
Abstract
Background
Cycloartane triterpenoids exhibited anticancer effects. This study aims to identify any potential novel anticancer cycloartane triterpenoids from Cimicifuga foetida L. rhizome (Sheng ma) and the mode of actions.
Methods
Cycloartane triterpenoids were isolated from the C. foetida rhizome by a series of column chromatography and identified by IR, MS and NMR. Their anticancer effects on several human cancer cell lines, MCF-7, HepG2, HepG2/ADM, HeLa, and PC3, and normal human mammary epithelial cells MCF10A were investigated by colony formation and MTT assays. Morphological analysis of apoptosis induction was performed by acridine orange/ethidium bromide dual-staining and Hoechst 33258 nuclear staining. The cell-cycle profile and annexin V staining were evaluated by flow cytometry. Apoptosis were investigated by measuring changes in mitochondrial membrane potential and analyzing expression of cell cycle- and apoptosis-related proteins in MCF-7 cells by Western blotting.
Results
A novel cycloartane triterpenoid, 25-O-acetyl-7,8-didehydrocimigenol-3-O-β-d-(2-acetyl)xylopyranoside (ADHC-AXpn), together with the known 7,8-didehydrocimigenol-3-O-β-d-xylopyranoside (DHC-Xpn) were isolated. MCF-7 growth was significantly inhibited by ADHC-AXpn in a dose- and time-dependent manner (IC50: 27.81 µM at 48 h; P = 0.004 vs. control at 25 μM for 48 h treatment), and ADHC-AXpn was selectively cytotoxic for cancerous cells (MCF-7, HepG2/ADM, HepG2 and HELA cells) based on its higher IC50 values for normal cells MCF10A (IC50: 78.63 µM at 48 h) than for tumor cells. In MCF-7 cells, ADHC-AXpn induced G2/M cell cycle arrest by mediating cyclin-B1, and CDK1 and its phosphorylation; and induced apoptosis through the mitochondrial-mediated apoptotic pathway, with inhibition of Akt activation. As ADHC-AXpn suppressed phosphorylation of ERK1/2, Raf and Akt proteins in MCF-7 cells, its apoptotic effect might be associated with Raf/MEK/ERK signaling and Akt activation.
Conclusions
ADHC-AXpn significantly suppressed the growth of MCF-7 cells, induced mitochondrial apoptosis and cell-cycle arrest, and inhibited Raf/MEK/ERK signaling pathway and Akt phosphorylation.
Similar content being viewed by others


Background
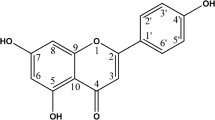
Chinese medicine (CM) is a rich source for the development of anticancer drugs and chemopreventive agents [1]. Cycloartane triterpenoid (CATP) are triterpene glycosides with a unique structural C-19 angular methyl (Fig. 1), are distributed in plants of several genuses, such as Astragalus, Cimicifuga, and Thalictrum [2, 3]. Several CATPs isolated from plants exhibited potential anticancer activities [4–7].

Chemical structures of components ADHC-AXpn and DHC-Xpn
The rhizome of Cimicifuga foetida L. (Sheng ma), has been used as an anti-inflammatory, analgesic, and antipyretic agent in CM [8]. C. foetida contains many CATPs [9, 10], and some CATPs from the Cimicifuga genus possessed anti-cancer effects in vivo and in vitro [4–6]. However, previous research focused on isolation and structure identification rather than biological activity. Our earlier research associated CATP anticancer activity with apoptosis induction and cell cycle arrest in tumor cells [4, 5], but respective biological pathways have not yet investigated.
This study aims to identify any potential novel anticancer CATP from C. foetida and evaluate their effects on the growth, colony formation, apoptosis induction and respective pathways in cancer cells.
Methods
Plant materials
Rhizome of C. foetida was collected in Emei Mountain, Sichuan province, China in October 2012 and identified by Dr. Sibao Chen by comparing its morphological features [8]. A voucher specimen (SZRI20121045) was deposited in the herbarium of state key laboratory of Chinese medicine and molecular pharmacology.
Equipment
Infrared spectra (IR) were recorded on a Shimadzu IR-450 spectrometer (Shimadzu, Kyoto, Japan). High resolution fast atom bombardment mass spectra (HR-FAB-MS) were recorded on a VG-Autospec-3000 spectrometer (Micromass, Manchester, UK), in m/z (percentage relative intensity of base peak). The nuclear magnetic resonance (NMR) spectra were measured in pyridine-d 5 on a Bruker AM-400 spectrometer (BrukerDaltonics, MA, USA), using tetramethyl silane as internal standard. Silica gel-60H (100–200 and 200–300 mesh) and silica gel GF254 sheets (0.20–0.25 mm; both from Qingdao Haiyang Chemical Group Co., Qingdao, China) were used for column chromatography (CCG) and thin layer chromatography (TLC), respectively.
Materials and reagents for pharmacological investigations
3-(4, 5)-Dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT), Hoechst 33258, propidium iodide (PI), doxorubicin (Dox) and carbonyl cyanide 3-c hlorophenylhydrazone were purchased from Sigma-Aldrich (St. Louis, MO, USA). 5, 5′, 6, 6′-tetrachloro-1, 1′, 3, 3′-tetraethyl benzimidazolyl-carbocyanine iodide (JC-1) and Alexa Fluor® 488 Annexin V/dead cell apoptosis kit were obtained from invitrogen (Carlsbad, CA, USA). BCA protein assay kit and electro chemiluminescence were obtained from Thermo Fisher Scientific (Rockford, IL, USA). Antibodies against cyclin B1, CDK1, p-CDK1(Thr161), caspase-9, cleaved caspase-9, PARP, cleaved PARP, Akt, p-Akt (Thr308 and Ser473), p-ERK1/2 and p-c-Raf were from cell signaling technology (Beverly, MA, USA). Antibodies against Bcl-2 and Bax were bought from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Protease inhibitor cocktail tablets and phosphatase inhibitor cocktail tablets were supplied by Roche (Roche Applied Science, Mannheim, Germany). Dimethylsulfoxide (DMSO); RPMI-1640 and Dulbecco’s modification of Eagle’s medium (DMEM), phosphate-buffered saline (PBS), fetal bovine serum (FBS), penicillin-streptomycin (P/S) and trypsin-ethylene diamine tetraacetic acid (EDTA) were purchased from life technologies (Grand Island, NY, USA).
Extraction and isolation
The rhizome of C. foetida (0.5 kg) was extracted three times with 95 % EtOH for 1 h under reflux. After combination of extractions and solvent removal, the residue (129 g) was suspended in water (1 L) and partitioned successively with 200 mL each of petroleum ether (60–80 ℃), ethyl acetate and n-BuOH. The ethyl acetate fraction (28 g) was subjected to CCG on silica gel-60H (100–200 mesh). Gradient elution with CHCl3–MeOH (1:0, 50:1, 20:1, 10:1 and 0:1), obtained five fractions: A (3.7 g), B (4.9 g), C (6.28 g), D (1.5 g) and E (1.8 g).
Fraction D was subjected to CCG on silica gel-60H (200–300 mesh), eluted with CHCl3–MeOH (70:30) and further purified by Sephadex G10 CCG, eluted with MeOH to obtain ADHC-AXpn (15 mg).
Fraction C was subjected to repeated CCG on silica gel-60H (200–300 mesh), eluted with CHCl3 and acetone (3:1) to give sub-fractions 1–5. Sub-fraction 5 was subjected to CCG on silica gel-60H (200–300 mesh), eluted in gradient with CHCl3-MeOH (90:10, 85:15 and 80:20). The fraction eluted by CHCl3-MeOH (80:20) was purified by Sephadex G10 CCG, and eluted with MeOH, to give DHC-Xpn (40 mg). Chemical structures of ADHC-AXpn and DHC-Xpn were elucidated by their spectral data (IR, MS and 1H, 13C-NMR) and by comparison with data in the literature. The purity of isolated components was determined over 98 % by peak area normalization method in HPLC analysis by an Agilent 1200 liquid chromatography system (HP Agilent Technologies, Palo Alto, CA, USA).
Cell culture
Cell lines MCF-7 (estrogen receptor-positive phenotype), HepG2, HeLa and PC3 cells were obtained from American type culture collection (Manassas, VA, USA). HepG2/ADM cells (multidrug resistance phenotype) were kindly provided by Prof. Kwok-Pui Fung (The Chinese University of Hong Kong, Hong Kong, China). Human MCF10A mammary epithelial cells were obtained from Invitrogen (Carlsbad, CA, USA). The MCF-7, HepG2, HepG2/ADM and PC3 cells were cultured in RPMI-1640 medium supplemented with 10 FBS and 1 % (v/v) P/S at 37 ℃ in a humidified incubator containing 5 % CO2. HeLa cells were cultured in DMEM medium with the same culture condition mentioned above. Human MCF10A mammary epithelial cells were cultured in a condition mentioned previously [11]. HepG2/ADM cells were cultured with 1.2 μM of Dox; during cell passage to keep their multidrug resistance property as compared with the corresponding parental cells.
Cell viability assay
The inhibitory effects of ADHC-AXpn and DHC-Xpn on the growth of tested cells were evaluated by MTT assay. Briefly, all tested cells (0.8 × 104/well) were seeded in 96-well plates, cultured for 24 h, then exposed to different concentrations of ADHC-AXpn and DHC-Xpn, respectively for another 24 or 48 h. Subsequently, 30 μL of 5 mg/mL MTT dissolved in PBS was added to each well and incubated for 4 h after removing the media. Formazan crystals were dissolved with 100 μL of DMSO, with absorbance at 490 nm measured by Bio-Rad 680 microplate reader (Bio-Rad, CA, USA). Cell viability was determined as a percentage of the control, with Dox as positive control.
Colony formation assay
MCF-7 cells (2.5 × 105/well) were seeded in 6-well plates, cultured for 24 h, then exposed to different concentrations of ADHC-AXpn (20, 30 or 35 μM) for 48 h before digestion in 0.25 % trypsin to reconstitute single-cell suspensions. Cell suspensions were then transferred into 6-well plates (600 cells/well) and incubated at 37 ℃ for 12 days. The formed colonies were fixed in 4 % paraformaldehyde for 30 min and stained with 1 % crystal violet for 5 min. These experiments were performed three times. Colonies with diameters larger than 0.5 mm were counted to calculate the colony formation rate.
Hoechst 33258 staining assay
The nuclear morphology of cells treated with ADHC-AXpn was observed with a Hoechst 33258 staining assay. MCF-7 cells (2.5 × 105/well) were seeded in 6-well plates, cultured for 24 h, and treated with different concentrations of ADHC-AXpn (20, 30 or 35 μM) for 48 h. The cells were then washed three times with PBS, fixed in 4 % paraformaldehyde at 4 ℃ for 30 min, and stained with 10 μg/mL of Hoechst 33258 for 15 min at 37 ℃. Changes in nuclear morphology were monitored with an Olympus IX51 inverted microscope (Olympus Corporation of the Americas, Inc., Central Valley, PA, USA).
Cell cycle analysis
Cell cycle distribution was analyzed by a fluorescent probe propidium iodide (PI) staining assay. MCF-7 cells (2.5 × 105/well) were seeded in 6-well plates, cultured for 24 h, then treated with different concentrations of ADHC-AXpn (20, 30 or 35 μM) for 24 or 48 h. Cells were then collected and fixed with 75 % ethanol at 4 ℃ overnight. After centrifugation in an Eppendorf 5417R centrifuge (Eppendorf Corp., Hamburg, Germany) at 800g for 5 min, the cells were incubated with PBS containing 0.02 mg/mL PI and 0.1 mg/mL RNase A in darkness at 37 ℃ for 30 min. The emitting fluorescence was measured by Guava Easy Cytometer (Guava Technologies, Millipore, Billerica, MA, USA); data were analyzed with MultiCycle AV software (Phoenix Flow Systems, San Diego, CA, USA).
Annexin V-FITC/PI double staining assay for apoptosis
MCF-7 cells (2.5 × 105/well) were seeded in 6-well plates, cultured for 24 h, and exposed to differing concentrations of ADHC-AXpn (20, 30 or 35 μM) for 36 h. Cells were then collected and stained by Alexa Fluor® 488 Annexin V/dead cell apoptosis kit according to manufacturer instructions. Green fluorescence emitted from Annexin V-FITC and red fluorescence from PI were detected by Guava Easy Cytometer (Guava Technologies, Millipore, Billerica, MA, USA). Data were analyzed by FlowJo 7.6 software (TreeStar, SanCarlos, CA, USA).
Measurement of mitochondrial membrane potential (MMP)
Changes in MMP were measured using a lipophilic cationic fluorescent probe JC-1. Briefly, MCF-7 cells (2.5 × 105/well) were seeded in 6-well plates, cultured for 24 h, then treated with ADHC-AXpn (20, 30 or 35 μM) for 24 h. The cells were collected and incubated with 5 μM JC-1 in darkness at 37 ℃ for 15 min, then assayed with a Guava Easy Cytometer (EasyCyte 8 HT, Millipore, USA) to evaluate changes in MMP..
Western blotting assay
MCF-7 cells (2.5 × 105/well) were seeded in 100-mm culture dishes, cultured for 24 h, then incubated with differing concentrations of ADHC-AXpn for various times. Cells were then collected by trypsinization with 0.25 % Trypsin-EDTA and transferred to centrifuge tubes. After centrifugation at 800g for 5 min, cells were washed twice with ice-cold PBS and then lysed in RIPA buffer containing 0.5 M DTT, 0.1 M PMSF and 20× phosphatase inhibitor for 30 min on ice. Protein samples were harvested by centrifugation at 12,000g at 4 ℃ for 12 min. Protein contents were quantified with BCA Protein Assay Kit (Thermo Scientific Pierce, Rockford, USA). The protein samples (35 μg) along with a rainbow-colored protein molecular marker (Amersham, Buckinghamshire, UK) were separated by 10 % SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA) by a semi-dry transfer apparatus (Bio-Rad, CA, USA). The membranes were blocked with 5 % nonfat dry milk in Tris-buffered saline containing 0.1 % Tween-20 (TBS-T) at room temperature for 1 h and incubated overnight with primary antibodies containing 5 % BSA at 4 ℃. Membranes were washed with TBS-T three times and appropriate secondary antibodies conjugated with horseradish peroxidase were added and incubated for another hour. Finally, immunoblots were visualized by enhanced chemiluminescence detection reagents, with β-actin as a loading control.
Statistical analysis
Each experiment was performed for three times and data were presented as mean ± standard deviation (SD). Analysis of variance (ANOVA) was performed to detect significant differences. In multiple comparisons, Tukey post-test was used. P value less than 0.05 was considered statistically significant. Analyses of dose-, concentration- and time-dependent effects were performed by sigmoidal non-linear regression. All statistical analysis tests were performed by Graph Pad Prism 5.0 for Windows (GraphPad Software, San Diego California, USA).
Results
Structure elucidation of ADHC-AXpn and DHC-Xpn
ADHC-AXpn was isolated as a white powder. The HR-FAB-MS showed a pseudo-molecular ion peak at m/z 725.3887 [M + Na]+ (calc. 725.3893), which supported the molecular formula as being C39H58O11. The IR spectrum displayed the presence of hydroxyl groups (3338 cm−1) and a carbonyl group (1730 cm−1). The 1H-NMR spectrum (Table 1) showed signals of cyclopropane methylene groups at δ 0.43 and 0.96 ppm (each 1H, d, J = 4.0 Hz), six methyl groups at δ 1.05, 1.08, 1.13, 1.20, 1.65 and 1.67 ppm; an aromatic proton at δ 4.82 ppm (d, J = 8.0 Hz), and a series of overlapped signals, which suggested that ADHC-AXpn was a cycloartane-type triterpene glycoside [12]. The 13C-NMR (Table 1) displayed the diagnostic signals of two oxygen-bearing alkyl carbons at δ 87.0 (C-24) and 72.1 ppm (C-23), and an oxygen-bearing quaternary carbon at δ 113.0 ppm (C-16), which implied that ADHC-AXpn was a cimigenol-type triterpene [13]. By fully comparing 1H and 13C NMR spectral data of ADHC-AXpn with those of the known component 25-O-acetyl-7,8-didehydrocimigenol-3-O-β-d-xylopyranoside (25-AC) [12], the structure of ADHC-AXpn was similar to that of 25-AC, excepting an additional acetyl moiety. When the 13C-NMR spectrum of ADHC-AXpn was compared with that of 25-AC, the signals of C-1′ and C-3′ were seen to shift to higher field by 2.6 and 2.0 ppm, while the signal of C-2′ shifted to lower field. These chemical shift variations suggested an acetyl moiety was linked to the C-2′ of the xylose unit [12]. Locations of the two acetyl groups were directly confirmed by hetero-nuclear multiple bond correlation (HMBC) experiments (Fig. 2). Briefly, significant correlation was observed between H-2′ (δH 5.56 ppm, dd, J = 7.5, 8.1 Hz) and the carbonyl signal at δ170.3 ppm, which indicated an acetyl group was assignable to C-2′. The methyl signals at δH 1.65 ppm (Me-26)/δH 1.67 ppm (Me-27) also showed correlations with a quaternary carbon signal at δC 83.44 ppm (C-25), the methine carbon signal at δC 87.05 ppm (C-24), and the methyl carbon signals at δC 21.58 ppm (C-27)/δC 24.14 ppm (C-26), which indicated that another acetyl group was located at C-25. A complete 1H and 13C-NMR spectral assignment of ADHC-AXpn was summarized in Tables 1 and 2.

Key HMBC for ADHC-AXpn
Taken together, the structure of ADHC-AXpn was formulated as 25-O-acetyl-7,8-didehydrocimigenol-3-O-β-d-(2-acetyl) xylopyranoside (Fig. 1). The known DHC-Xpn was determined as 7,8-didehydrocimigenol-3-O-β-d-xylopyranoside by comparison of its spectral data with those reported in the literature [14].
Growth inhibition effects of ADHC-AXpn and DHC-Xpn on carcinoma cells
The inhibitory effects of ADHC-AXpn and DHC-Xpn on the viability of various carcinoma cells were determined by MTT assay. ADHC-AXpn was much more potent than DHC-Xpn in decreasing the viability of the tested cells (MCF-7, HepG2/ADM, HepG2, HELA and PC3 cells) in both 24 and 48 h treatment periods apart from PC3 cells (Table 2). Among these cancer cells treated with ADHC-AXpn, MCF-7 cells appeared to be more sensitive to ADHC-AXpn than other cancer cells, as indicated by the IC50 values (Fig. 3a). As shown in Fig. 3b, the colony formation assay also confirmed that ADHC-AXpn (20, 30 or 35 μM) could inhibit proliferation of MCF-7 cells (P = 0.0078, P < 0.001, P < 0.0001). The IC50 values of ADHC-AXpn in human MCF10A mammary epithelial cells at 48 h were 78.63 ± 3.26 μM, which was much higher than for cancer cell line MCF-7. These data indicated that ADHC-AXpn was selectively cytotoxic for cancer cells.

Growth inhibitory activity of ADHC-AXpn in MCF-7 cells. a Cells were treated with increasing concentrations of ADHC-AXpn (10 to 50 μM) for 24 and 48 h; cell survival was then assessed by MTT assay. Doxorubicin (Dox) exposure at 5 μM was used as a positive control. b After exposure to ADHC-AXpn at concentrations of 20, 30 or 35 μM, cells were seeded onto 6-well plates (600 cells/well) for assessing colony formation. Data are expressed as mean ± SD of three independent experiments with **P = 0.0078; ***P < 0.001
ADHC-AXpn induces cell cycle arrest in MCF-7 cells
We investigated the effect of ADHC-AXpn on the MCF-7 cell cycle by flow cytometry. Treating MCF-7 cells with ADHC-AXpn for 24 h induced a G2/M arrest (Fig. 4a). The percentage of cells in subG1 phase increased when ADHC-AXpn exposure was extended to 48 h. To explore the mechanism of ADHC-AXpn-induced G2/M arrest, we investigated its effects on G2/M regulatory proteins. Western blotting showed ADHC-AXpn caused a time- and dose-dependent decrease in cyclin B1 (Fig. 4b). ADHC-AXpn decreased cyclin-dependent kinase 1 (CDK1), which regulates the cell cycle by forming the CDK-cyclin complex, and phospho-CDK1 (Thr161). ADHC-AXpn arrests the cell cycle in G2/M phase by decreasing expression of cyclin B1, CDK1 and phospho-CDK1 (Thr161).

Cell-cycle arrest induced by ADHC-AXpn in MCF-7 cells. a Cell cycle profiles of MCF-7 cells after treatment with ADHC-AXpn for 24 or 48 h. Cells exposed to indicated concentrations of ADHC-AXpn were collected, fixed with 75 % ethanol overnight, and stained with PI for 30 min; cell-cycle profiles were then determined by flow cytometry. Doxorubicin (dox) exposure at 5 μM was used as a positive control. b Changes in cell-cycle regulator protein expression after exposure to ADHC-AXpn in MCF-7 cells. After treatment with ADHC-AXpn, total cell lysates were immunoblotted to detect CDK1, p-CDK1 (Thr161) and cyclin B1 expression levels, using specific antibodies. β-actin served as a loading control
Induction of apoptosis by ADHC-AXpn in MCF-7 cells
Changes of cellular morphology were examined by fluorescent microscopy analysis (Fig. 5a). After treatment with different concentrations of ADHC-AXpn (20, 30 or 35 μM) for 48 h, cells stained with Hoechst 33258 showed apoptotic features, such as cytoplasmic shrinkage and condensation of nuclear chromatin. Consistently, PI/Annexin V double staining analysis (Fig. 5b) showed that treatment with ADHC-AXpn (30 or 35 μM) for 48 h led to significantly increased cell percentages in early and late apoptotic phases (P = 0.0094 and P = 0.0042 vs. control, respectively). Cleavage of PARP was also seen in ADHC-AXpn-treated MCF-7 cells (Fig. 5c).

Apoptosis induced by ADHC-AXpn in MCF-7 cells. a Representative image of MCF-7 cells stained with Hoechst 33258 after treatment with ADHC-AXpn for 48 h, under fluorescence microscope; red arrows: cell shrinkage, nuclear fragmentation. b Apoptosis quantification, using Annexin V/PI double staining assay after treatment with ADHC-AXpn for 36 h. MCF-7 cells were harvested and stained with PI and Annexin V-FITC in darkness for 15 min, followed by flow cytometry analysis. Lower right: Annexin V+/PI− (early apoptosis); upper right: Annexin V+/PI+ (late apoptosis). c Western blot showing PARP cleavage in MCF-7 cell lysates after treatment with ADHC-AXpn, using indicated antibodies. Loading control: β-actin; positive control: doxorubicin (Dox), 5 μM
Activation of mitochondrial apoptotic pathway and inhibition of Akt phosphorylation
Changes of MMP were measured by a fluorescent dye (JC-1) to further investigate the apoptotic pathway. MCF-7 cells treated with ADHC-AXpn (20, 30 or 35 μM) for 24 h showed a gradual decrease in the percentage of cells with high MMP from 94.1 (control) to 68.7 %, 63.0 and 55.9 %, respectively (Fig. 6a). We examined expression levels of pro-apoptotic protein Bax and anti-apoptotic protein Bcl-2. Western blot analysis showed that ADHC-AXpn treatment down-regulated Bcl-2 and clearly up-regulated Bax (Fig. 6b). The precursor form of caspase-9 was decreased, whereas expression of cleaved caspase-9 was increased. ADHC-AXpn exposure remarkably decreased p-Akt (Thr308 and Ser473), although total Akt showed no visible change.

Effect of ADHC-AXpn on mitochondrial function and Raf/MEK/ERK signaling pathway in MCF-7 cells. a Flow cytometry shows mitochondrial membrane depolarization induced by treatment with indicated concentrations of ADHC-AXpn for 24 h; detached cells were stained with JC-1; generic mitochondrial membrane depolarizer CCCP (20 μM), used as positive control, showed an expected decrease in red fluorescence. b Western blot shows caspase activation and changes in expression levels of Bcl-2 family proteins and Akt proteins in MCF-7 cells after treatment with ADHC-AXpn, using specific antibodies against caspase-9, cleaved caspase-9, Bcl-2 and Bax, Akt and p-Akt (Thr308 and Ser473), respectively. c Western blot shows inhibition of Raf/MEK/ERK signaling pathway related proteins in MCF-7 cells after treatment with ADHC-AXpn, using the indicated antibodies; loading control: β-actin
Raf/MEK/ERK pathway inhibition by ADHC-AXpn in MCF-7 cells
The Raf/MEK/ERK pathway is beneficial for survival of MCF-7 cells, and blockage of this pathway can trigger apoptosis [15]. To investigate whether the Raf/MEK/ERK pathway affects ADHC-AXpn-induced apoptosis, we assessed expression levels of p-ERK1/2 and p-c-Raf. Compared with the control group, ADHC-AXpn treatment decreased phosphorylation of ERK1/2 in both time- and dose-dependent manners (Fig. 6c). The upstream regulator of ERK1/2, p-c-Raf, was also down-regulated after exposure to ADHC-AXpn.
Discussion
In the present study, a novel cycloartane triterpenoid, ADHC-AXpn and the known DHC-Xpn were isolated and identified from C. foetia. The cytotoxic effects of these two isolated components on human cancer cells MCF-7, HepG2, HepG2/ADM, HeLa, PC3 and normal human mammary epithelial cells MCF10A were assessed by MTT assay and colony formation assay. ADHC-AXpn, which possesses acetyl groups at C-25 and C-2′ in its sugar groups, was more cytotoxic toward all tested cell lines than was DHC-Xpn, which bears hydroxyl groups at C-25 and C-2′ in its sugar group. Addition of a hydrophobic group (such as acetyl or halogen) linked to C-25 or other positions remarkably increased cytotoxic activity, which were consistent with a previous report on the structure–activity relationship of cycloartane triterpenoids in other cells [4]. The IC50 value of ADHC-AXpn for normal human mammary epithelial cells MCF10A was higher than those for cancer cells.
Activation of the apoptotic pathway in tumor cells is a major protective mechanism against the development and progression of cancer [16]. On the basis of the detected cytotoxic effects of ADHC-AXpn and DHC-Xpn on MCF-7, HepG2, HepG2/ADM, HeLa and PC3 cells (Table 2), further studies were performed to explore the mechanism of ADHC-AXpn-induced cell death in MCF-7 cells. To our knowledge, our study was the first one to demonstrate that ADHC-AXpn could induce MCF-7 cell death via apoptosis, accompanied with stereotypical apoptotic features that include cell shrinkage, phosphatidylserine (PS) externalization and PARP cleavage. ADHC-AXpn inhibited cyclin B1 and CDK1, and thereby led to clear G2/M arrest.
Apoptosis has two major pathways: the cell death receptor-mediated apoptotic pathway and the mitochondrial-mediated apoptotic pathway [17]. The latter is also referred to as the intrinsic apoptosis pathway, and it is involved in the caspase-dependent apoptotic pathway [18, 19]. Bcl-2 family proteins are major regulators of the mitochondrial pathway; Bcl-2 is a major anti-apoptosis protein, integrated in the mitochondrial membrane, and Bax is inactive in the cytosol or loosely attached to intracellular membranes as a monomer [20, 21]. The over-expression of Bax and lower-expression of Bcl-2 induce the MMP collapse and a subsequent cascade activation of caspases through homologous dimerization, and eventually lead to apoptosis. Four isolated cycloartane triterpenoids from Cimicifuga yunnanensis, another species of Cimicifuga genus, 25-O-acetylcimigenol-3-O-β-d-xylopyranoside, 25-chlorodeoxycimigenol-3-O-β-d-xylopyranoside, 25-O-acetylcimigenol-3-O-α-l-arabinopyranoside and 23-O-acetylcimigenol-3-O-β-d-xylopyranoside increased expression of p53 and Bax, resulting in mitochondria-mediated apoptosis [6]. Our results showed similar phenomena, including changes in Bcl-2 and Bax levels, and decreased MMP and caspase-9 activation (Fig. 6a, b), implying that mitochondrial dysfunction was involved in ADHC-AXpn-induced apoptosis in MCF-7 cells. In addition, decreased Akt phosphorylation might be involved in ADHC-AXpn-induced apoptosis, as Akt activation protects cells from apoptosis via inhibiting pro-apoptotic molecues [22].
The Raf/MEK/ERK pathway can transduce signals that originate from growth factor receptors to transcription factors, thus controlling cell survival and proliferation [15]. Raf, mediated cell cycle progression by regulating cell-cycle associated proteins such as Cdks, cyclins and p21Cip1 [23, 24]. The Raf/MEK/ERK pathway also affects apoptosis progression. Raf can induce transcription of anti-apoptotic genes such as Bcl-2, and activation of the Raf/MEK/ERK pathway, leading to phosphorylation and inactivation of the pro-apoptotic protein Bad [25–27]. ERK1/2 is required for survival signaling and ERK1/2 inhibition is a crucial part of the apoptotic mechanisms of various stimuli [28]. Our present studies on the regulatory effects of ADHC-AXpn on cyclin B1, CDK1, caspase-9, Bax and Bcl-2, suggested that the Raf/MEK/ERK pathway might be involved in ADHC-AXpn-induced apoptosis.
Conclusions
ADHC-AXpn significantly suppressed the growth of MCF-7 cells, induced mitochondrial apoptosis and cell-cycle arrest, and inhibited Raf/MEK/ERK signaling pathway and Akt phosphorylation.
Abbreviations
- Sheng ma :
-
the rhizome of Cimicifuga foetida L
- CM:
-
Chinese medicine
- ADHC-AXpn:
-
25-O-acetyl-7,8-didehydrocimigenol-3-O-β-d-(2-acetyl)xylopyranoside
- DHC-Xpn:
-
7,8-didehydrocimigenol-3-O-β-d-xylopyranoside
- 25-AC:
-
25-O-acetyl-7,8-didehydrocimigenol-3-O-β-d-xylopyranoside
- CATP:
-
cycloartane triterpenoid
- IR:
-
infrared spectra
- HR-FAB-MS:
-
high resolution fast atom bombardment mass spectra
- NMR:
-
nuclear magnetic resonance
- HMBC:
-
hetero-nuclear multiple bond correlation
- CCG:
-
column chromatography
- TLC:
-
thin layer chromatography
- MTT:
-
3-(4, 5)-Dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide
- PI:
-
propidium iodide
- Dox:
-
doxorubicin
- JC-1:
-
5, 5′, 6, 6′-tetrachloro-1, 1′, 3, 3′-tetraethyl benzimidazolyl-carbocyanine iodide
- DMSO:
-
dimethyl sulfoxide
- FBS:
-
fetal bovine serum
- PBS:
-
phosphate buffered saline
- PI:
-
peromide iodine
- P/S:
-
penicillin-streptomycin
References
Man SL, Gao WY, Wei CL, Liu CX. Anticancer drugs from traditional toxic Chinese medicines. Phytother Res. 2012;26:1449–65.
Dinda B, Debnath S, Mohanta BC, Harigaya Y. Naturally occurring triterpenoid saponins. Chem Biodivers. 2010;7:2327–580.
Tian Z, Sun Y, Xiao PG, Wu E: Recent advances in natural bioactive cycloartane triterpenoids. In: Awaad AS, Kaushik G, Govil JN, editors. Mechanism and action of phytoconstituents—recent progress in medicinal plants, vol. 31. New Delhi: Studium Press (India) Pvt. Ltd.; 2011. p. 49–63.
Tian Z, Yang MS, Huang F, Li K, Si JY, Shi L, Chen SB, Xiao PG. Cytotoxicity of three cycloartane triterpenoids from Cimicifuga dahurica. Cancer Lett. 2005;226:65–75.
Tian Z, Zhou L, Huang F, Chen SB, Yang JS, Wu E, Xiao PG, Yang MS. Anti-cancer activity and mechanisms of 25-anhydrocimigenol-3-O-β-d-xylopyranoside isolated from Souliea vaginata on hepatomas. Anti-Cancer Drug. 2006;17:545–51.
Fang ZZ, Nian Y, Li W, Wu JJ, Ge GB, Dong PP, Zhang YY, Qiu MH, Liu L, Yang L. Cycloartane triterpenoids from Cimicifuga yunnanensis induce apoptosis of breast cancer cells (MCF7) via p53-dependent mitochondrial signaling pathway. Phytother Res. 2011;25:17–24.
Nian Y, Wang HY, Zhou L, Su J, Li Y, Qiu MH. Cytotoxic cycloartane triterpenes of the traditional Chinese medicine “shengma” (Cimicifuga dahurica). Planta Med. 2013;79:60–9.
China Pharmacopoeia Committee. Pharmacopoeia of the People’s Republic of China, vol. 1. Beijing: Chinese medicine and technology publishing house; 2015. p. 73–4.
Nian Y, Zhang YL, Chen JC, Lu L, Qiu MH, Qing C. Cytotoxic chemical constituents from the roots of Cimicifuga foetida. J Nat Prod. 2010;73:93–8.
Nian Y, Zhang XM, Li Y, Wang YY, Chen JC, Lu L, Zhou L, Qiu MH. Cycloartane triterpenoids from the aerial parts of Cimicifuga foetida Linnaeus. Phytochemistry. 2011;72:1473–81.
Fu JJ, Ju DH, Xie YM. Low cell density induced Epithelial-Mesenchymal transition in MCF-10A cell is regulated by proteasome activity. J Oncobiomarkers. 2013;1:4–8.
Kusano A, Takahira M. ShibanoM, Miyase T, Kusano G: Studies on the constituents of Cimicifuga species. XXVI. Twelve new cyclolanostanol glycosides from the underground parts of Cimicifuga simplex Wormsk. Chem Pharm Bull. 1999;47:511–6.
Nian Y, Wang HY, Su J, Zhou L, Feng G, Li Y, Qiu MH. Cytotoxic cycloartane triterpenes from the roots of Cimicifuga heracleifolia. Tetrahedron. 2012;68:6521–7.
Zhang QW, Ye WC, Zhao SX, Che ZT. Studies on chemical constituents of Cimicifuga dahurica. Chin Trad Herb Drug. 2002;33:683–5.
Chang F, Steelman LS, Shelton JG, Lee JT, Navolanic PM, Blalock WL, Franklin R, McCubrey JA. Regulation of cell cycle progression and apoptosis by the Ras/Raf/MEK/ERK pathway (review). Int J Oncol. 2003;22:469–80.
Cheng L, Wang X, Zhang J, Zhang SB, Zheng SQ, Zheng J. Targeting apoptosis signaling pathways in cancer therapy. Chin J Pathol. 2009;38:639–42.
Soldatenkov VA, Smulson M. Poly (ADP-ribose) polymerase in DNA damage-response pathway: implications for radiation oncology. Int J Cancer. 2000;90:59–67.
Huang WW, Chiu YJ, Fan MJ, Lu HF, Yeh HF, Li KH, Chen PY, Chung JG, Yang JS. Kaempferol induced apoptosis via endoplasmic reticulum stress and mitochondria-dependent pathway in human osteosarcoma U-2 OS cells. Mol Nutr Food Res. 2010;54:1585–95.
Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90:1281–9.
Kowaltowski AJ, Vercesi AE, Fiskum G. Bcl-2 prevents mitochondrial permeability transition and cytochrome c release via maintenance of reduced pyridine nucleotides. Cell Death Differ. 2000;7:903–10.
Reed JC. Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006;13:1378–86.
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3 K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004.
Chang F, Steelman LS, McCubrey JA. Raf-induced cell cycle progression in human TF-1 hematopoietic cells. Cell Cycle. 2002;1:220–6.
Lee JTJ, McCubrey JA. The Raf/MEK/ERK signal transduction cascade as a target for chemotherapeutic intervention in leukemia. Leukemia. 2002;16:486–507.
Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–8.
Majewski M, Nieborowska-Skorska M, Salomoni P, Slupianek A, Reiss K, Trotta R, Calabretta B, Skorski T. Activation of mitochondrial Raf-1 is involved in the antiapoptotic effects of Akt. Cancer Res. 1999;59:2815–9.
Blagosklonny MV, Demidenko ZN, Fojo T. Inhibition of transcription results in accumulation of Wt p53 followed by delayed outburst of p53-inducible proteins: p53 as a sensor of transcriptional integrity. Cell Cycle. 2002;1:67–74.
Yuan L, Wang J, Xiao H, Wu W, Wang Y, Liu X. MAPK signaling pathways regulate mitochondrial-mediated apoptosis induced by isoorientin in human hepatoblastoma cancer cells. Food Chem Toxicol. 2013;53:62–8.
Authors’ contributions
SBC and DMZ designed the study. BBL isolated the tested compounds. WCY performed the structure elucidation of tested compounds. HYS, JYH and LJX performed the biological experiments. HYS and COC performed the data analysis and wrote the manuscript. DKWM and SWC revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81373953; 81303203) and Shenzhen Basic Research Program, China (JCYJ20120618173411244), and was part of the work of the Hong Kong Chinese Material Medica Standard Project.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sun, Hy., Liu, Bb., Hu, Jy. et al. Novel cycloartane triterpenoid from Cimicifuga foetida (Sheng ma) induces mitochondrial apoptosis via inhibiting Raf/MEK/ERK pathway and Akt phosphorylation in human breast carcinoma MCF-7 cells. Chin Med 11, 1 (2016). https://doi.org/10.1186/s13020-015-0073-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13020-015-0073-6