PAX6 were genotyped in family #12, and six STR markers flanking GCNT2 were genotyped in family #9 and sporadic case #5. PCR products were separated by electrophoresis on 8% denaturing polyacrylamide gel, and allele fragments were detected with routine silver staining. Haplotypes were determined based on each individual’s genotype and kinship. To examine the low-level mosaicism in the unaffected parents in family #12, allele-specific PCR was performed with primers designed for the mutant allele. PCR products were detected by agarose gel electrophoresis. The primer sequences are listed in Additional file 2: Table S2.
Results
Targeted region analysis
Next-generation sequencing (NGS) was performed on DNA from the 39 perdiatric cataract probands to detect variants. NGS yielded an output of 5.38G bases with an average of 5.3 M reads/ chip. At least 230,000 reads with a quality score of AQ20 were obtained per sample, with a coverage of approximately 98.15% for the targeted regions and an average depth of 160. The mean read length was 211 bp (Additional file 3: Table S3).
Identification of suspected causative variants
Twenty-three of the 39 cases tested harbored putative pathogenic variants (Table 1), with mutation detection rates in the familial and sporadic cases of 75% (12/16) and 47.8% (11/23) (Fig. 1), respectively. These variants were spread over 15 cataract-associated genes, with variations in crystallins (CRYAA, CRYBA1, CRYBA4, CRYBB1, CRYGC, CRYGD) accounting for 39.13% (9/23) of the cases. In addition, likely causative variants were found in MIP in three families; GCNT2, IARS2, and NHS in two families each; and BCOR, BFSP2, FYCO1, MAF, and PAX6 in one family each. Among the 23 causative variants identified in this study, 12 variants were novel, with the remaining 11 variants already reported. According to the ACMG mutation guidelines, all of the variants were classified as “pathogenic” or “likely pathogenic”. Additionally, four variants classified as “uncertain significance” were identified in two familial and two sporadic cases (Additional file 4: Table S4 and Additional file 5: Figure S1). Two familial cases and 10 individuals with sporadic pediatric cataract had no variants of interest found in the 80 cataract-associated genes screened through this study (Additional file 5: Figure S1).

Mutation spectrum of familial and sporadic pediatric cataract cases. The mutation detection rates in the familial and sporadic cases were 75 and 47.8%, respectively. Mutations were found in 15 different genes, with high-penetrance mutations distributed in crystallins, MIP, GCNT2, IARS2, and NHS
Variants in crystallin genes
Variants in the crystallin genes were the most frequent mutations found in this study, with nine patients presenting likely causative variants in crystallin genes, six in familial cases, and three in sporadic cases. All respective familial cases were caused by autosomal dominant mutations, with the results suggesting that the sporadic cases are new cases of autosomal dominant inheritance. Six of these were caused by missense mutations, two from frameshifts, and one from a nonsense mutation (Fig. 2). Additionally, four of the nine variants were novel: CRYBA1 c.552_557delinsGGAGG; p.(Cys185Glufs*33), CRYBA4 c.277 T > C; p.(Ser93Pro), CRYBB1 c.508G > T; p.(Asp170Tyr), and CRYGC c.233C > T; p.(Ser78Phe). All novel missense mutations occurred within a Greek Key motif and might impact on protein folding. The novel heterozygous deletion and insertion in CRYBA1 (c.552_557delinsGGAGG; p.(Cys185Glufs*33)) is predicted to lead to a premature stop codon, deleting three-fifths of the fourth Greek Key and all of the C-terminal domain of CRYBA3/A1. Five variations of crystallin (CRYBA1 c.552_557delinsGGAGG; p.(Cys185Glufs*33), CRYBA4 c.277 T > C; p.(Ser93Pro), CRYBB1 c.508G > T; p.(Asp170Tyr), CRYGD c.309dup; p.(Glu104Argfs*4), and CRYGD c.418C > T; p.(Arg140*)) caused total cataract with or without microphthalmia and nystagmus. CRYGC c.233C > T; p.(Ser78Phe) caused nuclear cataracts, and CRYAA c.61C > T; p.(Arg21Trp) produced perinuclear cataracts and microphthalmia. A hotspot mutation c.70C > A; p.(Pro24Thr) and a previously reported mutation c.134 T > C; p.(Leu45Pro) in CRYGD were identified in family #5 and sporadic case #2, with no phenotypic information available [10,11,12].

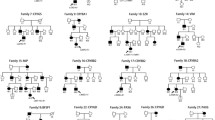
Pedigree and variants identified in crystallin genes These schematics show the encoded domain structure of CRYAA (a), CRYBA1 (b), CRYBA4 (c), CRYBB1 (d), CRYGC (e), and CRYGD (f). Mutations found in this study are illustrated above the schematics, with novel variants indicated in red characters. Probands are indicated by arrows, +/− indicates heterozygous individuals, −/− indicates individuals testing negative. WT: wild type, MT: mutant type
Variants in transcription factor genes MAF and PAX6
A likely de novo novel heterozygous missense mutation c. 950A > G; p.(Glu317Gly) in the bZIP domain of MAF was identified in the sporadic case #11(Fig. 3a) who was diagnosed with bilateral posterior polar cataracts.

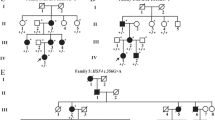
Pedigree and variants in transcription factor genes MAF and PAX6. The schematics show the encoded domain structure of MAF or PAX6, and the variants are illustrated above the schematics. MAF c.950A > G; p.(Glu317Gly) was identified in sporadic case #11 (a). PAX6 c.113G > A p.(Arg38Gln) was identified in family #12 (b). Patients II1 and II2 from family #12 inherited the same PAX6 allele from their unaffected mother. Allele specific PCR demonstrated that the variant was present in the asymptomatic mother. Probands are indicated by arrows. +/− indicates heterozygous individuals, −/− for individuals testing negative, −/G > A indicates a mosaic case besides the normal sequence “G” also chromosomes are found containing “A”. WT: Wild Type, MT: Mutant Type
Another novel variant, c.113G > A; p.(Arg38Gln), in the paired domain of PAX6, was found in both the proband of family #12 and his affected brother with cataract and nystagmus, but this variant was not observed via Sanger sequencing in either normal parent. The haplotype analysis demonstrated that both siblings inherited the same PAX6 allele from their mother, indicating that their mother may be gonadal mosaic for the disorder, and allele specific PCR confirmed that the variant was indeed present in the asymptomatic mother (Fig. 3b).
Variants in non-syndromal cataract genes BFSP2, FYCO1, GCNT2, and MIP
The BFSP2 gene encodes phakinin, a lens-specific intermediate filament-like protein. An in-frame deletion c.697_699del; p.(Glu233del) in the intermediate filament rod domain of BFSP2, previously reported by Jakobs PM and Zhang Q [13, 14], was identified in all affected individuals of family #11 with total cataract (Fig. 4a).

Pedigree and variants in BFSP2, FYCO1, GCNT2, and MIP. These schematics show the encoded domain structure of BFSP2 (a), exonic and protein domain structure of FYCO1 (b), encoded protein domain structure of GCNT2 (c) and MIP (d). Mutations found in this study are illustrated above the schematics, with novel variants indicated in red characters. Probands are indicated by arrows, +/− indicates heterozygous individuals, −/− for individuals testing negative. WT: wild type, MT: mutant type, W1: Wild Type 1, W2: Wild Type 2, M1: Mutant Type 1, M2: Mutant Type 2
Novel compound heterozygous variants c. 808C > T; p.(Gln270*) and c.3587 + 1G > T in FYCO1 were identified in the sporadic case #10, with parental segregation subsequently confirmed (Fig. 4b). The nonsense mutation c. 808C > T; p.(Gln270*) was predicted to truncate most of the coiled-coil region, as well as the entire FYVE zinc-finger and GOLD domain. Additionally, the G-to-T transversion located in the conserved intron 12 donor splice site (c.3587 + 1G > T) might affect splicing.
Homozygous or compound heterozygous mutations in GCNT2 caused cataract associated with the rare adult i blood group [15, 16]. The recurrent compound heterozygous mutations c.1043G > A; p.(Gly348Glu) and c.1148G > A; p.(Arg383His) in GCNT2, previously reported by Yu [15], were found in two patients from family #9 and sporadic case #5, and parental segregation was subsequently confirmed. Haplotype analysis revealed that the c.1043A allele of family #9 and sporadic case #5 was likely due to a founder effect, and the c.1148A allele origins of these two families were independent (Fig. 4c). I/i blood group typing was not performed since we had no access to fresh blood from the patients.
MIP is a less frequently investigated cataract-associated gene, but likely causative variants in MIP were identified in three patients, accounting for 13.04% (3/23) of the cases in this study. Two variants were familial and the third was a sporadic case of pediatric cataract. While the two missenses mutations, c.530A > G; p.(Tyr177Cys) and c.494G > A; p.(Gly165Asp), have been previously described [17, 18], the nonsense mutation c.612C > G; p.(Tyr204*) in family #8 is a novel variant (Fig. 4d), it might prevent MIP protein transport and reduce the formation of available water channels as well as p.(Lys228Glufs*4), which recently reported by Long X [19]. The cataract types of the patients harboring c.494G > A; p.(Gly165Asp) and c.612C > G; p.(Tyr204*) were not available, while the patient with mutation c.530A > G; p.(Tyr177Cys) had total cataract. In addition, all patients with MIP variants identified in this study had nystagmus.
Variants in syndromal cataract genes BCOR, IARS2, and NHS
In sporadic case #9, a likely de novo frameshift mutation c.4706dup; p.(Gly1570Argfs*7), was found in BCOR (Fig. 5a), the gene responsible for X-linked oculo-facio-cardio-dental (OFCD) syndrome [20]. Although c.4706dup; p.(Gly1570Argfs*7) was not reported in the literature, it is included in ClinVar database. The proband had bilateral total cataracts, microphthalmia, and microcornea, with additional dental and facial features consistent with OFCD syndrome. Her mother reported that she tired easily, but she did not undergo any cardiological tests. BCOR c.4706dup; p.(Gly1570Argfs*7) is predicted to delete part of the Ankyrin repeat-containing domain and the entire PCGF1 binding domain, which is necessary and sufficient for interaction with PCGF1, a component of the Polycomb group (PcG) multiprotein BCOR complex. This interaction is required to maintain the transcriptionally repressive state of BCL6 and CDKN1A [21].

Pedigree and variants in BCOR, IARS2, and NHS. These schematics show the encoded domain structure of BCOR (a), IARS2 (b), and NHS (c). BCOR and NHS are X-linked genes. The variants found in this study are illustrated above the schematics. Probands are indicated by arrows. A dotted circle indicates an obligate X-linked carrier. +/− indicates heterozygous individual, −/− indicates individual testing negative, +/0 indicates hemizygote testing positive, −/0 indicates hemizygote testing negative. W1: Wild Type 1, W2: Wild Type 2, W3: Wild Type 3, M1: Mutant Type 1, M2: Mutant Type 2, M3: Mutant Type 3
Two novel compound heterozygous mutations in IARS2, a nuclear gene encoding mitochondrial isoleucyl-tRNA synthetase [22], were found in family #10 and sporadic case #6 (Fig. 5b). The compound heterozygous variants c.607G > C; p.(Gly203Arg) and c.2575 T > C; p.(Phe859Leu) were identified in the sporadic case #6, who developed a sporadic case of perinuclear cataracts without other anomalies, and the mutations c.2446C > T; p.(Arg816*) and c.2575 T > C; p.(Phe859Leu) were identified in two affected brothers in family #10, both of which had bilateral cataracts without other anomalies. p.(Gly203Arg) lies in the class Ia aminoacyl-tRNA synthetases domain, p.(Phe859Leu) localizes to the anticodon-binding domain, and p.(Arg816*) would result in a truncated protein lacking the anticodon RNA-binding domain. Segregation studies revealed that both parents were heterozygous for the variants, confirming that these variants were in trans.
Two novel hemizygous frameshift mutations were identified in NHS, the gene responsible for X-linked Nance Horan syndrome (NHS) [23, 24]: c.3207_3208del; p. (Ala1070Phefs*16) and c.2739del; p.(Phe913Leufs*9), in sporadic cases #7 and #8, respectively (Fig. 5c). Both frameshift mutations in NHS were predicted to lead to protein truncations. While c.3207_3208del was a likely de novo mutation, c.2739del was heterozygous in the patient’s mother. The sporadic case #7 had bilateral cataracts, microphthalmia, microcornea, and nystagmus, as well as an asymmetric long narrow face, small nose, mild anteverted pinnae, and dental anomalies. His development and intelligence were normal. The sporadic case #8 had bilateral total cataracts, microphthalmia, microcornea, and nystagmus, along with characteristic facial features of a long narrow face, prominent nose, and large anteverted pinnae, dental anomalies of screwdriver-shaped incisors, and an intellectual delay consistent with NHS.
Discussion
In this study, we used target NGS to identify genetic variants in 39 Chinese probands with inherited pediatric cataract. Twenty-three cases were found to harbor putative pathogenic variants in 15 cataract-associated genes, including missense mutations, nonsense mutations, frameshift deletions/insertions, in-frame deletions, and splicing mutations. All missense mutations identified in this study were located at very important protein domains, and the frameshift deletions/insertions and nonsense mutations found resulted in premature termination codons or triggered a nonsense-mediated mRNA decay. The splicing mutation FYCO1 c.3587 + 1G > T changed the acceptor site of intron 12, which would generally cause exon skipping. Further functional studies are warranted to determine the physiological implications of each new mutation. The majority of these mutations appear to be autosomal dominant (15/23), with autosomal recessive (5/23) and X-linked changes (3/23) also detected. The most commonly mutated genes were those coding for crystalline, accounting for 39.13% of cases. MIP was mutated in three cases, representing the second most commonly mutated gene in our cohort. Interestingly, no gap junction protein-encoding genes were identified in our cohort, although they are frequently reported in non-syndromic pediatric cataract [25, 26].
The large number of genes known to cause pediatric cataract and the limited genotype-phenotype correlations complicate clinical testing using traditional sequencing technologies. These difficulties are especially evident in sporadic pediatric cataract cases, which make up the majority of pediatric cataract cases, and present diagnostic challenges when attempting to identify a genetic etiology [7]. Our study demonstrates that half of the mutations identified in sporadic pediatric cataract cases were due to likely de novo heterozygous mutations in autosomal-dominant genes (5/11), one-fourth were compound heterozygous mutations in autosomal recessive genes (3/11), and one-fourth were X-linked variants (3/11), two of which were likely de novo mutations. Similar difficulties exist in familial cases, as pedigree information alone may not accurately describe the inheritance risk. Family #12 was assumed to have a recessive form of pediatric cataract on the basis of family history, however, genetic testing revealed the presence of a PAX6 missense mutation in both affected brothers. Since all previously reported mutations in PAX6 have been dominant [27,28,29], this finding suggested that their asymptomatic mother was mosaic for the disorder. Moreover, parental mosaicism for mutated PAX6 in affected siblings has been reported recently [30]. Therefore, the NGS testing results dramatically altered the counseling of both the parents and the patients themselves. Thus, it is useful for parents of affected children, as well as the affected individuals themselves, to use a targeted NGS panel to provide accurate recurrence and transmission risk counseling.
Some syndromal pediatric cataracts may be subtle, with associated systemic features presenting or becoming apparent only in later childhood [8]. The sporadic case #9 carried a novel BCOR mutation associated with OFCD syndrome, but she presented with only subtle clinical features, yet her cardiac status should be monitored for signs of disease progression. Mutations in IARS2 are also commonly associated with syndromal pediatric cataract. While compound heterozygous mutations in IARS2 were identified in the patients of family #10 and sporadic case #6, these patients did not present with additional anomalies besides the cataract. Thus, growth hormone levels, neurotrophic keratitis, orbital myopathy, and skeletal dysplasia should be monitored through later follow-ups. Other genes, such as AGK and LONP1, are known to be mutated in syndromic forms of cataract and have also been reported to cause apparently non-syndromic cataract [12, 31]. IARS2 might be the third example of such genes that can be mutated in both syndromic and non-syndromic forms of pediatric cataract. Future cataract patients with different mutations in IARS2 will help clarify the phenotypic spectrum.
In recent years, multiple cataract-targeted gene panels have been developed with detection rates of 26–75% [8, 12, 25, 26, 32,33,34]. We investigated 16 familial and 23 sporadic cases with pediatric cataract in the Chinese population and achieved an overall mutation detection rate of 58.97%, which is almost identical to those reported in similar studies of patients from South Eastern Australia (62%) [25], China (62.96%) [32], and Saudi Arabia(58%) [12], including zero, 7.4 and 23% of sporadic cases, respectively. The mutation detection rate of the familial cases in our study was 75%, comparable to that published recently in familial patients from the UK (75%) [26] and another Australian cohort (73%) [8]. The mutation detection rate for familial cases in our cohort was much higher than that published recently in two studies in Chinese familial patients, with mutation pick-up rates of 50 and 64% [32, 33]. The mutation detection rate for sporadic cases in our study was 47.8%, lower than reported in sporadic patients from Australia (68%) and Saudi Arabia (62.5%) [8, 12]. If the new candidate genes are included, the mutation detection rate in the sporadic patients from Saudi Arabia increases to 75% [12]. A recently published article reported gene mutations screening in sporadic pediatric cataract in a Han Chinese population using target NGS, and identified pathogenic variants in 26% of cases [34], much lower than ours. The relatively low mutation detection rate in Chinese sporadic patients might be due to the number of target genes being different between different panels or different frequencies of mutations occurring different groups. Also, further clinical exome sequencing panels or whole exome sequencing were not performed for patients negative for mutations from the target NGS in our patients or another Chinese cohort.
One advantage in our study is that we obtained DNA samples from the parents of each proband (except sporadic patient #2; the DNA of his father was unavailable) regardless of family history. We also obtained DNA samples from at least two patients in familial cases and performed segregation analysis to confirm the disease-causing variations. Our study also has several limitations. Nearly all patients underwent cataract surgery prior to enrolment in this study, so phenotypic information was determined by reviewing medical records or recalled by the participants or their guardians. Medical records were not available for 11 probands, so the lens phenotype could not be ascertained in detail. The multi-gene panel was designed in October 2015, so cataract genes published after that date were not included. Four variants predicted to have uncertain significance under ACMG guideline were identified in two familial and two sporadic cases, additional studies will be needed to confirm their pathogenicity. In future efforts, clinical exome sequencing panels targeting all OMIM-identified disease genes, or whole exome sequencing analysis might be necessary for mutation negative cases regardless of family history.
Conclusion
In conclusion, we examined the clinical manifestations and molecular genetic characteristics of 39 Chinese patients with pediatric cataract. Twenty-three putative pathogenic variants were identified, with 12 novel and 11 recurrent. This has led to more accurate genetic diagnoses and recurrence risk counseling, impacting management for each family.
Abbreviations
- ACMG:
-
American college of medical genetics and genomics
- ExAC:
-
Exome aggregation consortium
- NGS:
-
Next-generation sequencing
- NHS:
-
Nance horan syndrome
- OFCD:
-
Oculo-facio-cardio-dental syndrome
- PCR:
-
Polymerase chain reaction
- STR:
-
Short tandem repeat
References
Apple DJ, Ram J, Foster A, Peng Q. Elimination of cataract blindness: a global perspective entering the new millenium. Surv Ophthalmol. 2000;45(Suppl 1):S1–196.
Gilbert C, Foster A. Childhood blindness in the context of VISION 2020--the right to sight. Bull World Health Organ. 2001;79(3):227–32.
Muhit M, Gilbert C. A review of the epidemiology and control of childhood blindness. Trop Dr. 2003;33(4):197–201.
Francis PJ, Ionides A, Berry V, Bhattacharya S, Moore AT. Visual outcome in patients with isolated autosomal dominant congenital cataract. Ophthalmology. 2001;108(6):1104–8.
Trumler AA. Evaluation of pediatric cataracts and systemic disorders. Curr Opin Ophthalmol. 2011;22(5):365–79.
Hejtmancik JF. Congenital cataracts and their molecular genetics. Semin Cell Dev Biol. 2008;19(2):134–49.
Wirth MG, Russell-Eggitt IM, Craig JE, Elder JE, Mackey DA. Aetiology of congenital and paediatric cataract in an Australian population. Br J Ophthalmol. 2002;86(7):782–6.
Ma AS, Grigg JR, Ho G, Prokudin I, Farnsworth E, Holman K, Cheng A, Billson FA, Martin F, Fraser C, Mowat D, Smith J, Christodoulou J, Flaherty M, Bennetts B, Sporadic JRV. Familial congenital cataracts: mutational Spectrum and new diagnoses using next-generation sequencing. Hum Mutat. 2016;37(4):371–84.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. ACMG laboratory quality assurance committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Santhiya ST, Shyam Manohar M, Rawlley D, Vijayalakshmi P, Namperumalsamy P, Gopinath PM, Löster J, Graw J. Novel mutations in the gamma-crystallin genes cause autosomal dominant congenital cataracts. J Med Genet. 2002;39(5):352–8.
Ma C, Zheng G, Hao L. Analysis of disease-causing gene mutation in three Chinese families with congenital inherited cataract. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2018;35(2):165–8.
Patel N, Anand D, Monies D, Maddirevula S, Khan AO, Algoufi T, Alowain M, Faqeih E, Alshammari M, Qudair A, Alsharif H, Aljubran F, Alsaif HS, Ibrahim N, Abdulwahab FM, Hashem M, Alsedairy H, Aldahmesh MA, Lachke SA, Alkuraya FS. Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Hum Genet. 2017;136(2):205–25.
Jakobs PM, Hess JF, FitzGerald PG, Kramer P, Weleber RG, Litt M. Autosomal-dominant congenital cataract associated with a deletion mutation in the human beaded filament protein gene BFSP2. Am J Hum Genet. 2000;66(4):1432–6.
Zhang Q, Guo X, Xiao X, Yi J, Jia X, Hejtmancik JF. Clinical description and genome wide linkage study of Y-sutural cataract and myopia in a Chinese family. Mol Vis. 2004;10:890–900.
Yu LC, Twu YC, Chang CY, Lin M. Molecular basis of the adult i phenotype and the gene responsible for the expression of the human blood group I antigen. Blood. 2001;98(13):3840–5.
Borck G, Kakar N, Hoch J, Friedrich K, Freudenberg J, Nürnberg G, Yilmaz R, Daud S, Baloch DM, Nürnberg P, Oldenburg J, Ahmad J, Kubisch C. An Alu repeat-mediated genomic GCNT2 deletion underlies congenital cataracts and adult i blood group. Hum Genet. 2012;131(2):209–16.
Senthil Kumar G, Kyle JW, Minogue PJ, Dinesh Kumar K, Vasantha K, Berthoud VM, Beyer EC, Santhiya ST, An MIP. AQP0 mutation with impaired trafficking and function underlies an autosomal dominant congenital lamellar cataract. Exp Eye Res. 2013;110:136–41.
Yang G, Zhang G, Wu Q, Zhao J. A novel mutation in the MIP gene is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Mol Vis. 2011;17:1320–3.
Long X, Huang Y, Tan H, Li Z, Zhang R, Linpeng S, Lv W, Cao Y, Li H, Liang D, Wu L. Identification of a novel MIP frameshift mutation associated with congenital cataract in a Chinese family by whole-exome sequencing and functional analysis. Eye (Lond). 2018; doi: https://doi.org/10.1038/s41433-018-0084-5.
Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A, Hadley DW, Tifft C, Zhang L, Wilkie AO, van der Smagt JJ, Gorlin RJ, Burgess SM, Bardwell VJ, Black GC, Biesecker LG. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet. 2004;36(4):411–6.
Junco SE, Wang R, Gaipa JC, Taylor AB, Schirf V, Gearhart MD, Bardwell VJ, Demeler B, Hart PJ, Kim CA. Structure of the polycomb group protein PCGF1 in complex with BCOR reveals basis for binding selectivity of PCGF homologs. Structure. 2013;21(4):665–71.
Schwartzentruber J, Buhas D, Majewski J, Sasarman F, Papillon-Cavanagh S, Thiffault I, Sheldon KM, Massicotte C, Patry L, Simon M, Zare AS, McKernan KJ, FORGE Canada Consortium, Michaud J, Boles RG, Deal CL, Desilets V, Shoubridge EA, Samuels ME. Mutation in the nuclear-encoded mitochondrial isoleucyl-tRNA synthetase IARS2 in patients with cataracts, growth hormone deficiency with short stature, partial sensorineural deafness, and peripheral neuropathy or with Leigh syndrome. Hum Mutat. 2014;35(11):1285–9.
Burdon KP, McKay JD, Sale MM, Russell-Eggitt IM, Mackey DA, Wirth MG, Elder JE, Nicoll A, Clarke MP, FitzGerald LM, Stankovich JM, Shaw MA, Sharma S, Gajovic S, Gruss P, Ross S, Thomas P, Voss AK, Thomas T, Gécz J, Craig JE. Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am J Hum Genet. 2003;73(5):1120–30.
Kammoun M, Brady P, De Catte L, Deprest J, Devriendt K, Vermeesch JR. Congenital diaphragmatic hernia as a part of Nance-Horan syndrome? Eur J Hum Genet. 2018 Mar;26(3):359–66.
Javadiyan S, Craig JE, Souzeau E, Sharma S, Lower KM, Mackey DA, Staffieri SE, Elder JE, Taranath D, Straga T, Black J, Pater J, Casey T, Hewitt AW, Burdon KP. High-throughput genetic screening of 51 pediatric cataract genes identifies causative mutations in inherited pediatric cataract in south eastern Australia. G3 (Bethesda). 2017;7(10):3257–68.
Gillespie RL, O'Sullivan J, Ashworth J, Bhaskar S, Williams S, Biswas S, Kehdi E, Ramsden SC, Clayton-Smith J, Black GC, Lloyd IC. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology. 2014;121(11):2124–37.
Hanson I, Churchill A, Love J, Axton R, Moore T, Clarke M, Meire F, van Heyningen V. Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations. Hum Mol Genet. 1999;8(2):165–72.
Wolf MT, Lorenz B, Winterpacht A, Drechsler M, Schumacher V, Royer-Pokora B, Blankenagel A, Zabel B, Wildhardt G. Ten novel mutations found in Aniridia. Hum Mutat. 1998;12(5):304–13.
Grønskov K, Rosenberg T, Sand A, Brøndum-Nielsen K. Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur J Hum Genet. 1999;7(3):274–86.
Deml B, Reis LM, Lemyre E, Clark RD, Kariminejad A, Semina EV. Novel mutations in PAX6, OTX2 and NDP in anophthalmia. microphthalmia and coloboma Eur J Hum Genet. 2016;24(4):535–41.
Aldahmesh MA, Khan AO, Mohamed JY, Alghamdi MH, Alkuraya FS. Identification of a truncation mutation of acylglycerol kinase (AGK) gene in a novel autosomal recessive cataract locus. Hum Mutat. 2012;33(6):960–2.
Zhai Y, Li J, Yu W, Zhu S, Yu Y, Wu M, Sun G, Gong X, Yao K. Targeted exome sequencing of congenital cataracts related genes: broadening the mutation Spectrum and genotype-phenotype correlations in 27 Chinese Han families. Sci Rep. 2017;7(1):1219.
Sun W, Xiao X, Li S, Guo X, Zhang Q. Exome sequencing of 18 Chinese families with congenital cataracts: a new sight of the NHS gene. PLoS One. 2014;9(6):e100455.
Li D, Wang S, Ye H, Tang Y, Qiu X, Fan Q, Rong X, Liu X, Chen Y, Yang J, Lu Y. Distribution of gene mutations in sporadic congenital cataract in a Han Chinese population. Mol Vis. 2016;22:589–98.
Hansen L, Yao W, Eiberg H, Kjaer KW, Baggesen K, Hejtmancik JF, Rosenberg T. Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest Ophthalmol Vis Sci. 2007;48(9):3937–44.
Yang G, Chen Z, Zhang W, Liu Z, Zhao J. Novel mutations in CRYGD are associated with congenital cataracts in Chinese families. Sci Rep. 2016;6:18912.
Devi RR, Yao W, Vijayalakshmi P, Sergeev YV, Sundaresan P, Hejtmancik JF. Crystallin gene mutations in Indian families with inherited pediatric cataract. Mol Vis. 2008;14:1157–70.
Acknowledgments
We thank all of the patients and families for their contributions to this work.
Funding
The research was supported by the National Natural Science Foundation of China (81230015), CAMS Innovation Found for Medical Sciences (2016-I2M-1-002), the Beijing Municipal Science and Technology Commission (Z151100003915078), the National Key Research and Development Program of China (2016YFC0905100) to Xue Zhang; and The National Natural Science Foundation of China (81670896), the Liaoning Provincial Education Department Science and Technology Research Project (LK201653) to Lihua Cao.
Availability of data and materials
Not applicable. Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Contributions
LC and XZ designed the study; LC and YL wrote the manuscript; JL and YL participated in molecular genetic studies; SH performed the target NGS; LY collected the patients’ samples; YL analyzed the clinical data; CL analyzed the target NGS data. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study adhered to the tenets of the Declaration of Helsinki and was approved by the China Medical University Institutional Review Board, written informed consent forms were obtained from all investigated participants or their guardians if they were under 18 years old.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. List of 80 pediatric cataract genes. (XLS 36 kb)
Additional file 2:
Table S2. The primers for haplotype analysis and allele specific PCR. (XLS 25 kb)
Additional file 3:
Table S3. Target NGS analysis. (XLS 28 kb)
Additional file 4:
Table S4. Pediatric cataract families with uncertain significance variants. (XLS 29 kb)
Additional file 5:
Figure S1. Pedigrees of the families without suspected causative variants. (TIF 8488 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, J., Leng, Y., Han, S. et al. Clinical and genetic characteristics of Chinese patients with familial or sporadic pediatric cataract. Orphanet J Rare Dis 13, 94 (2018). https://doi.org/10.1186/s13023-018-0828-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-018-0828-0