
Abstract
Background
According to current clinical guidelines mutational analysis for KRAS and NRAS is recommended prior to EGFR-directed therapy of colorectal cancer (CRC) in the metastatic setting. Therefore, reliable, fast, sensitive and cost-effective methods for routine tissue based molecular diagnostics are required that allow the assessment of the CRC mutational status in a high throughput fashion.
Methods
We have developed a custom designed assay for routine mass-spectrometric (MS) (MassARRAY®, Agena Bioscience) analysis to test the presence/absence of 18 KRAS, 14 NRAS and 4 BRAF mutations. We have applied this assay to 93 samples from patients with CRC and have compared the results with Sanger sequencing and a chip hybridization assay (KRAS LCD-array Kit, Chipron). In cases with discordant results, next-generation sequencing (NGS) was performed.
Results
MS detected a KRAS mutation in 46/93 (49 %), a NRAS mutation in 2/93 (2 %) and a BRAF mutation in 1/93 (1 %) of the cases. MS results were in agreement with results obtained by combination of the two other methods in 92 (99 %) of 93 cases.
In 1/93 (1 %) of the cases a G12V mutation has been detected by Sanger sequencing and MS, but not by the chip assay. In this case, NGS has confirmed the G12V mutation in KRAS.
Conclusions
Mutational analysis by MS is a reliable method for routine diagnostic use, which can be easily extended for testing of additional mutations.
Similar content being viewed by others

Background
Colorectal carcinoma (CRC) is the third most common cancer in men and the second most common cancer in woman worldwide with approximately 694.000 deaths reported in 2012 [1]. It has long been recognized that the epidermal growth factor receptor (EGFR) pathway is frequently activated in CRC [2]. This results in promotion of tumor growth, inhibition of apoptosis, vascular proliferation, invasion, and metastasis.
Therefore, targeted therapies against EGFR such as cetuximab and panitumumab have been developed and are currently approved for the treatment of metastatic disease (mCRC), irrespective of whether applied in combination with conventional chemotherapy or as single agents.
Downstream signalling of EGFR activates RAS- and RAF-genes that are members of this pathway and can harbour oncogenic mutations in 30–60 % (KRAS) [3–6], 5–20 % (BRAF) [7–9] and 1–3 % (NRAS) [10, 11] of cases respectively.
It has been shown that activating mutations of KRAS or NRAS lead to a consecutive activation of the RAS-RAF pathway downstream of EGFR and consequently result in resistance to anti-EGFR therapy [12–14]. For this reason, currently the American Society of Clinical Oncology (ASCO) [15] and the European Society for Medical Oncology (ESMO) [16] recommend treatment with anti-EGFR antibodies only in mCRC patients with RAS wild-type tumors. This approach is reasonable since patients with mutated RAS have no benefit from this therapy. Additionally, it reduces overall treatment costs and prevents patients from unnecessary side effects. In 2009, when the recommendations were published, the definition of a RAS wild type tumor was based on a negative result, when the most common mutations in codon 12 and 13 (97 %) of exon 2 were analyzed. However, since recent studies suggest that resistance to anti-EGFR therapy might also be mediated by less frequent mutations of KRAS [10, 17–19] or NRAS [10, 11, 18] in codon 61 of exon 3 as well as codon 117 and 146 of exon 4 (3 %), it is now mandatory to include those mutational hotspots in the genetic testing, as well. This fact is underlined by a recent study where approximately 20 % of tumors originally classified as having no KRAS mutations in exon 2, harboured another mutation in one of the RAS genes [11].
As with KRAS mutations (especially codon 12), BRAF mutations have also been linked to a worse patient prognosis in CRC [8, 20]. However, it is important to note that the prognostic impact of the presence of a BRAF mutation is dependent on the microsatellite status: whereas microsatellite-stable BRAF mutated CRC are associated with a worse, microsatellite-instable BRAF mutated tumors are associated with a better prognosis than BRAF wild type CRC [21]. However, the presence of BRAF mutations currently is believed not to be predictive for the response to anti-EGFR therapies [22].
Furthermore, recent studies showed that acetylsalicylic acid and other nonsteroidal anti-inflammatory drugs are associated with reduced disease recurrence and improved outcome in CRC and that these benefits are limited to patients with PIK3CA-mutated cancers [17, 19, 23]. Also other genetic mutations that are frequently altered in CRC such as p53 [24] or PTEN [25] may serve as prognostic or predictive biomarkers. While very promising, all lack clinical significance at present.
When the first anti-EGFR therapies for mCRC entered the arena, institutes of pathology expanded their expertise in molecular techniques in order to be able to evaluate the mutational status of KRAS, NRAS and BRAF.
Accurate mutation assessment depends on several factors such as available tissue, DNA-quality, percentage of tumor cells as well as the specificity and sensitivity of various test systems [26].
When choosing an assay for routine diagnostics, additional factors such as workload, time-to-results, hands-on time, equipment, assay costs, assay flexibility and robustness of the technique applied needed to be addressed [27].
Methods currently available for KRAS, NRAS and BRAF mutational analysis include Sanger sequencing, which is still regarded as the gold standard, and numerous alternatives as allele-specific PCR [28], single nucleotide primer extension assays [27, 29], pyrosequencing [30], real-time PCR [31], high resolution melting curve analysis [30, 32], amplification refractory mutation system (ARMS)-Scorpion assay [33], strip or chip assay combining PCR followed by hybridization to a KRAS or NRAS-specific probe [27, 34], next-generation sequencing (NGS) [34] and matrix assisted laser desorption/ionization mass spectrometry (MALDI MS) [17]. However, past studies with MALDI MS mainly applied genetic panels provided by the manufacturer that cover many mutations that are not recommended to assess. In consequence these panels are not cost-effective for routine use.
Therefore, we developed a MALDI MS test assay for the simultaneous detection of KRAS, NRAS and BRAF mutations in a routine high throughput setting and implemented an optimised workflow. We compared the data generated by this assay to data from standard RAS mutation detection methods, specifically the KRAS 1.4 LCD Array Kit (Chipron, Berlin) and Sanger Sequencing. Cases with discordant mutation results were subjected to NGS analysis to definitely clarify the mutational status.
Methods
Patients
93 tumor samples of patients with CRC were analysed using a custom panel of mutation assays across the KRAS, NRAS and BRAF oncogenes with the Agena Bioscience MALDI MS platform.
Sample preparation and DNA extraction
Hematoxylin and Eosin (H&E)-stained slides from formalin-fixed paraffin-embedded (FFPE) tumor samples were reviewed by a pathologist and the tumor area was selected for analysis. A serial unstained tissue section was manually dissected and subjected to Tissue lysis buffer (Qiagen, Hilden, Germany). Following Proteinase-K digestion and a decrosslink-step, DNA was automatically isolated using a QiaSymphony® device (Qiagen, Hilden, Germany) and subsequently quantified by OD 260 nm.
Workflow
A simple workflow for mutational testing has been employed in routine diagnostics, which is depicted in Fig. 1.

The study workflow. All cases were tested by the KRAS LCD-array Kit and MALDI MS. If a mutation was found by the KRAS LCD-array Kit, a report was signed out. If no mutation was detected by the KRAS LCD-array Kit, that covers exclusively exon 2, Sanger sequencing has been performed for KRAS, NRAS and BRAF in exon 2–4 and a report was signed out accordingly
KRAS LCD-Array kit
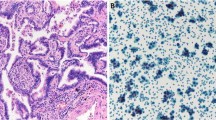
The KRAS LCD-Array kit for detection of KRAS mutations in codon 12 and 13 (Exon 2) is based on the amplification of a short PCR fragment and the subsequent identification of point mutations by amplicon hybridization to immobilized capture probes. Biotin labelling of the generated 170 bp PCR fragment occurs during PCR amplification. Following a short hybridization to wild type and mutation specific capture probes immobilized on the surface of the LCD-Array, bound PCR fragments are visualized using the sensitive streptavidin-enzyme-substrate cascade (Fig. 2).

Examples of KRAS mutations. a A representative example of a chromatogram. A KRAS mutation (c.183A > C; p.Q61H) in exon 3 as detected by Sanger sequencing is depicted (arrow). Each mutation was observed on the forward and the reverse strand. b An example of a chip-hybridization result from one patient. On top 3 type-specific double signals (arrowhead). On the left, the mutation-specific double signal (c35G > T; p.G12V) (arrow)
To detect even small amounts of mutated KRAS sequences within an excess amount of wild type background, amplification is carried out in the presence of the KRAS Wildtype Supressor Compound (K-RAS WSC). This molecule preferentially suppresses wild type-sequence amplification and therefore allows sequence-specific detection of smallest amounts of KRAS mutations in codon 12 and 13.
Sanger sequencing
PCR primers were bought from Metabion (Munich, Germany). PCR amplification products were purified by ethanol precipitation and were bidirectionally sequenced using Big Dye® v3.1 reagents (Thermo Fisher Scientific, Waltham, USA) according to the manufacturer’s protocol. Sequencing products were purified using XTerminatorTM beads (Thermo Fisher Scientific, Waltham, USA) and automated sequencing performed by capillary electrophoresis on an ABI3500 (Applied Biosystems). Sequences were aligned and examined by visual inspection of the electropherogram (Fig. 2).
MALDI MS assay design
Relevant mutations of KRAS, NRAS and BRAF have been identified from the COSMIC database and the respective literature. DNA sequences were extracted from the UCSC Genome Browser. These sequences were subsequently utilized to build our multiplex assay with Assay Design (v.3.0.0) covering KRAS mutations as rare as 0.002 % according to the COSMIC database (Table 1).
MALDI MS mutation detection
After semi-automated DNA-Isolation (QIAsymphony®, Qiagen, Hilden, Germany), DNA content was calculated by NanoDropTM spectrophotometry (Peqlab, Erlangen, Germany). PCR and downstream reactions were performed according to the iPLEX Pro Kit® (Agena Bioscience, Hamburg, Germany) datasheet. In brief, a multiplex PCR-reaction (primers by Metabion, Munich, Germany) was performed at a final volume of 5 μl containing 10–100 ng template DNA leading to amplicon sizes ranging from 88 to 127 bp.
To dephosphorylate unincorporated dNTPs, 2 μL of a Shrimp-Alkaline-Phosphatase (SAP) Mix (iPlex® Pro Kit) was added to each PCR reaction. After incubation steps according to the manufacturer’s protocol, an extension primer reaction was performed to hybridize and elongate the extension primers at the nucleotide position of interest.
Finally, the sample volume was increased by addition of 42 μl ultrapure water and free ions were removed by a resin cleanup step. 10–20 nl of the reaction products were dispensed onto a matrix-precoated 96- well SpectroCHIP® bioarray by application of a nanodispenser (RS1000, Agena Bioscience). MS experiments were conducted on a MassArray® Analyser 4 system according to the manufacturer’s protocol (Agena Bioscience, San Diego). This system is specifically designed for the detection of genetic mutations and not for the detection of other molecules. Results were analysed by MassArray® Workstation software (v.3.3) (Agena Bioscience) (Fig. 3).

Representative examples of MALDI MS results. On the left, 16 cases plus wild type controle and no-template control tested for a KRAS mutation (p.Q61H). 15 cases are KRAS wild-type (including the wild-type control); one of the cases is highlighted with an arrow (a). In this specific case the sequencing primer (first arrow) is completely elongated by an Adenin indicated by a high peak in the mass range of the wild-type allele (second arrow) (b). One case shows a mutation in KRAS (c.183A > C, p.Q61H) (c). The reverse sequence-specific primer is completely consumed and elongated by either Thymine or Guanine (wild-type and mutated allele; arrows) (d)
The general principle is based on amplification of the DNA by PCR, resulting in copies of both mutant and wildtype alleles. Primer extension performed using terminator nucleotides A, C, T, G, each with distinct masses leads to different masses of the amplicons depending on the mutational status that can subsequently be detected by mass spectrometry. If there is a mutant allele, three mass peaks may be seen: the unextended primer peak, the amplified wild-type allele peak and the amplified mutant allele peak (Fig. 3). The ratio of the areas under the curve of the wild-type allele and the mutant allele peaks are a quantitative measure of the percentage of mutant alleles.
Next-generation sequencing (IonTorrent™)
For library preparation, the multiplex PCR-based Ion Torrent AmpliSeq™ technology (Thermo Fisher Scientific, Waltham, USA) was used. Amplicon library preparation was performed with the Ion AmpliSeq Library Kit v2.0 using 10 ng of DNA. Briefly, the DNA was mixed with the primer pool, containing all primers for generating the 180 amplicons and the Ampliseq HiFi Master Mix and transferred to a PCR cycler (BioRad, Munich, Germany). After the end of the PCR reaction, primer end sequences were partially digested using FuPa reagent, followed by the ligation of barcoded sequencing adapters (Ion Xpress Barcode Adapters 1–16, Thermo Fisher Scientific, Waltham, USA). The final library was purified using AMPure® XP magnetic beads (Beckman Coulter, Krefeld, Germany) and quantified using qPCR (Ion Library Quantitation Kit, Thermo Fisher Scientific, Waltham, USA) on a StepOne® qPCR machine (Thermo Fisher Scientific, Waltham, USA). The individual libraries were diluted to a final concentration of 100pM and eight to ten libraries were pooled and processed to library amplification on Ion Spheres using Ion PGM™ template OT2 200Kit. Unenriched libraries were quality-controlled using Ion Sphere quality control measurement on a QuBit® instrument. After library enrichment (Ion OneTouch® ES), the library was processed for sequencing using the Ion Torrent 200 bp sequencing v2 chemistry and loaded onto a chip. Data analyses were performed using the Ion Torrent Suite Software (v.4.2) as described previously. We use a custom designed colon cancer panel that includes KRAS and NRAS mutations in exon 2, 3 and 4.
Results
KRAS and NRAS
A total of 93 samples from patients with CRC were analysed by MALDI MS and the KRAS LCD-array Kit. Since the latter kit only detects KRAS mutations in exon 2, all cases with KRAS wild-type status were further analysed by Sanger sequencing of exons 3 and 4 of KRAS and exons 2,3 and 4 of NRAS (Fig. 1).
MALDI MS detected a KRAS mutation in 46/93 (49 %) tissue probes, from which 38/93 (41 %) were in exon 2, 3/93 (3 %) were in exon 3 and 5/93 (5 %) were in exon 4.
37/93 (40 %) of the KRAS mutations were already confirmed by the KRAS LCD-array kit, while additional 9/93 (10 %) of KRAS mutations were confirmed by subsequent Sanger sequencing.
A mutation in the NRAS gene was identified in 2/93 (2 %) of cases. MALDI MS and Sanger sequencing were in agreement in 2/2 (100 %) cases.
In total, MALDI MS results were in agreement to results obtained by the combination of the other two methods in 92/93 (99 %) cases.
RAS-wild type tumors
Wild type tumors were analysed by three methods. Sanger sequencing identified 44/93 (47 %), the KRAS LCD-array kit 56/93 (60 %) and MALDI MS 44/93 (47 %) of patients with wild type tumors.
One disconcordant case
In 1/93 (1 %) case a G12V mutation has been shown by MALDI MS and Sanger sequencing, but not by the KRAS LCD-array kit. In this single case, NGS had been performed and confirmed the presence of a G12V mutation with an allele frequency of 15 %. Thus, MALDI MS results were in agreement to results obtained by the combination of three methods in 93/93 (100 %) of the cases.
BRAF
BRAF was mutated in 1/93 (1 %) case respectively. Again, MALDI MS and Sanger sequencing results were in perfect agreement (100 %).
All mutations found occurred exclusively. The results are summarized (Table 2).
Hands-on time and turnaround time
Overall hands-on time was shortest for the KRAS LCD-array Kit and MALDI MS (45 min), intermediate for NGS (70 min) and longest for Sanger sequencing (120 min).
The overall time-to-results was shortest for the KRAS LCD-array Kit (220 min), followed by MALDI MS (430 min), Sanger sequencing (715 min) and NGS (1020 min).
Approximated times to perform each step of the different methods namely KRAS LCD-array Kit, Sanger sequencing, MALDI MS and NGS are summarized (Table 3).
Discussion
According to recent clinical guidelines it is mandatory to evaluate the mutational status of oncogenes that act downstream of EGFR specifically KRAS and NRAS in mCRC in order to select a patient population that is most likely to benefit from anti-EGFR therapy [5, 8, 14, 25]. Additionally, genes such as BRAF that provide prognostic information may be included in the testing. Besides classical Sanger sequencing, new emerging techniques for the detection of genetic mutations are available. Among them MALDI MS has prompted particular interest among scientists and pathologists, since it combines high sensitivity and specificity with low cost per test, fast turnaround time and easy sample handling [17].
Sensitivity and specificity
Sanger sequencing is generally considered to be the gold standard for the detection of mutations in KRAS, NRAS and BRAF. Specificity is generally high with all methods applied for the detection of genetic mutations [30, 32, 34–36]. However, sensitivity has been reported to differ [34, 37].
Whereas direct sequencing has been reproducibly shown to have a detection limit of >10 % mutant alleles, high resolution melting analysis has a lower detection limit of 10 % [38] that is similar to SnP shot assays (10 %) [27] and can further be improved by the cobasR test (5 %) [39], the TheraScreen® test (1 %) [33, 38, 40–42] or Strip assays (1 %) [27, 38]. Also, NGS [34] and MALDI MS have similarly low detection limits of 1–5 % mutant allels [28, 33, 34, 38–44]. Altimari et al. [34] state that NGS was superior in terms of sensitivity and specificity compared to other techniques in detecting KRAS mutations in FFPE material, but MALDI MS was not included in their analysis.
To improve both, sensitivity and specificity, in direct sequencing, especially in specimen with low amount of DNA, it was recommended to increase the number of PCR cycles. Also to avoid false –positive errors, duplication of the test was expected to be effective [33]. Amplifiable DNA amounts are often limited when FFPE samples are used as a source, since DNA is highly fragmented by formalin treatment [33].
It has also been shown that various methods tested in different laboratories showed a decreasing correct mutational allele frequency proportionally with decreasing percentage of tumor cell content [26].
Therefore macro- or micro-dissection of the tumor area is usually done, which improves the test results and is therefore strongly recommended [45]. Tumor cell enrichment correlated significantly with the abundance of KRAS-mutated DNA [34].
Discordant results of different methods were attributed to tumor heterogeneity, contamination of the tumor sample with normal tissue, analytic factors affecting assay sensitivity and lack of experience with the respective method [19, 34, 35].
However, despite efforts to improve Sanger sequencing it has been shown that the sensitivity the specificity of mass spectrometrical methods exceeds that of traditional Sanger sequencing and is highly concordant with pyrosequencing, allelee-specific PCR [17, 46–48] and NGS [49]. In our study DNA quality was sufficient in all cases and none of the MALDI MS, NGS or Sanger sequencing reactions had to be repeated.
Cost
When estimating the cost of a test, three parameters have to be considered, cost of instrumentation, consumable cost per test and hands-on-time. In the literature relatively few statements about costs of the different methods could be obtained.
Sarasqueta et al. reported low costs per test for direct sequencing and SNap shot® compared to a Strip assay [27]. But the advantage of strip or chip hybridisation after PCR is low cost for technical equipment [45].
High performance of the PNA clamp PCR assay and low cost compared to TheraScreen® test assay was reported by Norgard et al. [43]. The reagent costs for pyrosequencing and Sanger sequencing were comparable but higher than that of melting curve analysis in one study [30].
Likewise, costs per sample in Sanger sequencing is higher compared to MALDI MS, especially when complex testing of numerous mutations was performed [17]. In a previous study, we reported equipment cost to be highest with Sanger and pyrosequencing, followed by real-time- and array-based systems. Costs per sample were lowest for Sanger and pyrosequencing, two-fold higher for the array and three-fold higher for high resolution melting curve analysis [45].
NGS and MALDI MS have comparable costs for the technical equipment, but costs per test are much lower in MALDI MS especially when a customized assay for mutation detection is build. It is important to mention, that local prices for equipment and tests may vary considerably.
Hands-on-time is an important cost factor and has been reported to be around 2 h for Sanger sequencing, SNap shot® assays, Therascreen, high resolution melting curve analysis, NGS and MALDI MS [27, 38, 50]. The StripAssays may be conducted within 1,5 h [27]. Melting curve analysis has the shortest hands on time compared to the other mentioned methods [30, 45]. In our study hands-on time was around 45 min for performing the KRAS LCD-Kit and MALDI MS, 70 min for NGS and around 2 h for Sanger sequencing.
In the authors opinion MALDI MS is the most cost effective method to detect clinically relevant mutations in CRC. Since it is an open platform more mutational hotspots for testing may be easily added. However, this holds only true for high throughput laboratories, since the equipment costs are rather high compared to array- or real-time PCR based methods or high resolution melting curve analysis. For laboratories with low throughput, techniques with low equipment costs and high costs per sample may be more cost effective on the short and also on the long term. Therefore definite conclusions regarding cost-effectiveness cannot easily be generalized.
Turnaround time
Besides hands-on-time turnaround time/time-to-result is important and has been reported to be 2 working days for Sanger sequencing, 1.5 working days for SNaP shot and pyrosequencing and 1 working day for Strip- and chip assays [27, 38, 45, 51] which is in line with our results. With respect to turnaround time high resolution melting curve analysis has been reported to outperform the former mentioned methods [45]. We perform MALDI MS mutational testing within 1.5 working days. In our opinion a time-to-result of around 2 working days seems reasonable, but if the time line is critical one might choose high resolution melting curve analysis as the preferred method. However, as previously mentioned this method harbours some disadvantages as false-positive results may occur more frequently and costs per test are high compared to the other methods [38, 45].
KRAS and NRAS
We detected KRAS mutations by MALDI MS in 49 % of all samples analysed which is in perfect accordance with other reports [17, 41, 45]. In one case MALDI MS and Sanger sequencing detected a mutation but the KRAS LCD-array kit failed to do so. At first, it was unclear whether the detection of the G12V mutation could be attributed to lower detection limits of MALDI MS compared to the chip-assay or if this case represented a false-positive result. Since it has been proven in the past that MALDI MS and NGS yield similar results [49], a NGS experiment has been performed. Indeed, the very same mutation could be confirmed at an allelic frequency of approximately 15 % by NGS. Taken all three alternative methods together, MALDI MS showed concordant results in 100 % of the cases. This proves the perfect reliability of MALDI MS mutational testing. It is recommended by several authors to test for all codons of KRAS. We included mutations in our panel that occurred in frequencies as low as 0.002 % of cases in CRC according to the COSMIC database because in our opinion this represents a reasonable trade-off between sensitivity and wise handling of resources. NRAS mutations were detected in 3 % of our cases, again well in accordance with other reports [8, 10, 11]. Concordance of NRAS testing was 100 % between the three methods applied. As all KRAS and NRAS mutations have been shown to have lower response rates to anti-EGFR therapy compared to RAS wild-types [10], mutational testing for both should be standard of care for all mCRC.
BRAF
Besides mutations of KRAS and NRAS, the BRAF gene plays a critical role in CRC. It has been shown that BRAF mutations are frequent in sporadic CRC with MSI (32–74 %) and in serrated polyps (up to 90 % in sessile serrated adenomas [52]). The improved classification of serrated lesions by BRAF mutation testing may bet the key to identify lesions with a higher potential to progression into BRAF V600E mutated CRC [53].
This subset of tumors is characterized by right-sided location in the colon, prevalence of mucin, high levels of promoter methylation of CpG islands (CIMP) and a good prognosis compared to its BRAF wild type counterparts with 5-year survival rates over 70 % [17, 54, 55]. In contrast a small subset of BRAF mutated CRC harbour MSS (4 %) and have a significantly worse prognosis with 5-year survival rates of only 16,7 % [21]. Testing for a BRAF mutation alone is therefore not sufficient but adds significant prognostic information in combination with MSI/MSS testing. Of note is that hereditary non-poliposis colorectal carcinomas (HNPCC) generally do not exhibit BRAF mutations, therefore it might be tested to exclude such a hereditary form of CRC.
Thus we added testing for BRAF mutations to our customized panel in order to be able to provide improved prognostic information in conjunction with microsatellite testing.
In our cohort one case with BRAF mutation has been found by MALDI-MS and Sanger Sequencing. Our incidence of only 1 % BRAF mutations is probably due to the rather low sample size considering that BRAF mutations have been reported in the literature at a frequency of 5–20 % of all CRC.
MALDI MS
The MALDI MS technology has the advantage that tests with adequate quality standards may be designed and that it is an open platform which allows fast inclusion of complex mutations of various gens that may be important in the future. Although the clinical significance of mutations with low allelic frequency in relation to prognosis and therapeutic benefit has yet not fully understood, MALDI MS is very specific, significantly more sensitive than Sanger sequencing and reaches detection limits comparable to other modern technologies such as NGS. In addition, for laboratories with a high throughput it combines the advantage of low hands-on time, fast turnaround time and cost effectiveness.
Conclusion
Taken together evaluation of KRAS and NRAS mutational status for therapeutic requirements and BRAF mutational analysis by MALDI MS for prognostic and classification purposes is clearly an attractive approach for routine diagnostics.
Abbreviations
- ARMS:
-
Amplification refractory mutation system
- ASCO:
-
American Society of Clinical Oncology
- Bp:
-
Basepair
- BRAF:
-
V-Raf murine sarcoma viral oncogene homolog B
- CRC:
-
Colorectal cancer
- COSMIC:
-
Catalogue Of Somatic Mutations In Cancer
- mCRC:
-
Metastasized Colorectal Cancer
- DNA:
-
Deoxyribonucleic acid
- EGFR:
-
Epidermal growth factor receptor
- ESMO:
-
European Society for Medical Oncology
- H&E:
-
Hematoxylin and Eosin
- HNPCC:
-
Hereditary non-poliposis colorectal carcinomas
- KRAS:
-
V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- MALDI MS:
-
Matrix assisted laser desorption/ionization mass spectrometry
- MSI:
-
Microsatellite instable
- MSS:
-
Microsatellite stable
- NGS:
-
Next-Generation Sequencing
- NRAS:
-
Neuroblastoma RAS viral oncogene homolog
- NTP:
-
Nucleoside triphosphate
- PCR:
-
Polymerase chain reaction
- PIK3CA:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit
- PTEN:
-
Phosphatase and tensin homolog
- RAF:
-
Rapidly Accelerated Fibrosarcoma (protein family)
- RAS:
-
Rat sarcoma (protein family)
- SAP:
-
Shrimp-Alkaline-Phosphatase
- UCSC:
-
University of California, Santa Cruz
- WSC:
-
Wildtype Supressor Compound
References
Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray, F. GLOBOCAN 2012 v1.1, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11, Lyon. 2014. http://globocan.iarc.fr. Accessed: 20 Apr 2015.
Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. The Lancet Oncology. 2005;6(5):322–7. doi:10.1016/S1470-2045(05)70168-6.
Tougeron D, Lecomte T, Pages JC, Villalva C, Collin C, Ferru A, et al. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Annals of Oncology : official Journal of the European Society for Medical Oncology / ESMO. 2013;24(5):1267–73. doi:10.1093/annonc/mds620.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. Journal of Clinical oncology: Official Journal of the American Society of Clinical Oncology. 2008;26(10):1626–34. doi:10.1200/JCO.2007.14.7116.
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. The New England Journal of Medicine. 2008;359(17):1757–65. doi:10.1056/NEJMoa0804385.
Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. The New England Journal of Medicine. 2009;360(14):1408–17. doi:10.1056/NEJMoa0805019.
Yuan ZX, Wang XY, Qin QY, Chen DF, Zhong QH, Wang L, et al. The prognostic role of BRAF mutation in metastatic colorectal cancer receiving anti-EGFR monoclonal antibodies: a meta-analysis. PloS One. 2013;8(6):e65995. doi:10.1371/journal.pone.0065995.
Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. The New England Journal of Medicine. 2009;361(1):98–9. doi:10.1056/NEJMc0904160.
Lin CC, Lin JK, Lin TC, Chen WS, Yang SH, Wang HS, et al. The prognostic role of microsatellite instability, codon-specific KRAS, and BRAF mutations in colon cancer. Journal of Surgical Oncology. 2014;110(4):451–7. doi:10.1002/jso.23675.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. The Lancet Oncology. 2010;11(8):753–62. doi:10.1016/S1470-2045(10)70130-3.
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. The New England Journal of Medicine. 2013;369(11):1023–34. doi:10.1056/NEJMoa1305275.
Banck MS, Grothey A. Biomarkers of Resistance to Epidermal Growth Factor Receptor Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clinical Cancer Research: an Official Journal of the American Association for Cancer Research. 2009;15(24):7492–501. doi:10.1158/1078-0432.CCR-09-0188.
Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Research. 2006;66(8):3992–5. doi:10.1158/0008-5472.CAN-06-0191.
Lievre A, Laurent-Puig P. Genetics: Predictive value of KRAS mutations in chemoresistant CRC. Nature Reviews Clinical Oncology. 2009;6(6):306–7. doi:10.1038/nrclinonc.2009.69.
Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. Journal of Clinical Oncology: official Journal of the American Society of Clinical Oncology. 2009;27(12):2091–6. doi:10.1200/JCO.2009.21.9170.
Van Cutsem E, Oliveira J, Group EGW. Advanced colorectal cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Annals of Oncology: official Journal of the European Society for Medical Oncology/ESMO. 2009;20 Suppl 4:61–3. doi:10.1093/annonc/mdp130.
Fumagalli D, Gavin PG, Taniyama Y, Kim SI, Choi HJ, Paik S, et al. A rapid, sensitive, reproducible and cost-effective method for mutation profiling of colon cancer and metastatic lymph nodes. BMC Cancer. 2010;10:101. doi:10.1186/1471-2407-10-101.
Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. British Journal of Cancer. 2009;101(4):715–21. doi:10.1038/sj.bjc.6605177.
Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE. Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clinical Cancer Research: an official Journal of the American Association for Cancer Research. 2010;16(3):790–9. doi:10.1158/1078-0432.CCR-09-2446.
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. Journal of Clinical Oncology: official Journal of the American Society of Clinical Oncology. 2008;26(35):5705–12. doi:10.1200/JCO.2008.18.0786.
Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Research. 2005;65(14):6063–9. doi:10.1158/0008-5472.CAN-05-0404.
Ogino S, Shima K, Meyerhardt JA, McCleary NJ, Ng K, Hollis D, et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: results from intergroup trial CALGB 89803. Clinical Cancer Research: an official Journal of the American Association for Cancer Research. 2012;18(3):890–900. doi:10.1158/1078-0432.CCR-11-2246.
Domingo E, Church DN, Sieber O, Ramamoorthy R, Yanagisawa Y, Johnstone E, et al. Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. Journal of Clinical Oncology: official journal of the American Society of Clinical Oncology. 2013;31(34):4297–305. doi:10.1200/JCO.2013.50.0322.
Oden-Gangloff A, Di Fiore F, Bibeau F, Lamy A, Bougeard G, Charbonnier F, et al. TP53 mutations predict disease control in metastatic colorectal cancer treated with cetuximab-based chemotherapy. British Journal of Cancer. 2009;100(8):1330–5. doi:10.1038/sj.bjc.6605008.
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. Journal of Clinical Oncology: official Journal of the American Society of Clinical Oncology. 2009;27(35):5924–30. doi:10.1200/JCO.2008.21.6796.
Dijkstra JR, Heideman DA, Meijer GA, Boers JE, t Hart NA, Diebold J, et al. KRAS mutation analysis on low percentage of colon cancer cells: the importance of quality assurance. Virchows Archiv: an International Journal of Pathology. 2013;462(1):39–46. doi:10.1007/s00428-012-1356-2.
Farina Sarasqueta A, Moerland E, de Bruyne H, de Graaf H, Vrancken T, van Lijnschoten G, et al. SNaPshot and StripAssay as valuable alternatives to direct sequencing for KRAS mutation detection in colon cancer routine diagnostics. The Journal of Molecular Diagnostics: JMD. 2011;13(2):199–205. doi:10.1016/j.jmoldx.2010.10.006.
Cushman-Vokoun AM, Stover DG, Zhao Z, Koehler EA, Berlin JD, Vnencak-Jones CL. Clinical utility of KRAS and BRAF mutations in a cohort of patients with colorectal neoplasms submitted for microsatellite instability testing. Clinical Colorectal Cancer. 2013;12(3):168–78. doi:10.1016/j.clcc.2013.04.005.
Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. British Journal of Cancer. 2007;96(8):1166–9. doi:10.1038/sj.bjc.6603685.
Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, et al. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. The Journal of Molecular Diagnostics: JMD. 2010;12(4):425–32. doi:10.2353/jmoldx.2010.090188.
Tol J, Dijkstra JR, Vink-Borger ME, Nagtegaal ID, Punt CJ, Van Krieken JH, et al. High sensitivity of both sequencing and real-time PCR analysis of KRAS mutations in colorectal cancer tissue. Journal of Cellular and Molecular Medicine. 2010;14(8):2122–31. doi:10.1111/j.1582-4934.2009.00788.x.
Ma ES, Wong CL, Law FB, Chan WK, Siu D. Detection of KRAS mutations in colorectal cancer by high-resolution melting analysis. Journal of Clinical Pathology. 2009;62(10):886–91. doi:10.1136/jcp.2008.063677.
Bando H, Tsuchihara K, Yoshino T, Kojima M, Ogasawara N, Fukushima H, et al. Biased discordance of KRAS mutation detection in archived colorectal cancer specimens between the ARMS-Scorpion method and direct sequencing. Japanese Journal of Clinical Oncology. 2011;41(2):239–44. doi:10.1093/jjco/hyq216.
Altimari A, de Biase D, De Maglio G, Gruppioni E, Capizzi E, Degiovanni A, et al. 454 next generation-sequencing outperforms allele-specific PCR, Sanger sequencing, and pyrosequencing for routine KRAS mutation analysis of formalin-fixed, paraffin-embedded samples. OncoTargets and therapy. 2013;6:1057–64. doi:10.2147/OTT.S42369.
Feigelson HS, Goddard KA, Johnson MA, Funk KC, Rahm AK, Kauffman TL, et al. Reliability of KRAS mutation testing in metastatic colorectal cancer patients across five laboratories. BMC Research Notes. 2012;5:196. doi:10.1186/1756-0500-5-196.
Ogino S, Kawasaki T, Brahmandam M, Yan L, Cantor M, Namgyal C, et al. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. The Journal of Molecular Diagnostics: JMD. 2005;7(3):413–21. doi:10.1016/S1525-1578(10)60571-5.
Harle A, Busser B, Rouyer M, Harter V, Genin P, Leroux A, et al. Comparison of COBAS 4800 KRAS, TaqMan PCR and high resolution melting PCR assays for the detection of KRAS somatic mutations in formalin-fixed paraffin embedded colorectal carcinomas. Virchows Archiv: an International Journal of Pathology. 2013;462(3):329–35. doi:10.1007/s00428-013-1380-x.
Jancik S, Drabek J, Berkovcova J, Xu YZ, Stankova M, Klein J, et al. A comparison of Direct sequencing, Pyrosequencing, High resolution melting analysis, TheraScreen DxS, and the K-ras StripAssay for detecting KRAS mutations in non small cell lung carcinomas. Journal of Experimental & Clinical Cancer Research: CR. 2012;31:79. doi:10.1186/1756-9966-31-79.
Lee S, Brophy VH, Cao J, Velez M, Hoeppner C, Soviero S, et al. Analytical performance of a PCR assay for the detection of KRAS mutations (codons 12/13 and 61) in formalin-fixed paraffin-embedded tissue samples of colorectal carcinoma. Virchows Archiv: an International Journal of Pathology. 2012;460(2):141–9. doi:10.1007/s00428-011-1180-0.
Kotoula V, Charalambous E, Biesmans B, Malousi A, Vrettou E, Fountzilas G, et al. Targeted KRAS mutation assessment on patient tumor histologic material in real time diagnostics. PloS One. 2009;4(11):e7746. doi:10.1371/journal.pone.0007746.
Franklin WA, Haney J, Sugita M, Bemis L, Jimeno A, Messersmith WA. KRAS mutation: comparison of testing methods and tissue sampling techniques in colon cancer. The Journal of Molecular Diagnostics: JMD. 2010;12(1):43–50. doi:10.2353/jmoldx.2010.080131.
Angulo B, Garcia-Garcia E, Martinez R, Suarez-Gauthier A, Conde E, Hidalgo M, et al. A commercial real-time PCR kit provides greater sensitivity than direct sequencing to detect KRAS mutations: a morphology-based approach in colorectal carcinoma. The Journal of Molecular Diagnostics: JMD. 2010;12(3):292–9. doi:10.2353/jmoldx.2010.090139.
Nordgard O, Oltedal S, Janssen EA, Gilje B, Korner H, Tjensvoll K, et al. Comparison of a PNA clamp PCR and an ARMS/Scorpion PCR assay for the detection of K-ras mutations. Diagnostic Molecular Pathology: the American Journal of Surgical Pathology, Part B. 2012;21(1):9–13. doi:10.1097/PDM.0b013e31821e59dc.
Pinto P, Rocha P, Veiga I, Guedes J, Pinheiro M, Peixoto A, et al. Comparison of methodologies for KRAS mutation detection in metastatic colorectal cancer. Cancer Genetics. 2011;204(8):439–46. doi:10.1016/j.cancergen.2011.07.003.
Weichert W, Schewe C, Lehmann A, Sers C, Denkert C, Budczies J, et al. KRAS genotyping of paraffin-embedded colorectal cancer tissue in routine diagnostics: comparison of methods and impact of histology. The Journal of Molecular Diagnostics: JMD. 2010;12(1):35–42. doi:10.2353/jmoldx.2010.090079.
Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, et al. High-throughput oncogene mutation profiling in human cancer. Nature Genetics. 2007;39(3):347–51. doi:10.1038/ng1975.
Vivante A, Amariglio N, Koren-Michowitz M, Ashur-Fabian O, Nagler A, Rechavi G, et al. High-throughput, sensitive and quantitative assay for the detection of BCR-ABL kinase domain mutations. Leukemia. 2007;21(6):1318–21. doi:10.1038/sj.leu.2404635.
van Puijenbroek M, Dierssen JW, Stanssens P, van Eijk R, Cleton-Jansen AM, van Wezel T, et al. Mass spectrometry-based loss of heterozygosity analysis of single-nucleotide polymorphism loci in paraffin embedded tumors using the MassEXTEND assay: single-nucleotide polymorphism loss of heterozygosity analysis of the protein tyrosine phosphatase receptor type J in familial colorectal cancer. The Journal of Molecular Diagnostics: JMD. 2005;7(5):623–30. doi:10.1016/S1525-1578(10)60596-X.
Portier BP, Kanagal-Shamanna R, Luthra R, Singh R, Routbort MJ, Handal B, et al. Quantitative assessment of mutant allele burden in solid tumors by semiconductor-based next-generation sequencing. American Journal of Clinical Pathology. 2014;141(4):559–72. doi:10.1309/AJCP1JUGQMW7ZNTL.
Al-Ahmadie HA, Iyer G, Janakiraman M, Lin O, Heguy A, Tickoo SK, et al. Somatic mutation of fibroblast growth factor receptor-3 (FGFR3) defines a distinct morphological subtype of high-grade urothelial carcinoma. The Journal of Pathology. 2011;224(2):270–9. doi:10.1002/path.2892.
Querings S, Altmuller J, Ansen S, Zander T, Seidel D, Gabler F, et al. Benchmarking of mutation diagnostics in clinical lung cancer specimens. PloS One. 2011;6(5):e19601. doi:10.1371/journal.pone.0019601.
Rosenberg DW, Yang S, Pleau DC, Greenspan EJ, Stevens RG, Rajan TV, et al. Mutations in BRAF and KRAS differentially distinguish serrated versus non-serrated hyperplastic aberrant crypt foci in humans. Cancer Research. 2007;67(8):3551–4. doi:10.1158/0008-5472.CAN-07-0343.
Mesteri I, Bayer G, Meyer J, Capper D, Schoppmann SF, von Deimling A, et al. Improved molecular classification of serrated lesions of the colon by immunohistochemical detection of BRAF V600E. Modern Pathology: an official Journal of the United States and Canadian Academy of Pathology, Inc. 2014;27(1):135–44. doi:10.1038/modpathol.2013.126.
Tanaka H, Deng G, Matsuzaki K, Kakar S, Kim GE, Miura S, et al. BRAF mutation, CpG island methylator phenotype and microsatellite instability occur more frequently and concordantly in mucinous than non-mucinous colorectal cancer. International Journal of Cancer Journal International du cancer. 2006;118(11):2765–71. doi:10.1002/ijc.21701.
Li WQ, Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Iacopetta B. BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Molecular Cancer. 2006;5:2. doi:10.1186/1476-4598-5-2.
Acknowledgements
We thank Dr. Nicole Pfarr and Katrin Steinhausser for excellent technical assistance. We also acknowledge the financial support of the Deutsche Forschungsgemeinschaft and Ruprecht-Karls-Universität Heidelberg within the funding programme Open Access Publishing.
Financial disclosure
No extramural funding has been received.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Dr. MK drafted the manuscript and was involved in interpretation of the data. Dr. NA and Dr. VE have acquired the data and revised the manuscript for important intellectual content. Prof. WW was involved in the interpretation of the data and in the revision of the manuscript. Prof. JK was responsible for the conception of the study and revised the manuscript for important intellectual content. All authors agreed to the final version of the manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kriegsmann, M., Arens, N., Endris, V. et al. Detection of KRAS, NRAS and BRAF by mass spectrometry - a sensitive, reliable, fast and cost-effective technique. Diagn Pathol 10, 132 (2015). https://doi.org/10.1186/s13000-015-0364-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-015-0364-3