
Abstract
Gene fusion events have been linked to oncogenesis in many cancers. However, gene fusions in meningioma are understudied compared to somatic mutations, chromosomal gains/losses, and epigenetic changes. Fusions involving B-raf proto-oncogene, serine/threonine kinase (BRAF) are subtypes of oncogenic BRAF genetic abnormalities that have been reported in certain cases of brain tumors, such as pilocytic astrocytomas. However, BRAF fusions have not been recognized in meningioma. We present the case of an adult female presenting with episodic partial seizures characterized by déjà vu, confusion, and cognitive changes. Brain imaging revealed a cavernous sinus and sphenoid wing mass and she underwent resection. Histopathology revealed a World Health Organization (WHO) grade 1 meningioma. Genetic profiling with next generation sequencing and microarray analysis revealed an in-frame BRAF::PTPRN2 fusion affecting the BRAF kinase domain as well as chromothripsis of chromosome 7q resulting in multiple segmental gains and losses including amplifications of cyclin dependent kinase 6 (CDK6), tyrosine protein-kinase Met (MET), and smoothened (SMO). Elevated pERK staining in tumor cells provided evidence of activated mitogen-activated protein kinase (MAPK) signaling. This report raises the possibility that gene fusion events may be involved in meningioma pathogenesis and warrant further investigation.
Similar content being viewed by others

Introduction
Meningiomas are the most common primary brain tumors comprising one third of all central nervous system tumors [1]. The World Health Organization (WHO) groups meningiomas into three grades based on their histopathological and molecular characteristics. Distinguishing between grades of meningiomas has important implications for treatment, management, and prognosis [2]. Approximately 80% of meningiomas are WHO grade 1 and can be managed with surgery or stereotactic radiosurgery (SRS) with low recurrence risk, while about 20% are atypical or anaplastic subtypes (WHO grade 2 and 3, respectively) with associated increased recurrence and malignant potential [3,4,5].
While the WHO grading provides a framework to manage and prognosticate meningioma behavior, some meningiomas may not behave according to their WHO grade [6]. Molecular and genetic analysis of meningiomas through next generation sequencing (NGS) and DNA methylation profiling are increasingly used to refine our understanding of meningioma recurrence risk [7,8,9,10,11,12,13]. Recent studies using RNA sequencing and DNA methylation profiling have shown that meningiomas can be categorized according to their methylation or gene expression patterns [12, 14, 15]. NGS has led to the identification of novel driver gene mutations in meningioma in addition to NF2, including SMO, PI3KCA, TRAF7, KLF4, AKT1, POLR2A, and SMARCE1 [7, 16,17,18,19,20]. Discovery of driver mutations can offer additional routes of personalized therapy for surgically challenging or recurrent meningiomas, as in the Alliance A071401 Clinical Trial [21,22,23]. Identification of genetic markers in meningioma such as TERT and CDKN2A provides insight into prognosis and potential recurrence [24, 25]. Molecular sequencing has also revealed potential therapeutic targets in meningioma including SMO, AKT/PI3K, and BAP-1 [26]. Incorporating molecular and genetic insights with histopathological analysis is advancing characterization and treatment of meningioma; however, limited studies have investigated the role of gene fusions in meningioma.
Here, we report the case of an adult female presenting with episodic partial seizures who was found to have a WHO grade 1 left cavernous sinus/sphenoid wing meningioma. Genetic profiling of her resected tumor with NGS revealed no pathologic mutations. Gene fusion panel identified a 7q34 B-raf proto-oncogene, serine/threonine kinase (BRAF) fusion with a 7q36.3 protein tyrosine phosphatase receptor type N2 (PTPRN2). Copy number profiling revealed chromothripsis of chromosome 7q resulting in multiple segmental gains and losses including amplifications of 7q21.2–7q22.1 containing cyclin dependent kinase 6 (CDK6), 7q31.1–7q31.31 containing tyrosine protein-kinase Met (MET), 7q31.33 - q32.1 containing smoothened (SMO), as well as amplifications of both 7q33, 7q36.1 and 7q36.3. Additional immunostaining revealed pERK positivity in tumor cells, indicating increased mitogen-activated protein kinase (MAPK) pathway activation. Methylation profiling matched to a Merlin-intact meningioma, consistent with the copy number profile of this tumor. To our knowledge, this is the first BRAF fusion reported in meningioma. Future investigations into the significance of BRAF fusions in meningioma may help select good candidates for targeted anti-BRAF therapy.
Case presentation
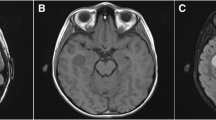
The patient was a healthy 30-year-old right-handed female with no significant past medical history who presented with one month of multiple acute episodes of déjà vu, phantosmia (odorant perceived in the absence of stimulus), and Broca’s aphasia followed by sensations of panic and dizziness. She occasionally had difficulty finding words for a short period after the episodes. She had some mild decreased left facial sensation. The patient was referred to the epilepsy team for work up of her partial seizures. Electroencephalogram (EEG) did not show epileptiform activity. Brain magnetic resonance imaging (MRI) revealed an avidly enhancing 5.9 × 4.8 cm left cavernous sinus and sphenoid wing mass with cerebral edema, significant compression of the left temporal lobe and left frontal lobe, 3 mm midline shift (Fig. 1a), and invasion into the left cavernous sinus, left anterior clinoid, and the left superior orbital fissure. Edema of the left hippocampus and temporal gyri was present on brain MRI. The patient was started on 500 mg of Levetiracetam twice a day for management of the partial seizures.

A) Pre-operative MRI showing a large middle cranial fossa mass with surrounding edema, significant compression of the left temporal lobe and effacement of the lateral ventricle. B) Post-operative MRI showing residual meningioma in the cavernous sinus following planned subtotal resection to preserve neurological function
Given the size and location of the mass as well as the patient’s young age, the patient underwent a left cranio-orbital craniotomy with planned subtotal resection (Fig. 1b), with intentional residual left inside the cavernous sinus to preserve cranial nerve function. The procedure was completed without intraoperative complication.
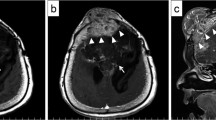
Histopathology of the tumor sample showed an epithelioid neoplasm composed of oval to round, bland nuclei with occasional nuclear pseudoinclusions arranged in whorls and fascicles (Fig. 2a). Hypercellularity, microcystic features, and focal clear cell change were present. The sample was absent of necrosis, sheeting, prominent macro-nucleoli, and small cell change. There were no chordoid, papillary, or rhabdoid features present. Immunohistochemistry (IHC) stains for somatostain receptor 2a (SSTR2a) and progesterone receptor (PR) were positive in the tumor cells (Fig. 2b and e). Additionally, pERK staining of the sample showed strong positivity in a subset of the tumor cells, indicating increased MAPK activation (Fig. 2c). The DNA methylation profile was analyzed using the Illumina EPIC 850k platform, and matched with meningioma (confidence score 1.0). Post-hoc analysis revealed the tumor was in the Merlin-intact methylation group [12]. The sample was negative for TERT promoter mutation based on targeted sequencing. Standard workup of meningioma at our institution involves in-depth molecular analysis for recurrent tumors, WHO grades 2 or 3, and select WHO grade 1 tumors at the treating physician’s discretion. Due to the patient’s young age and very large size at presentation, molecular analysis was performed on her tumor to inform risk profiling and future treatments if necessary. The tumor sample was analyzed with PGDx Solid Tumor NGS panel, FusionPlex Solid Tumor NGS panel, and Oncoscan Solid Tumor Microarray. Interestingly, an in-frame fusion transcript BRAF::PTPRN2 was detected along with chromothripsis of chromosome 7q, resulting in multiple segmental gains and losses, including amplifications of 7q21.2q22.1 (CDK6), 7q31.1q31.31 (MET), and 7q31.33q31.1 (SMO) (demonstrated based on DNA methylation profile in Fig. 3a). This BRAF::PTPRN2 fusion is an in-frame fusion between BRAF Exon 16 on chromosome 7q34 and PTPRN2 Exon 14 on chromosome 7q36.3, affecting the BRAF kinase domain (Fig. 4a). Oncoscan also detected upregulation of the fused regions 7q34 (5 times copy number state) and 7q36.3 (8 times copy number state). The combined histologic, immunohistochemical, and molecular features supported the preoperative diagnosis of WHO grade 1 meningioma.

A) H&E stain of resected tumor showing oval to round bland nuclei with occasional nuclear pseudoinclusions arranged in whorls and fascicles. B) Immunohistochemical staining showing positive expression of somatostatin receptor 2a. C) pERK stain showing positive staining in a large subset of our patient’s tumor cells. D) Negative control pERK stain showing negative staining in a comparative WHO grade 1 meningioma. E) Immunohistochemical staining showing positive expression of progesterone receptor (PR). All photomicrographs taken at 200x magnification

A) Copy number profile from DNA methylation sequencing reveals chromothripsis of chromosome 7. B) tSNE plot showing the current case clustering with meningioma

A) Gene fusion diagram showing BRAF gene on chromosome 7q34 and PTPRN2 gene on chromosome 7q36.3, resulting in an in-frame fusion between BRAF exon 16 and PTPRN2 exon 14, affecting the BRAF kinase domain. B) Comparative gene fusion diagram of a pilocytic astrocytoma fusion showing a BRAF::KIAA1549 gene fusion resulting from a duplication of BRAF on chromosome 7q34
Postoperatively, the patient was transferred to the neurosurgery intensive care unit for recovery. The patient had mild diplopia, decreased sensation to pressure in the left V3 dermatome, and mild ptosis on the left, but was otherwise neurologically intact and doing well. The patient was discharged on postoperative day 2 and continued prophylactic anticoagulation therapy as well as continued Levetiracetam. At eight-week follow-up in clinic, the patient continued to do well with no further seizures, resolution of diplopia and improved facial sensation.
Discussion and conclusions
The BRAF gene, located at chromosome 7q34, encodes the B-Raf proto-oncogene serine/threonine kinase, which regulates cell growth, division, and differentiation [27, 28]. Activating BRAF alterations can lead to a constitutively active B-Raf protein, resulting in neoplastic growth. The most common BRAF alteration is a V600E mutation, where the substitution of valine with glutamic acid at position 600 causes hyperactivation of the protein [29, 30]. Previous studies have reported different BRAF mutations in colorectal, lung, thyroid cancers, and melanoma [28, 31,32,33]. BRAF mutations in meningioma, though rare, have been reported in the literature with the majority being BRAF V600E point mutations in rhabdoid meningioma [34, 35].
Targeted therapies have been developed to inhibit constitutively activated BRAF proteins. BRAF inhibitors (BRAFi), such as vemurafenib and dabrafenib, have shown significant efficacy in targeted treatment of patients with BRAF positive cancers such as melanoma [36, 37]. BRAFi therapy may also have potential efficacy for treating BRAF-activated mutant primary brain tumors, such as gliomas and astrocytomas, based on initial data [38, 39]. However, resistance to BRAFi therapy remains a significant challenge in the treatment of BRAF-activated primary brain tumors [39,40,41].
BRAF gene fusions are a less common subtype of BRAF genetic alteration that have been reported primarily in brain tumors. Several studies have reported on the BRAF::KIAA1549 fusion in pilocytic astrocytomas as well as diffuse leptomeningeal glioneuronal tumors (DLGNT) [42,43,44]. The resultant protein, containing the N-terminal region of KIAA1549 and the kinase domain of BRAF, leads to activation of the MAPK signaling pathway and contributes to oncogenesis [45].
Given the availability of clinical inhibitors, BRAF alterations have been investigated as a potential treatment target in multiple cancers. However, to the best of our knowledge, the fusion of BRAF with PTPRN2 in a WHO grade 1 meningioma has not been previously reported. PTPRN2, also found on chromosome 7, encodes the protein Islet Antigen-2β (IA-2β) which belongs to the protein tyrosine phosphatase (PTP) family and plays an important role in insulin secretion [46]. IA-2β may play a role in glucose intolerance in insulin-dependent diabetes [47]. However, the full activity of IA-2β has not yet been elucidated and its tyrosine phosphatase activity has not been experimentally validated. PTPRN2 is upregulated in breast cancer and has been shown to increase tumor growth and metastatic potential in mouse models [48]. Additionally, epigenetic modification PTPRN2 has been suggested to play a role in the oncogenesis of hepatocellular carcinoma [49], and it is hypomethylated in some glioblastomas, glioblastoma stem cells and primary xenografts [50]; however, it has never been reported to be altered in meningioma. In our patient, upregulation of the sites of BRAF::PTPRN2 fusion at 7q34 and 7q36.3, along with positive pERK staining in tumor cells, raise the possibility that this novel fusion may contribute to oncogenesis by increased MAPK activation, but further experimental validation would be necessary to define the impact of the fusion.
Chromothripsis of chromosome 7q was also identified in our patient, resulting in amplifications of CDK6, MET, and SMO. Chromothripsis is a single, large event that involves complex chromosomal rearrangements, including deletions, duplications, inversions, and translocations resulting in reorganization of genes [51, 52]. While the frequency of chromothripsis in meningiomas is not well-established, cases have been reported [16, 53]. Some studies have suggested that cancers with complex chromosomal rearrangements, such as chromothripsis, may exhibit more aggressive behavior, progression to higher grade, increased recurrence rates, or altered response to treatment [51, 54, 55]. However, further research is needed to better understand these associations.
The identification of the BRAF::PTPRN2 fusion in meningiomas is novel, and highlights the need for further interrogation of gene fusions in meningioma tumorigenesis and progression. While the clinical and biological implications of the BRAF::PTPRN2 fusion are not fully known, further investigation could offer insight into the mechanisms of meningioma development and recurrence after surgical treatment. Further mechanistic and clinical research of this novel fusion is needed to determine its potential oncogenesis and whether patients with such fusions would be good candidates for anti-BRAF therapy.
In conclusion, we report a novel BRAF::PTPRN2 fusion with associated chromothripsis of chromosome 7q in a case of a WHO grade 1 meningioma. These findings contribute to a growing literature of the genomic profiling in meningiomas. Further studies are needed to detail the clinical and pathological significance of BRAF fusions as well as to explore the potential of targeted therapies for meningioma.
Data availability
Not applicable.
Abbreviations
- BRAF:
-
V-raf murine sarcoma viral oncogene homolog B1
- WHO:
-
World Health Organization
- PTPRN2:
-
protein tyrosine phosphatase receptor type N2
- CDK6:
-
cyclin dependent kinase 6
- MET:
-
tyrosine protein kinase Met
- SMO:
-
smoothened
- MAPK:
-
mitogen-activated protein kinase
- SRS:
-
stereotactic radiosurgery
- NGS:
-
next generation sequencing
- EEG:
-
Electroencephalogram
- MRI:
-
magnetic resonance imaging
- IHC:
-
immunohistochemistry
- BRAFi:
-
BRAF inhibitor
- DLGNT:
-
diffuse leptomeningeal glioneuronal tumor
- IA-2β:
-
islet antigen-2β
- PTP:
-
protein tyrosine phosphatase
References
Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C et al (2022) CBTRUS Statistical Report: primary brain and other Central Nervous System tumors diagnosed in the United States in 2015–2019. Neuro-Oncol 24:v1–95. https://doi.org/10.1093/neuonc/noac202
Yang S-Y, Park C-K, Park S-H, Kim DG, Chung YS, Jung H-W (2008) Atypical and anaplastic meningiomas: prognostic implications of clinicopathological features. J Neurol Neurosurg Psychiatry 79:574–580. https://doi.org/10.1136/jnnp.2007.121582
Sahm F, Reuss DE, Giannini C (2018) WHO 2016 classification: changes and advancements in the diagnosis of miscellaneous primary CNS tumours. Neuropathol Appl Neurobiol 44:163–171. https://doi.org/10.1111/nan.12397
Cornelius JF, Slotty PJ, Steiger HJ, Hänggi D, Polivka M, George B (2013) Malignant potential of skull base versus non-skull base meningiomas: clinical series of 1,663 cases. Acta Neurochir (Wien) 155:407–413. https://doi.org/10.1007/s00701-012-1611-y
Maggio I, Franceschi E, Tosoni A, Nunno VD, Gatto L, Lodi R et al (2021) Meningioma: not always a benign Tumor. A review of advances in the treatment of meningiomas. CNS Oncol 10:CNS72. https://doi.org/10.2217/cns-2021-0003
Goldbrunner R, Minniti G, Preusser M, Jenkinson MD, Sallabanda K, Houdart E et al (2016) EANO guidelines for the diagnosis and treatment of meningiomas. Lancet Oncol 17:e383–391. https://doi.org/10.1016/S1470-2045(16)30321-7
Pepe F, Pisapia P, Del Basso de Caro ML, Conticelli F, Malapelle U, Troncone G et al (2020) Next generation sequencing identifies novel potential actionable mutations for grade I meningioma treatment. Histol Histopathol 35:741–749. https://doi.org/10.14670/HH-18-195
Nassiri F, Liu J, Patil V, Mamatjan Y, Wang JZ, Hugh-White R et al (2021) A clinically applicable integrative molecular classification of meningiomas. Nature 597:119–125. https://doi.org/10.1038/s41586-021-03850-3
Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S et al (2017) DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol 18:682–694. https://doi.org/10.1016/S1470-2045(17)30155-9
Youngblood MW, Miyagishima DF, Jin L, Gupte T, Li C, Duran D et al (2021) Associations of meningioma molecular subgroup and Tumor recurrence. Neuro-Oncol 23:783–794. https://doi.org/10.1093/neuonc/noaa226
Patel AJ, Wan Y-W, Al-Ouran R, Revelli J-P, Cardenas MF, Oneissi M et al (2019) Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc Natl Acad Sci U S A 116:21715–21726. https://doi.org/10.1073/pnas.1912858116
Choudhury A, Magill ST, Eaton CD, Prager BC, Chen WC, Cady MA et al (2022) Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities. Nat Genet 54:649–659. https://doi.org/10.1038/s41588-022-01061-8
Driver J, Hoffman SE, Tavakol S, Woodward E, Maury EA, Bhave V et al (2022) A molecularly integrated grade for meningioma. Neuro-Oncol 24:796–808. https://doi.org/10.1093/neuonc/noab213
Choudhury A, Chen WC, Lucas C-HG, Bayley JC, Harmanci AS, Maas SLN et al (2023) Hypermitotic meningiomas harbor DNA methylation subgroups with distinct biological and clinical features. Neuro-Oncol 25:520–530. https://doi.org/10.1093/neuonc/noac224
Raleigh D, Chen W, Choudhury A, Youngblood M, Polley M-Y, Lucas C-H et al (2023) Targeted gene expression profiling predicts meningioma outcomes and radiotherapy responses. Res Sq. https://doi.org/10.21203/rs.3.rs-2663611/v1. rs.3.rs-2663611
Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G et al (2013) Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet 45:285–289. https://doi.org/10.1038/ng.2526
Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Özduman K et al (2013) Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 339:1077–1080. https://doi.org/10.1126/science.1233009
Smith MJ, O’Sullivan J, Bhaskar SS, Hadfield KD, Poke G, Caird J et al (2013) Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet 45:295–298. https://doi.org/10.1038/ng.2552
Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C et al (1993) Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363:515–521. https://doi.org/10.1038/363515a0
Clark VE, Harmancı AS, Bai H, Youngblood MW, Lee TI, Baranoski JF et al (2016) Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet 48:1253–1259. https://doi.org/10.1038/ng.3651
Bi WL, Santagata S (2022) Skull Base tumors: Neuropathology and Clinical implications. Neurosurgery 90:243. https://doi.org/10.1093/neuros/nyab209
Bujko M, Kober P, Tysarowski A, Matyja E, Mandat T, Bonicki W et al (2014) EGFR, PIK3CA, KRAS and BRAF mutations in meningiomas. Oncol Lett 7:2019–2022. https://doi.org/10.3892/ol.2014.2042
Alliance for Clinical Trials in Oncology (2023) Phase II trial of SMO/ AKT/ NF2/CDK inhibitors in Progressive Meningiomas with SMO/ AKT/ NF2/CDK pathway mutations. clinicaltrials.gov
Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J et al (2015) TERT promoter mutations and risk of recurrence in Meningioma. JNCI J Natl Cancer Inst 108:djv377. https://doi.org/10.1093/jnci/djv377
Wang JZ, Patil V, Liu J, Dogan H, Tabatabai G, Yefet LS et al (2023) Increased mRNA expression of CDKN2A is a transcriptomic marker of clinically aggressive meningiomas. Acta Neuropathol (Berl) 146:145–162. https://doi.org/10.1007/s00401-023-02571-3
Graillon T, Tabouret E, Chinot O (2021) Chemotherapy and targeted therapies for meningiomas: what is the evidence? Curr Opin Neurol 34:857. https://doi.org/10.1097/WCO.0000000000001002
Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR (2018) Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene 37:3183–3199. https://doi.org/10.1038/s41388-018-0171-x
Jarkowski A, Khushalani NI (2014) BRAF and beyond: tailoring strategies for the individual Melanoma patient. J Carcinog 13:1. https://doi.org/10.4103/1477-3163.126759
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. https://doi.org/10.1038/nature00766
Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A et al (2011) Raf family kinases: old dogs have learned new tricks. Genes Cancer 2:232–260. https://doi.org/10.1177/1947601911407323
Pan J-H, Zhou H, Zhu S-B, Huang J-L, Zhao X-X, Ding H et al (2018) Development of small-molecule therapeutics and strategies for targeting RAF kinase in BRAF-mutant Colorectal cancer. Cancer Manag Res 10:2289–2301. https://doi.org/10.2147/CMAR.S170105
Leonetti A, Facchinetti F, Rossi G, Minari R, Conti A, Friboulet L et al (2018) BRAF in non-small cell Lung cancer (NSCLC): pickaxing another brick in the wall. Cancer Treat Rev 66:82–94. https://doi.org/10.1016/j.ctrv.2018.04.006
Namba H, Nakashima M, Hayashi T, Hayashida N, Maeda S, Rogounovitch TI et al (2003) Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J Clin Endocrinol Metab 88:4393–4397. https://doi.org/10.1210/jc.2003-030305
Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol (Berl) 121:397–405. https://doi.org/10.1007/s00401-011-0802-6
Behling F, Barrantes-Freer A, Skardelly M, Nieser M, Christians A, Stockhammer F et al (2016) Frequency of BRAF V600E mutations in 969 central nervous system Neoplasms. Diagn Pathol 11:55. https://doi.org/10.1186/s13000-016-0506-2
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J et al (2011) Improved survival with vemurafenib in Melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516. https://doi.org/10.1056/NEJMoa1103782
Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M et al (2012) Dabrafenib in BRAF-mutated metastatic Melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet Lond Engl 380:358–365. https://doi.org/10.1016/S0140-6736(12)60868-X
Nicolaides TP, Li H, Solomon DA, Hariono S, Hashizume R, Barkovich K et al (2011) Targeted therapy for BRAFV600E malignant astrocytoma. Clin Cancer Res 17:7595–7604. https://doi.org/10.1158/1078-0432.ccr-11-1456
Bautista F, Paci A, Minard-Colin V, Dufour C, Grill J, Lacroix L et al (2014) Vemurafenib in pediatric patients with BRAFV600E mutated high-grade gliomas. Pediatr Blood Cancer 61:1101–1103. https://doi.org/10.1002/pbc.24891
Chamberlain MC (2016) Recurrent ganglioglioma in adults treated with BRAF inhibitors. CNS Oncol 5:27–29. https://doi.org/10.2217/cns.15.40
Hofer S, Berthod G, Riklin C, Rushing E, Feilchenfeldt J (2016) BRAF V600E mutation: a treatable driver mutation in pleomorphic xanthoastrocytoma (PXA). Acta Oncol Stockh Swed 55:122–123. https://doi.org/10.3109/0284186X.2015.1021428
Kurani H, Gurav M, Shetty O, Chinnaswamy G, Moiyadi A, Gupta T et al (2019) Pilocytic astrocytomas: BRAFV600E and BRAF fusion expression patterns in pediatric and adult age groups. Childs Nerv Syst ChNS off J Int Soc Pediatr Neurosurg 35:1525–1536. https://doi.org/10.1007/s00381-019-04282-1
Faulkner C, Ellis HP, Shaw A, Penman C, Palmer A, Wragg C et al (2015) BRAF Fusion Analysis in Pilocytic Astrocytomas: KIAA1549-BRAF 15 – 9 fusions are more frequent in the Midline Than within the Cerebellum. J Neuropathol Exp Neurol 74:867–872. https://doi.org/10.1097/NEN.0000000000000226
Manoharan N, Ajuyah P, Senapati A, Wong M, Mullins A, Rodriguez M et al (2021) Diffuse leptomeningeal glioneuronal tumour (DLGNT) in children: the emerging role of genomic analysis. Acta Neuropathol Commun 9:147. https://doi.org/10.1186/s40478-021-01248-w
Collins VP, Jones DTW, Giannini C (2015) Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol (Berl) 129:775–788. https://doi.org/10.1007/s00401-015-1410-7
Li Q, Borovitskaya AE, DeSilva MG, Wasserfall C, Maclaren NK, Notkins AL et al (1997) Autoantigens in insulin-dependent Diabetes Mellitus: molecular cloning and characterization of human IA-2 beta. Proc Assoc Am Physicians 109:429–439
Lee S (2019) The association of genetically controlled CpG methylation (cg158269415) of protein tyrosine phosphatase, receptor type N2 (PTPRN2) with childhood obesity. Sci Rep 9:4855. https://doi.org/10.1038/s41598-019-40486-w
Sengelaub CA, Navrazhina K, Ross JB, Halberg N, Tavazoie SF (2016) PTPRN2 and PLCβ1 promote metastatic Breast cancer cell migration through PI(4,5)P2-dependent actin remodeling. EMBO J 35:62–76. https://doi.org/10.15252/embj.201591973
Gentilini D, Scala S, Gaudenzi G, Garagnani P, Capri M, Cescon M et al (2017) Epigenome-wide association study in hepatocellular carcinoma: identification of stochastic epigenetic mutations through an innovative statistical approach. Oncotarget 8:41890–41902. https://doi.org/10.18632/oncotarget.17462
Lee E-J, Rath P, Liu J, Ryu D, Pei L, Noonepalle SK et al (2015) Identification of global DNA methylation signatures in Glioblastoma-Derived Cancer Stem cells. J Genet Genomics Yi Chuan Xue Bao 42:355–371. https://doi.org/10.1016/j.jgg.2015.06.003
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ et al (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144:27–40. https://doi.org/10.1016/j.cell.2010.11.055
Shorokhova M, Nikolsky N, Grinchuk T (2021) Chromothripsis—Explosion in Genetic Science Cells 10:1102. https://doi.org/10.3390/cells10051102
Baltus C, Toffoli S, London F, Delrée P, Gilliard C, Gustin T (2019) Chromothripsis in an early recurrent Chordoid Meningioma. World Neurosurg 130:380–385. https://doi.org/10.1016/j.wneu.2019.07.003
Bi WL, Greenwald NF, Abedalthagafi M, Wala J, Gibson WJ, Agarwalla PK et al (2017) Genomic landscape of high-grade meningiomas. NPJ Genomic Med 2:15. https://doi.org/10.1038/s41525-017-0014-7
Maher CA, Wilson RK (2012) Chromothripsis and human Disease: piecing together the shattering process. Cell 148:29–32. https://doi.org/10.1016/j.cell.2012.01.006
Acknowledgements
Not applicable.
Funding
No funding was received for this work.
Author information
Authors and Affiliations
Contributions
NSS, KRN, JTA, and STM drafted the manuscript. NSS, KRN, JTA, and STM collected and interpreted clinical data. STM performed the tumor resection. JTA performed the pathological examination. NSS, MWY, CMH, JTA, and STM performed the literature review. KRN and JTA created the figures. NSS, KRN, MWY, CMH, JTA, and STM edited and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Disclosures
The authors have no disclosures of relevance to this work. S.T.M. is supported by the Malnati Brain Tumor Institute of the Lurie Cancer Center.
Ethics approval and consent to participate
This study was approved by our Institutional Review Board and patient gave consent.
Consent for publication
Written informed consent regarding the submission and potential publication of this manuscript was obtained from the case study patient. Consent for treatment and surgery was obtained during the patient’s hospitalization.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sadagopan, N.S., Nandoliya, K.R., Youngblood, M.W. et al. A novel BRAF::PTPRN2 fusion in meningioma: a case report. acta neuropathol commun 11, 194 (2023). https://doi.org/10.1186/s40478-023-01668-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-023-01668-w