
Abstract
Ambient and indoor air pollution contributes annually to approximately seven million premature deaths. Air pollution is a complex mixture of gaseous and particulate materials. In particular, fine particulate matter (PM2.5) plays a major mortality risk factor particularly on cardiovascular diseases through mechanisms of atherosclerosis, thrombosis and inflammation. A review on the PM2.5-induced atherosclerosis is needed to better understand the involved mechanisms. In this review, we summarized epidemiology and animal studies of PM2.5-induced atherosclerosis. Vascular endothelial injury is a critical early predictor of atherosclerosis. The evidence of mechanisms of PM2.5-induced atherosclerosis supports effects on vascular function. Thus, we summarized the main mechanisms of PM2.5-triggered vascular endothelial injury, which mainly involved three aspects, including vascular endothelial permeability, vasomotor function and vascular reparative capacity. Then we reviewed the relationship between PM2.5-induced endothelial injury and atherosclerosis. PM2.5-induced endothelial injury associated with inflammation, pro-coagulation and lipid deposition. Although the evidence of PM2.5-induced atherosclerosis is undergoing continual refinement, the mechanisms of PM2.5-triggered atherosclerosis are still limited, especially indoor PM2.5. Subsequent efforts of researchers are needed to improve the understanding of PM2.5 and atherosclerosis. Preventing or avoiding PM2.5-induced endothelial damage may greatly reduce the occurrence and development of atherosclerosis.
Similar content being viewed by others

Background
The World Health Organization (WHO) reported that approximately 91% of people worldwide live in unhealthy environments where air quality levels exceed WHO limits. The combined effects of indoor and ambient air pollution result in approximately 7 million premature deaths from noncommunicable diseases every year [1]. Chemicals in the air initiate or potentiate a wide range of noncommunicable diseases [2]. Fine particulate matter (PM2.5, the aerodynamic diameter ≤ 2.5 μm) in air pollution became the fifth death risk factor in 2015 [3]. PM2.5 is a complex mixture, and its major source is combustion, such as traffic-related diesel exhaust particles (DEPs), industry, indoor cooking activities, and bushfires [4]. For example, the Australian bushfires in 2019-2020 had extreme impacts on air quality throughout the region and even the globally [5]. Thus, the global burden of cardiovascular disease caused by PM2.5 may be much greater than that previously reported by WHO. Evidence has indicated that PM2.5 induces lipid metabolism dysregulation and increases hypertension and the prevalence of cardiac arrhythmias, thus accelerating the progression of atherosclerosis, and increasing the risk of cardiovascular disease- and stroke-related mortality [6,7,8,9].
Atherosclerosis is a chronic inflammatory disease of large and medium-sized arteries. The causes of atherosclerosis are inflammation, hemodynamic damage and abnormal lipid metabolism in early-stage atherosclerosis [10]. When endothelial cells are activated, they express inflammatory cytokines (such as interleukin (IL)-8, monocyte chemoattractant protein (MCP)-1) and adhesion molecules (such as intercellular adhesion molecule (ICAM-1) and vascular cell adhesion molecule (VCAM-1)), attracting blood monocytes that bind to the activated endothelial monolayer and infiltrate the arterial wall. Important biomarkers of the development of atherosclerotic inflammation include C-reactive protein (CRP), IL-6, adhesion molecules, and matrix metalloproteinases (MMPs) [10]. These factors induce macrophage polarization into the pro-inflammatory M1-like or anti-inflammatory M2-like phenotype [11]. The scavenger receptors of macrophages, such as low-density lipoprotein receptor-related protein 1 (LRP1), cluster of differentiation 36 (CD36) and class B type 1 (SR-B1), play a key role in lipid uptake, deposition and the development of atherosclerotic plaques [12,13,14]. Liver X receptor α (LXR-α)/ATP-binding cassette transporter A1 (ABCA1)/ABCG1-dependent cholesterol efflux is a crucial event in the suppression of lipid accumulation during the transformation of macrophage foam cells [15]. Vascular smooth muscle cells (VSMCs) migrate from the media to the intima, synthesize extracellular matrix macromolecules such as elastin, proteoglycans and collagen, and form fibrous caps formation. The death of foam cells and VSMCs leads to the release of extracellular lipids in atherosclerotic lesions, leading to the formation of a necrotic core [11, 16]. MMPs (such as MMP-9) are highly expressed in atherosclerotic plaques, leading to substantial enhancement of elastin degradation and inducing plaque rupture [17]. Currently, several imaging techniques can be used to investigate plaques and signs of vulnerability, such as CT, magnetic resonance imaging (MRI) and ultrasound [18]. Molecular imaging is an innovative technique for the detection of plaque inflammation. The utility of several nanoparticles, such as sodium fluoride, iron oxide and polyethylene glycol molecules, for the molecular imaging of atherosclerosis in animal models and patients has been investigated [19,20,21]. In the past few decades, treatment strategies for atherosclerosis have mainly focused on lowering lipid levels with high-intensity statins. However, only approximately 25% of patients who receive high-intensity statins as a lipid-lowering therapy achieve the recommended level of low-density lipoprotein cholesterol (LDL-C, ≤ 1.8 mmol/L) [22]. Approximately 75% of patients do not respond to statin therapy sufficiently; therefore, novel therapeutic strategies are needed.
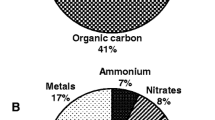
PM2.5 is a complex mixture, and a review had comprehensively summarized the chemical composition and characteristics of PM2.5, including inorganic elements, water-soluble ions, carbonaceous aerosols and organic compounds (polycyclic aromatic hydrocarbons (PAHs) and volatile organic compounds (VOCs) etc) [4].. A study showed that coal combustion and vehicular emissions are the main sources of PAHs and VOCs in PM2.5 [23, 24]. Evidence has demonstrated that DEPs accelerate the development or exacerbation of atherosclerosis [25, 26]. Organic chemicals from DEPs, such as PAHs adhere to the carbon cores of particles, and certain PAHs can trigger Ca2+ signaling and increase inflammation in endothelial cells [27,28,29,30]. Evidence has shown that the levels of urinary PAH biomarkers are associated with cardiovascular disease [31]. Due to the antagonistic and synergic effects of complex VOC mixtures, the toxic effects of VOCs are difficult to estimate [32]. In addition, the surface of particles may bind reactive copollutants, including biomolecules (such as endotoxins), redox-active transition metals, and reactive quinones/aldehydes, which may be carried by particles and enter lung tissue and the circulation, inducing secondary toxicity [33, 34]. The ions and metal components of PM2.5, including SO42−, K+, Cl−, K, Si, As, Zn, Se and Pb, could be mainly responsible for systemic inflammation [35]. Evidence has shown that the binding of endotoxins to the surface of PM2.5 particles plays a critical role in the inflammatory response. Endotoxin neutralizer (polymyxin B) and knockout of toll-like receptor 4 (TLR4) strongly inhibit the PM2.5-triggered inflammatory response [36]. However, the understanding of the major toxic effects exerted by the specific components of PM2.5 is limited. Further investigating the toxicity of PM2.5 components will contribute to a comprehensive understanding of PM2.5, which may be a key area of future research.
Endothelial cells cover the internal surface of blood vessels, and the integral endothelial cell layer maintains a complex functional balance to inhibit the inflammatory response or thrombosis. Ambient PM2.5 exposure elicits the deterioration of endothelial function, systemic inflammation and coagulation [37, 38]. Evidence has shown that a 10 μg/m3 increase in the PM2.5 concentration at a 1-day lag was associated with increased brachial-ankle pulse wave velocity (baPWV, a physiological indicator of arterial stiffness), but not with high-sensitivity C-reactive protein (hsCRP, a biomarker of vascular inflammation) levels; thus, arterial stiffness might be more sensitive to ambient PM2.5 exposure than inflammation [39]. Accordingly, indoor PM2.5 also induces endothelial dysfunction and inhibits blood vessel formation but has no significant association with arterial stiffness [40, 41]. Endothelial dysfunction disrupts anti-inflammatory processes, anti-platelet aggregation, anti-thrombotic processes and vascular repair in vivo [42]. Alterations in vascular function might be the earliest pathophysiological mechanism contributing to air pollution-mediated cardiovascular diseases, and indeed, such changes are a critical early predictor of atherosclerosis [43, 44]. However, there is a lack of systematic understanding of the mechanism of PM2.5-induced endothelial dysfunction. Moreover, a review focused on scientific evidence that DEPs induce endothelial dysfunction, including bioavailability and mechanisms, and is related to cardiovascular injury [45].
Therefore, this review mainly provides an overview of the literature related to PM2.5 and atherosclerosis, and discusses the mechanism of PM2.5-induced vascular endothelial injury. Approximately 30% of ambient PM2.5 is attributable to traffic sources [46,47,48]. Thus, we also review evidence for the role DEPs in atherosclerosis and endothelial dysfunction in this review. Fig. 1 summarizes the main mechanisms of PM2.5-triggered atherosclerosis. We review the main mechanisms of endothelial dysfunction after exposure to ambient or indoor PM2.5. Furthermore, we discuss the role of ambient PM2.5-induced vascular endothelial injury in the development of atherosclerosis. Targeting endothelial injury, as the initial pathological process of atherosclerosis, is the key to preventing the occurrence of atherosclerotic cardiovascular disease. Therefore, a scientific understanding of the mechanism of PM2.5-induced endothelial dysfunction will play a critical role in the prevention and treatment of atherosclerosis and other cardiovascular-related diseases.

Summarized the main pathogenic mechanisms of PM2.5-triggered atherosclerosis. Four main hypotheses have proposed by which inhaled particulate matter effect on cardiovascular system [49]: a. inflammatory mediators; b. unidentified mediators; c. autonomic imbalance; d. direct particle translocation. PM2.5 increased endothelial permeability, declined vascular tone and vascular reparative capacity, thus induced vascular endothelial injury. The initial step of atherosclerosis is vascular endothelial dysfunction, and then activated endothelial cells promoted monocytes recruited and maturation of monocytes into macrophages. Lipid accumulation and continued uptake by macrophages lead to foam cell formation and then developed into atherosclerotic lesion
PM2.5 and atherosclerosis
As shown in Fig. 1, four main hypotheses by which inhaled particulate matter affects the cardiovascular system have been proposed: a. Inhaled particulate matter reaches the terminal bronchioles and enters alveoli, inducing an inflammatory response in the lung; b. Released inflammatory mediators and unidentified mediators enter the circulation; c. A small proportion of particles reach the circulation; and d. Inhaled particulate matter activates alveoli sensory receptors, leading to autonomic imbalance [49]. Fig. 1 summarizes the main mechanisms of PM2.5-triggered atherosclerosis. Endothelial injury increases the release of IL-6, VCAM-1, ICAM-1, and other inflammatory cytokines, recruiting blood monocytes that bind to the activated endothelial monolayer. The bound monocytes migrated directly into the intima and mature into macrophages. PM2.5 increases the expression of CD36 in plaque macrophages and mediates the abnormal accumulation of oxidized lipids (such as 7-ketocholesterol, 7-KCh), finally promoting foam cell formation [12]. TLR4 recognizes modified lipoprotein, which mediates lipoprotein accumulation in macrophages [50]. Jin Geng et al. showed that PM2.5 can trigger foam cell formation via the TLR4/MyD88/NF-κB pathway [51]. Oxidized low-density lipoprotein (ox-LDL) primes and activates the NOD-like receptor protein 3 (NLRP3) inflammasome by binding to TLR4 or CD36 in macrophages and increases the release of inflammatory cytokines (IL-1β and IL-18) and pyroptosis [11]. PM2.5 induces oxidative stress, increasing the apoptosis of foam cells via the mitochondrial apoptosis pathway [52]. PM2.5 impairs HDL functions such as HDL-mediated cholesterol efflux, thus facilitating foam cell formation and accumulation [53]. Apoptotic cells are not quickly and efficiently engulfed and decomposed by phagocytes, resulting in secondary necrosis and the release of a large amount of pro-inflammatory cytokines and thus contributing to the development of the necrotic core [54]. However, currently, there is a lack of evidence concerning the efferocytosis of phagocytes in atherosclerosis induced by PM2.5. In addition, oxidative stress induced by PM2.5 can increase the proliferation and foam cell formation in VSMCs; however, future research is required to demonstrate the role of oxidative stress in mediating PM-triggered foam cell formation [55, 56]. VSMCs migrate from the media to the intima and synthesize extracellular matrix macromolecules such as elastin, proteoglycans and collagen, and fibrous cap formation. However, reports concerning the migration of VSMCs triggered by PM2.5 are lacking. The death of foam cells and the release of extracellular lipids in atherosclerotic lesions lead to the formation of a necrotic core [11, 16].
Table 1 summarizes epidemiological studies on the association between PM2.5 exposure and atherosclerosis. One of the major sources of ambient PM2.5 is traffic-derived emissions. Traffic noise is an important confounding factor in the effects of air pollutant exposure from traffic, and evidence has demonstrated that night-time traffic noise and PM2.5 are both associated with a 3.9% (95% CI: 0.0 - 8.0%) increase in thoracic aortic calcification (TAC)-burden per 5 dB(A) night-time traffic noise and an 18.1% (95% CI: 6.6 - 30.9%) increase in TAC burden per 2.4 μg/m3 PM2.5. Importantly, both are independently associated with the development and progression of subclinical atherosclerosis [59, 71]. Carotid intima-media thickness (CIMT) is defined as the distance between the lumen-intima and media-adventitia borders of the common carotid artery and can be measured by vascular ultrasound; an increase in the CIMT is a marker of subclinical atherosclerosis [93]. PM2.5 exposure is associated with an increase in the CIMT; moreover, increased or slowed CIMT progression is associated with PM2.5 concentration [68]. The PM2.5 components sulfur, elemental carbon (EC) and organic carbon (OC), but not silicon, are associated with increased CIMT, and OC has the strongest association [69, 78]. Long-term exposure to PM2.5 can increase systematic inflammation, the levels of fibrofatty and necrotic core components, and total plaque volume [38, 79]. Short-term exposure to PM2.5 is associated with inflammation, coagulation, endothelial activation and ox-LDL levels [38, 63]. Table 2 summarizes animal studies of PM2.5-induced atherosclerosis. PM2.5 promotes the progression of advanced atherosclerotic lesions. Concentrated ambient PM2.5, rather than whole diesel exhaust and diesel exhaust gases, is mainly responsible for plaque exacerbation [99]. However, an improved understanding of which components of PM2.5 induce or promote the development of atherosclerosis, which may be a direction of future research, is needed. Mechanistically, changes in oxidative stress, systematic inflammation and lipid metabolism are the most common mechanisms of PM2.5-induced atherosclerosis. The mechanisms of atherosclerosis induced by PM2.5 have been reviewed [105]. Oxidative stress induced by particulate matter may be key to triggering the development of atherosclerosis, coagulation and thrombosis [106, 107]. Endothelial dysfunction is a critical initiating event that promotes the development of atherosclerosis. However, a systematic understanding of the mechanisms underlying PM2.5-induced endothelial dysfunction leading to atherosclerosis is lacking.
Ambient PM2.5 induces endothelial dysfunction
The endothelial cell monolayers of blood vessels play a role in exchanging macromolecules between the blood and tissues. Mechanical stimuli such as shear stress, inflammatory cytokines and angiotensin-II (Ang-II) affect endothelial permeability [108, 109]. We concentrated on studies indicating that PM2.5 impacts vascular endothelial dysfunction. Many studies have demonstrated that PM2.5 increases the vascular permeability, impairs endothelial vasomotor function and vascular reparative capacity via different mechanisms and occurs before vascular diseases such as atherosclerosis. Traffic-derived emissions are a major source of ambient PM2.5; therefore, literature on the toxicity of traffic emissions (mainly DEPs) on blood vessels and endothelial cells and the similarities between PM2.5 and DEPs are also discussed in this review. The effects of PM2.5 and DEPs on endothelial cells are shown in Table 3. In brief, the existing evidence shows that PM2.5 and DEPs both consistently induces endothelial cytotoxicity through similar mechanisms, such as by increasing endothelial cellular apoptosis via oxidative stress or autophagy, reducing the migration of endothelial cells and enhancing vascular endothelial permeability [111, 117, 118, 122, 123]. The detailed mechanisms of endothelial cytotoxicity induced by PM2.5 or traffic-derived pollutants are discussed below. In addition, coal-fired PM2.5 also decreases endothelial viability; however, the detailed mechanisms are limited to increases in DNA methylation and oxidative DNA damage in EA.hy926 cells [128].
Ambient PM2.5 increases vascular endothelial permeability
The proposed mechanism of PM2.5-triggered vascular endothelial permeability increase is presented in Fig. 2a. In Balb/c mice exposed to low-dose (1.27 mg/kg) and high-dose (6.34 mg/kg) PM2.5 through the tail vein for 48 h, PM2.5 destroyed the integrity of vessels, as assessed by the Evans blue infiltration assay, and the results confirmed that PM2.5 increased vascular permeability in vivo [114]. DEPs increase vascular endothelial permeability by downregulating the expression of zonula occludens-1 (ZO-1, a tight junction protein) [123]. Ambient PM2.5 exposure disrupts the balance between antioxidation and oxidation in vascular endothelial cells, leading to increased permeability of the endothelial monolayer. Epidemiological evidence has shown that exposure to PM2.5 reduces the anti-inflammatory and antioxidant capacity of high-density lipoprotein (HDL), and decreases the expression of antioxidant markers such as glutathione peroxidase (GSH) and superoxide dismutase (SOD) [67, 129]. A review showed that particulate matter impairs HDL function via oxidative pathways [53]. Importantly, evidence has shown that exercise training enhances HDL functions, including cholesterol efflux capacity and antioxidant capacity, and protects against endothelial dysfunction induced by PM2.5 [130]. PM2.5 decreases the mitochondrial membrane potential, increases reactive oxygen species (ROS) generation, and causes oxidative stress, inflammation and apoptosis in EA.hy926 cells and human umbilical vein endothelial cells (HUVECs) [110, 115, 124]. ROS play important roles in inflammatory responses, apoptosis, and cell growth, as well as in the oxidation of LDL cholesterol [131]. PM2.5 induces ROS generation and endothelial cell apoptosis through the mitochondrial pathway in EA.hy926 cells [132]. PM2.5 induces cell autophagy and apoptosis via endoplasmic reticulum (ER) stress in EA.hy926 cells and HUVECs [111]. Although normal autophagy seems to protect cells from PM2.5-triggered apoptosis, PM2.5 blocks autophagic flux and then robustly aggravates endothelial cell apoptosis [111, 122, 127]. Effective inhibition of ER stress using 4-PBA (an ER stress inhibitor) contributes to the alleviation of PM2.5 induced cell apoptosis and the expression of LC3II [111]. Bafi A1 (an autolysosome inhibitor) aggravates PM2.5-induced cell apoptosis by disrupting autophagic flux [111]. Exposure to PM2.5 induces activation of the inflammatory cyclooxygenase-2 (COX-2)/prostaglandin E synthase (PGES)/prostaglandin E 2 (PGE2) axis and promotes the inflammatory response and apoptosis in mouse aorta endothelial cells (MAECs) [121]. Excessive apoptosis triggers an increase in transcellular permeability in the vascular endothelial monolayer [133]. Ambient PM2.5 disrupts iron uptake and storage by regulating the expression of transferrin receptor (TFRC), ferritin light chain (FTL) and heavy chain (FTH1), causing intracellular iron overload and subsequently provoking ferroptosis in EA.hy926 cells and HUVECs [112]. PM2.5 induces ROS production and lipid peroxidation in endothelial cells and increases membrane permeability [112, 134]. Furthermore, PM2.5 induces senescence associated-β galactosidase (SA-β-gal) activation via redox sensitivity of the local angiotensin system in premature coronary arterial endothelial cells (PCAECs), leading to endothelial senescence [113]. The presence of senescent endothelial cells in a nonsenescent monolayer disrupts the tight junction morphology of surrounding young cells and increases the permeability of the monolayer [135].

Summary of the main mechanisms of PM2.5-caused vascular endothelial injury. Mainly involved three aspects: a. PM2.5 increased vascular endothelial permeability; b. PM2.5 impaired vasomotor function; c. PM2.5 declined vascular reparative capacity
Vascular endothelial (VE)-cadherin is largely expressed on endothelial cell membranes. The extracellular domain of VE-cadherin mediates endothelial cell-to-cell adhesion through hemophilic trans interactions, whereas its cytoplasmic tail associates with the actin cytoskeleton, strengthening the adhesion junction between endothelial cells [136]. Thus, VE-cadherin plays a key role in maintaining endothelial barrier integrity, controlling the transmembrane movement of macromolecule substances such as blood cells. PM2.5 induces the phosphorylation of vascular endothelial growth factor receptor 2 (VEGFR2) on the endothelial cell membrane and activates downstream mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling, leading to the shedding of adhesion connexin VE-cadherin [114]. PM2.5 induces endothelial cell cytoskeleton rearrangement via Rho-dependent pathways to facilitate vascular hyperpermeability [137]. Targeting of tissue inhibitor of metalloproteinase 3 (TIMP3)/MMP9 and VE-cadherin by miR-21 in response to PM2.5 increases vascular endothelial cell permeability in HUVECs [108]. Inflammation stimulates a series of signaling pathways that reduce the level of VE-cadherin expressed or induces VE-cadherin phosphorylation and then destroys the adhesion structure [138].
Ambient PM2.5 impairs vasomotor function
Endothelin-1 (ET-1) is a protein primarily produced by endothelial cells that regulates cell proliferation and vascular tone by activating its receptors, including type A (ETA) and type B (ETB) [139]. Evidence has indicated that inflammation, ischemia and hypoxia stimulate the expression of ET-1 and its receptors [140]. Qinghua Sun et al. exposed ApoE-/- mice to concentrated ambient PM2.5 for 6 months and assessed the vasoconstriction of aortic rings in response to phenylephrine and serotonin and vasorelaxation in response to acetylcholine. The results demonstrated that PM2.5 significantly increased the constriction of the aorta, especially in high-fat diet-fed mice [101]. The proposed mechanism of by which PM2.5 causes endothelial vasomotor function impairment is presented in Fig. 2b. PM2.5 elevates the circulating levels of AngII, locally activates the AngII/AngII type 1 receptor (AT1R) axis and activates phospholipase C (PLC) and protein kinase C (PKC), promoting ET-1 biosynthesis in HUVECs [116]. ET-1 is released from endothelial cells, acts on the endothelial ETB receptor and increases nitric oxide (NO) production [141]. The production of NO by endothelial cells contributes to regulating vasomotor tone. NO cats on circulating blood platelets, leukocytes and adjacent smooth muscle cells and reduces smooth muscle cell contractility [142]. PM2.5 impairs the balance of vasorelaxation by oxidative stress, and superoxide radicals combine with NO to form peroxynitrite, thus reducing NO bioavailability in the vessel wall [143]. Furthermore, PM2.5 upregulates the expression of ETB and ETA receptors in rat coronary arteries [144]. ETB in vascular endothelial cells mediates the vasodilation, while ETA and ETB in vascular smooth muscle cells mediate the contractility, especially ETA activation, which plays a greater role in coronary vasoconstriction [141]. ET-1 in the vasculature causes brief vessel relaxation due to ETB activation in endothelial cells. However, this effect is quickly reversed by ET-1 binding to ETA, which reduces NO production in vascular smooth muscle cells and leads to the well-known constrictive effects of ET-1 in the vasculature [145]. Therefore, PM2.5 upregulates the expression of ETB and ETA receptors in coronary arteries, but PM2.5 mainly increases vasoconstriction and contributes to the progression of atherosclerosis. AngII enhances ET-1-mediated vasoconstriction by upregulating the expression of ETA in VSMCs [146]. In addition, PM2.5 exposure causes vascular insulin resistance and suppresses insulin-stimulated endothelial nitric oxide synthase (eNOS) phosphorylation (likely an endothelial-specific event) [147]. Insulin stimulates the phosphorylation of eNOS and increases eNOS activity and NO production [148,149,150]. Therefore, PM2.5-provoked endothelial insulin resistance could be a key event in regulating vascular tone. In brief, PM2.5 shifts the balance of vasomotor tone towards vasoconstriction by increasing the levels of ET-1 and its receptors, as well as decreasing NO production and bioavailability. Exercise training effectively prevents the imbalance in vasomotor function triggered by PM2.5 [130].
Ambient PM2.5 suppresses vascular endothelial repair
Endothelial progenitor cells (EPCs), a group of stem cells/progenitor cells, settle in the adult bone marrow and can mobilize to the peripheral blood, home to sites of vascular injury, proliferate and differentiate into endothelial cells, and facilitate vascular recovery [151]. In addition to exerting antioxidative and anti-inflammatory effects, HDL protects EPCs by increasing eNOS levels and decreasing MMP9 levels, thereby reducing the apoptosis of EPCs [152]. Bone marrow-derived EPCs from C57BL/6 mice exposed to PM2.5 inhalation for 9 or 30 days were injected into unexposed mice subjected to hind limb ischemia and vascular perfusion was assessed by laser Doppler perfusion imaging (LDPI). The results confirmed that PM2.5 significantly impaired angiogenesis and that bone marrow-derived EPCs have vascular reparative capacity in vivo [153]. The proposed mechanism of PM2.5-triggered vascular repair suppression is presented in Fig. 2c. In C57BL/6 mice exposed to concentrated PM2.5 inhalation for 9 or 30 consecutive days (6 h/day), PM2.5 impaired endothelial progenitor cellular differentiation and mobilization through vascular insulin resistance and nuclear factor kappa-B (NF-κB) and inflammasome activation, while insulin sensitizers prevented PM2.5-triggered vascular insulin resistance and inflammation and decreased circulating EPCs [154]. ROS and inflammation suppress the proliferation of EPCs and enhance the apoptosis of EPCs [155]. Furthermore, PM2.5 decreases the abundance of EPCs, and impairs EPC functions and prevents EPC-mediated vascular endothelial recovery associated with vascular endothelial growth factor (VEGF) resistance and a decrease in NO bioavailability [153]. In addition to EPCs, the proliferation and migration abilities of mature endothelial cells are additional important factors in endothelial injury repair. Endothelial cell proliferation and migration are initially required for arterial repair after injury [156]. PM2.5 decreases the viability and suppresses the proliferation and migration of HUVECs and human microvascular endothelial cells through oxidative stress [117]. In brief, PM2.5 disrupts two major factors involved in repairing vascular endothelial damage: a. the abundance and function of EPCs and b. the proliferation and migration abilities of endothelial cells.
Ambient PM2.5-triggered endothelial injury and atherosclerosis
Ambient PM2.5 causes a pro-coagulant state
Atherosclerosis is characterized by the accumulation of inflammatory cells and lipids in the walls of arteries. The intact endothelial cell layer exerts the first defense to hinder the development of atherosclerosis. Endothelial cell-derived mediators take part in hemostasis, including tissue factor (TF), tissue factor pathway inhibitors (TFPI), thrombomodulin (TM), and von Willebrand factor (vWF) [157, 158]. For example, normal vascular endothelium-synthesized TFPI regulates the balance between coagulation and fibrinolysis [44]. Exposure to air pollution affects each of these dynamic processes, and increasing evidence suggests that the balance of between platelet activation, coagulation and fibrinolysis shifts towards a pro-coagulant and anti-fibrinolytic state [8]. Sprague-Dawley (SD) rats were exposed to PM2.5 (1.8, 5.4, 16.2 mg/kg bw) by intratracheal instillation every three days for 30 days, and the results showed that PM2.5 increased the expression of TF in the vessel wall and activated coagulation factors VII and X and the formation of thrombin [158,159,160]. PM2.5 reduces the expression of TM in the vascular endothelial monolayer, thereby decreasing anti-coagulant function [160]. vWF is mainly released by activated endothelial cells, which bridge platelets and aggregates in the injured vessel walls [161]. PM2.5 downregulates the expression of vWF in the serum and promotes the adherence of platelets to injured endothelial layers, implying that vWF is consumed during the process of platelet aggregation after exposure to PM2.5 [104, 160]. Platelet adhesion, aggregation and coagulation are implicated in inflammatory pathologies of atherosclerosis [162, 163].
Ambient PM2.5 induces an inflammatory response
Short-term exposure to PM2.5 induces systemic inflammation and increases the circulation levels of inflammatory biomarkers such as CRP, tumor necrosis factor α (TNF-α), IL-6, IL-8 and MCP-1 [35, 164]. PM2.5 triggers the secretion of IL-6 and IL-1β by activating the TLR-mediated pathway in HUVECs, while TLR2 and TLR4 inhibitors reduce the PM2.5-triggered inflammatory response [120]. PM2.5 triggers endothelial activation, increases the expression of adhesion molecules (ICAM-1 and VCAM-1) and induces THP-1 cell adhesion to endothelial cells through the ERK/AKT/NF-κB-dependent pathway in EA.hy926 cells; moreover, ERK/AKT/NF-κB inhibitors have been used to demonstrate the abovementioned effects [118]. Monocytes in the blood adhere to endothelial cells through adhesion molecules and then migrate into the vascular wall [126, 165]. Monocytes that enter the blood vessel wall transform into macrophages, which clear lipids and dead or dying cells [166]. Exposure of ApoE-/- or LDLR-/- mice to concentrated ambient PM2.5 for 6 months (6 h/day, 5 days/week) showed that PM2.5 significantly increases the expression of CD36 in plaque macrophages, increases the internalization of ox-LDL and mediates macrophage-derived foam cell formation [12]. Both macrophage-derived foam cells and necrotic cells release various inflammatory factors (such as TNF, IL-1, and IL-6), thereby expanding the inflammatory response cascade and inducing persistent inflammation in local blood vessels [166]. Moreover, evidence from animal experiments has shown that inflammation is significantly unregulated in ApoE-/- mice after exposure to PM2.5 (10 mg/kg bw) for two months and that inflammation increases even if PM2.5 exposure is stopped [6]. The above evidence focuses on the effects of PM2.5 on inflammation and atherosclerosis.
Ambient PM2.5 promotes lipid deposition
Lipid deposition is one of the key factors promoting the development of atherosclerosis, especially ox-LDL deposition, which contributes to necrotic core formation. In the past few decades, lowering lipid levels has been the main strategy for the treatment of atherosclerosis [167]. However, currently, two views about how lipids enter and deposit in the vascular wall are held. It has been believed that damaged vascular walls cause LDL infiltration into the vascular wall and induce the development of atherosclerosis [168]. Professor Shaul holds a different view, suggesting that the receptor scavenger receptor type B1 (SR-B1) on endothelial cells has a transcytosis effect on LDL and promotes the accumulation of LDL in the vascular wall [169]. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) is expressed in endothelial cells, monocytes/macrophages and vascular smooth muscle cells and is essential for binding to oxLDL [170]. Pro-inflammatory and pro-oxidant, and mechanical stimuli including ox-LDL, TNF-α, Ang II and shear stress, can rapidly activate the expression of LOX-1 [171]. In endothelial cells, LOX-1 activation induces inflammation, reduces eNOS activation and NO availability, and triggers endothelial dysfunction [172]. Ox-LDL induces the release of the soluble form of LOX-1 (sLOX-1) from endothelial cells into the circulation and the level of sLOX-1 correlates with carotid plaque inflammation and risk for ischemic stroke [173]. Inhaled vehicle emissions trigger significant increases in plasma sLOX-1 levels in humans and mediate the upregulation of ET-1 and MMP9 expression via ox-LDL-LOX-1 receptor signaling, further inducing vascular effects [174]. LOX-1 protein levels are increased in the aorta after coexposure to ozone and DEPs [175]. Moreover, in ApoE-/- mice exposed to a mixture of gasoline and diesel engine exhaust (MVE), the expression of LOX-1 was increased in cerebral microvascular endothelial cells, and at least in part, MVE altered the structure and integrity of the brain microvasculature via LOX-1 signaling [176]. Many studies have demonstrated that PM2.5 contributes to lipid dysregulation in the sera of ApoE-/- mice and promotes macrophage engulfment of ox-LDL through surface scavenger receptors to induce foam cell formation [6, 12, 177]. However, studies on the effect of PM2.5 on lipid uptake and transport in endothelial cells are lacking, although limited evidence has shown that traffic-derived pollutants increase LOX-1 signaling in endothelial cells. In addition to inflammation, lipid uptake and transport are key factors in the development of atherosclerosis. Thus, the effect of PM2.5 on the binding of ox-LDL to endothelial cells is an area of intense investigation.
Indoor PM2.5 elicits endothelial dysfunction and atherosclerosis
WHO data has shown that more than 41% of households are still using solid fuels and kerosene for cooking, producing harmful smoke in the home and causing the death of approximately 916 thousand individuals from cardiovascular disease [178]. CIMT is a marker of subclinical atherosclerosis. Recently, epidemiological evidence showed that PM2.5 emissions from biomass cooking fuel in peri-urban villages of India were positively associated with increased CIMT [61]. Consistent with this study, Matthew S Painschab et al. also showed that indoor PM2.5 sourced from biomass fuel in Puno, Peru, was associated with CIMT, an enhanced prevalence of atherosclerotic plaque and increased blood pressure [88]. In the rural villages of Sichuan, China, household PM2.5 from biomass stoves is associated with central hemodynamics and increased blood pressure; however, it is not associated with pulse wave velocity (PWV, a marker of arterial stiffness) [87]. Indoor particles impair microvascular function through inflammation and oxidative stress [179]. Cooking oil fumes (COFs), the main pollutants in kitchen air, can significantly reduce cellular viability, and inhibit angiogenesis in HUVECs through the ROS-mediated NLRP3 inflammasome pathway or VEGF/VEGFR2/MEK1/2/ERK1/2/mTOR pathway [40, 119]. COF-derived PM2.5 mediates autophagy via the ROS/AKT/mTOR axis in HUVECs [180]. Evidence for the mediating role of indoor PM2.5 in vascular endothelial dysfunction is limited.
Conclusion
In summary, PM2.5 exposure is positively associated with atherosclerosis based on epidemiological evidence. Epidemiological and animal experimental evidence has established that PM2.5-induced atherosclerosis is mainly mediated by inflammation and lipid metabolism alterations. On the basis of in vivo and in vitro studies, PM2.5 induces vascular endothelial dysfunction and a procoagulant state and increases inflammation and lipid abnormalities, thus promoting the development of atherosclerosis. However, only a few studies have tried to explore preventive measures. It would be meaningful to explore measures or targets that can contribute to the prevention of PM2.5-induced endothelial dysfunction or atherosclerosis, which remains to be solved before the environment improves. Changes occur at the molecular level significantly earlier than histopathology and clinical symptoms. Consequently, improving the understanding of molecular mechanisms will be helpful in preventing the occurrence or development of atherosclerosis, or identifying potential therapeutic targets for atherosclerosis treatment.
Availability of data and materials
Databases/repositories and materials is not applicable in this review.
Abbreviations
- AAC:
-
Abdominal aortic calcium agatston score
- ABCA1:
-
ATP-binding cassette transporter A1
- AngII:
-
Angiotensin II
- aHR:
-
Adjusted hazard ratio
- AT1R:
-
AngII type 1 receptor
- AIx:
-
Augmentation index
- ApoA1:
-
Apolipoprotein A1
- ApoB:
-
Apolipoprotein B
- AP-1:
-
Activation protein-1
- ASC:
-
Apoptosis associated speck like protein
- Bafi A1:
-
Bafilomycin A1
- BC:
-
Black carbon
- BP:
-
Blood pressure
- baPWV:
-
Brachial-ankle pulse wave velocity
- CAC:
-
Coronary artery calcification
- CCS:
-
Coronary artery calcium score
- CD36:
-
Cluster of differentiation 36
- cfPWV:
-
Carotid-femoral PWV
- CI:
-
Confidence interval
- CIMT:
-
Carotid intima-media thickness
- COFs:
-
Cooking oil fumes
- COX-2:
-
Cyclooxygenase -2
- CRP:
-
C-reactive protein
- DEG:
-
Diesel exhaust gases
- DFOM:
-
Deferoamine mesylate
- EA.hy926:
-
Human umbilical vein cell line
- EC:
-
Elemental carbon
- eNOS:
-
Endothelial nitric oxide synthase
- EPC:
-
Endothelial progenitor cells
- ET-1:
-
Endothelin-1
- ETA :
-
Endothelin type A
- ETB :
-
Endothelin type B
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- FA:
-
Filtered air
- Fer-1:
-
Ferrostatin-1
- Foxp3:
-
Forkhead box transcription factor P3
- FTL:
-
Ferritin light chain
- FTH1:
-
Ferritin heavy chain
- GSH:
-
Glutathione peroxidase
- GSH-Px:
-
Glutathione peroxidase
- HDL:
-
High-density lipoprotein
- HDL-C:
-
High density lipoprotein-cholesterol
- HDL-P:
-
High-density lipoprotein cholesterol particle matter
- HOI:
-
HDL oxidant index
- Hs-CRP:
-
High sensitive C-reactive protein
- HRP:
-
High-risk plaque
- HUVECs:
-
Human umbilical vein endothelial cells
- IL:
-
Interleukin
- IAD:
-
Inter-adventitial diameter
- IMT:
-
Intima-media thickness
- iNOS:
-
Inducible nitric oxide synthase
- IQR:
-
Interquartile
- 7-KCh:
-
7-ketocholesterol
- LDH:
-
Lactate dehydrogenase
- LDL:
-
Low-density lipoprotein
- LDL-C:
-
Low density lipoprotein-cholesterol
- LDPI:
-
Laser Doppler perfusion imaging
- LOX-1:
-
Lectin-like oxidized low-density lipoprotein receptor-1
- LXR-α:
-
Liver X receptor α
- MAECs:
-
Mouse aorta endothelial cells
- 3-MA:
-
3-Methyadenine
- MAPK:
-
Mitogen-activated protein kinase
- MCP:
-
Monocyte chemoattractant protein
- MDA:
-
Malondialdehyde
- MMP:
-
Matrix metalloproteinase
- MMP:
-
Mitochondrial membrane potential
- MRI:
-
Magnetic resonance imaging
- NAC:
-
N-acetyl-L-cysteine
- NF-κB:
-
Nuclear factor kappa-B
- NO:
-
Nitric oxide
- NIST:
-
National Institute of Standards and Technology
- 8-OHdG:
-
8-hydroxy-2’-deoxyguanosine
- NLRP3:
-
NOD-like receptor protein 3
- O3 :
-
Ozone
- OC:
-
Organic carbon
- OR:
-
Odds ratio
- ox-LDL:
-
Oxidized low-density lipoprotein
- PAH:
-
Polycyclic aromatic hydrocarbon
- 4-PBA:
-
4-phenylbutyrate
- PCAECs:
-
Premature coronary arterial endothelial cells
- PGES:
-
Prostaglandin E synthase
- PGE2:
-
Prostaglandin E 2
- PM2.5 :
-
Fine particulate matter
- PM2.5abs :
-
Absorbance levels of PM2.5
- PMA:
-
Phorbol-myristate-acetate
- PLC:
-
Phospholipase C
- PKC:
-
Protein kinase C
- PNacc:
-
Particle number of accumulation mode particles
- PP:
-
Pilse pressure
- PWV:
-
Pulse wave velocity
- Rapa/Rap:
-
Rapamycin
- ROS:
-
Reactive oxygen species
- SA-β-gal:
-
Senescence associated-β galactosidase
- SBP:
-
Systolic blood pressure
- SO2 :
-
Sulfur dioxide
- SOD:
-
Superoxide dismutase
- sICAM-1:
-
Soluble Intercellular Adhesion Molecule-1
- SR-BI:
-
Scavenger receptor type B1
- TAC:
-
Thoracic aortic calcium agatston score
- T-AOC:
-
Total antioxidant capacity
- TC:
-
Total cholesterol
- TF:
-
Tissue factor
- TFPI:
-
Tissue factor pathway inhibitors
- TFRC:
-
Transferrin receptor
- TG:
-
Triglycerides
- TGF-β:
-
Ttransforming growth factor-β
- TM:
-
Thrombomodulin
- TIMP3:
-
Tissue inhibitor of metalloproteinase 3
- TNF-α:
-
Tumor necrosis factor α
- TLR:
-
Toll-like receptor
- UFP:
-
Ultrafine particles
- VCAM-1:
-
Vascular cell adhesion molecule-1
- VE:
-
Vascular endothelial
- VEGF:
-
Vascular endothelial growth factor
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
- VOC:
-
Volatile organic compounds
- VSMCs:
-
Vascular smooth muscle cells
- vWF:
-
Von willebrand factor
- WDE:
-
Whole diesel exhaust
- WHO:
-
World Health Organization
- ZO-1:
-
zonula occludens-1
References
Mannucci PM, Harari S, Franchini M. Novel evidence for a greater burden of ambient air pollution on cardiovascular disease. Haematologica. 2019;104(12):2349–57. https://doi.org/10.3324/haematol.2019.225086.
Benziger CP, Roth GA, Moran AE. The Global Burden of Disease Study and the Preventable Burden of NCD. Glob Heart. 2016;11(4):393–7. https://doi.org/10.1016/j.gheart.2016.10.024.
Cohen AJ, Brauer M, Burnett R, Anderson HR, Frostad J, Estep K, et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet. 2017;389(10082):1907–18. https://doi.org/10.1016/s0140-6736(17)30505-6.
Liang CS, Duan FK, He KB, Ma YL. Review on recent progress in observations, source identifications and countermeasures of PM2.5. Environ Int. 2016;86:150–70. https://doi.org/10.1016/j.envint.2015.10.016.
Walter CM, Schneider-Futschik EK, Knibbs LD, Irving LB. Health impacts of bushfire smoke exposure in Australia. Respirology. 2020. https://doi.org/10.1111/resp.13798.
Zhang J, Liang S, Ning R, Jiang J, Zhang J, Shen H, et al. PM2.5-induced inflammation and lipidome alteration associated with the development of atherosclerosis based on a targeted lipidomic analysis. Environ Int. 2020;136:105444. https://doi.org/10.1016/j.envint.2019.105444.
Park SK, Auchincloss AH, O'Neill MS, Prineas R, Correa JC, Keeler J, et al. Particulate air pollution, metabolic syndrome, and heart rate variability: the multi-ethnic study of atherosclerosis (MESA). Environ Health Perspect. 2010;118(10):1406–11. https://doi.org/10.1289/ehp.0901778.
Brook RD, Rajagopalan S, Pope CA 3rd, Brook JR, Bhatnagar A, Diez-Roux AV, et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331–78. https://doi.org/10.1161/CIR.0b013e3181dbece1.
Jiang Y, Lu H, Man Q, Liu Z, Wang L, Wang Y, et al. Stroke burden and mortality attributable to ambient fine particulate matter pollution in 195 countries and territories and trend analysis from 1990 to 2017. Environ Res. 2020;184:109327. https://doi.org/10.1016/j.envres.2020.109327.
Zhu YH, Xian XM, Wang ZZ, Bi YC, Chen QG, Han XF, et al. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules. 2018;8:3 doi: ARTN 8010.3390/biom8030080.
Bäck M, Yurdagul A Jr, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16(7):389–406. https://doi.org/10.1038/s41569-019-0169-2.
Rao X, Zhong J, Maiseyeu A, Gopalakrishnan B, Villamena FA, Chen LC, et al. CD36-dependent 7-ketocholesterol accumulation in macrophages mediates progression of atherosclerosis in response to chronic air pollution exposure. Circ Res. 2014;115(9):770–80. https://doi.org/10.1161/circresaha.115.304666.
Mueller PA, Zhu L, Tavori H, Huynh K, Giunzioni I, Stafford JM, et al. Deletion of Macrophage Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) Accelerates Atherosclerosis Regression and Increases C-C Chemokine Receptor Type 7 (CCR7) Expression in Plaque Macrophages. Circulation. 2018;138(17):1850–63. https://doi.org/10.1161/circulationaha.117.031702.
Tang SL, Chen WJ, Yin K, Zhao GJ, Mo ZC, Lv YC, et al. PAPP-A negatively regulates ABCA1, ABCG1 and SR-B1 expression by inhibiting LXRα through the IGF-I-mediated signaling pathway. Atherosclerosis. 2012;222 2:344-354; doi: https://doi.org/10.1016/j.atherosclerosis.2012.03.005.
Zeng Y, Peng Y, Tang K, Wang YQ, Zhao ZY, Wei XY, et al. Dihydromyricetin ameliorates foam cell formation via LXRα-ABCA1/ABCG1-dependent cholesterol efflux in macrophages. Biomed Pharmacother. 2018;101:543–52. https://doi.org/10.1016/j.biopha.2018.02.124.
Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–25. https://doi.org/10.1038/nature10146.
Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116(1):59–69. https://doi.org/10.1172/jci25074.
Saba L, Saam T, Jager HR, Yuan C, Hatsukami TS, Saloner D, et al. Imaging biomarkers of vulnerable carotid plaques for stroke risk prediction and their potential clinical implications. Lancet Neurol. 2019;18(6):559–72. https://doi.org/10.1016/S1474-4422(19)30035-3.
Sharkey J, Starkey Lewis PJ, Barrow M, Alwahsh SM, Noble J, Livingstone E, et al. Functionalized superparamagnetic iron oxide nanoparticles provide highly efficient iron-labeling in macrophages for magnetic resonance-based detection in vivo. Cytotherapy. 2017;19(4):555–69. https://doi.org/10.1016/j.jcyt.2017.01.003.
Robson PM, Dweck MR, Trivieri MG, Abgral R, Karakatsanis NA, Contreras J, et al. Coronary Artery PET/MR Imaging: Feasibility, Limitations, and Solutions. JACC Cardiovasc Imaging. 2017;10(10 Pt A):1103–12. https://doi.org/10.1016/j.jcmg.2016.09.029.
Hop H, de Boer SA, Reijrink M, Kamphuisen PW, de Borst MH, Pol RA, et al. (18)F-sodium fluoride positron emission tomography assessed microcalcifications in culprit and non-culprit human carotid plaques. J Nucl Cardiol. 2019;26(4):1064–75. https://doi.org/10.1007/s12350-018-1325-5..
Sundboll J, Larsen AP, Veres K, Adelborg K, Sorensen HT. Cardiovascular event rates and trajectories of LDL-cholesterol levels and lipid-lowering therapy in patients with atherosclerotic cardiovascular disease: A population-based cohort study. Thromb Res. 2019;183:124–30. https://doi.org/10.1016/j.thromres.2019.09.034.
Wang J, Geng NB, Xu YF, Zhang WD, Tang XY, Zhang RQ. PAHs in PM2.5 in Zhengzhou: concentration, carcinogenic risk analysis, and source apportionment. Environ Monit Assess. 2014;186(11):7461–73. https://doi.org/10.1007/s10661-014-3940-1.
Hellén H, Hakola H, Haaparanta S, Pietarila H, Kauhaniemi M. Influence of residential wood combustion on local air quality. Sci Total Environ 2008;393 2-3:283-90; doi: https://doi.org/10.1016/j.scitotenv.2008.01.019.
Miller MR, McLean SG, Duffin R, Lawal AO, Araujo JA, Shaw CA, et al. Diesel exhaust particulate increases the size and complexity of lesions in atherosclerotic mice. Part Fibre Toxicol. 2013;10:61. https://doi.org/10.1186/1743-8977-10-61.
Pöss J, Lorenz D, Werner C, Pavlikova V, Gensch C, Speer T, et al. Diesel exhaust particles impair endothelial progenitor cells, compromise endothelial integrity, reduce neoangiogenesis, and increase atherogenesis in mice. Cardiovasc Toxicol. 2013;13(3):290–300. https://doi.org/10.1007/s12012-013-9208-0.
Brinchmann BC, Le Ferrec E, Podechard N, Lagadic-Gossmann D, Holme JA, Øvrevik J. Organic chemicals from diesel exhaust particles affects intracellular calcium, inflammation and β-adrenoceptors in endothelial cells. Toxicol Lett. 2019;302:18–27. https://doi.org/10.1016/j.toxlet.2018.11.009.
Holme JA, Brinchmann BC, Le Ferrec E, Lagadic-Gossmann D, Øvrevik J. Combustion Particle-Induced Changes in Calcium Homeostasis: A Contributing Factor to Vascular Disease? Cardiovasc Toxicol. 2019;19(3):198–209. https://doi.org/10.1007/s12012-019-09518-9.
Brinchmann BC, Skuland T, Rambøl MH, Szoke K, Brinchmann JE, Gutleb AC, et al. Lipophilic components of diesel exhaust particles induce pro-inflammatory responses in human endothelial cells through AhR dependent pathway(s). Part Fibre Toxicol. 2018;15(1):21. https://doi.org/10.1186/s12989-018-0257-1.
Brinchmann BC, Le Ferrec E, Podechard N, Lagadic-Gossmann D, Shoji KF, Penna A. Lipophilic Chemicals from Diesel Exhaust Particles Trigger Calcium Response in Human Endothelial Cells via Aryl Hydrocarbon Receptor Non-Genomic Signalling. Intern J Mol Sci. 2018;19:5. https://doi.org/10.3390/ijms19051429.
Alshaarawy O, Elbaz HA, Andrew ME. The association of urinary polycyclic aromatic hydrocarbon biomarkers and cardiovascular disease in the US population. Environ Int. 2016;89-90:174-178; doi: 10.1016/j.envint.2016.02.006.
Manisalidis I, Stavropoulou E, Stavropoulos A, Bezirtzoglou E. Environmental and Health Impacts of Air Pollution: A Review. Front Public Health. 2020;8:14. https://doi.org/10.3389/fpubh.2020.00014.
Munzel T, Gori T, Al-Kindi S, Deanfield J, Lelieveld J, Daiber A, et al. Effects of gaseous and solid constituents of air pollution on endothelial function. Eur Heart J. 2018;39(38):3543–50. https://doi.org/10.1093/eurheartj/ehy481.
Rao X, Zhong J, Brook RD, Rajagopalan S. Effect of Particulate Matter Air Pollution on Cardiovascular Oxidative Stress Pathways. Antioxid Redox Signal. 2018;28(9):797–818. https://doi.org/10.1089/ars.2017.7394.
Zhang Q, Niu Y, Xia Y, Lei X, Wang W, Huo J, et al. The acute effects of fine particulate matter constituents on circulating inflammatory biomarkers in healthy adults. Sci Total Environ. 2020;707:135989. https://doi.org/10.1016/j.scitotenv.2019.135989.
Bekki K, Ito T, Yoshida Y, He C, Arashidani K, He M, et al. PM2.5 collected in China causes inflammatory and oxidative stress responses in macrophages through the multiple pathways. Environ Toxicol Pharmacol. 2016;45:362–9. https://doi.org/10.1016/j.etap.2016.06.022.
Pope CA 3rd, Bhatnagar A, McCracken JP, Abplanalp W, Conklin DJ, O'Toole T. Exposure to Fine Particulate Air Pollution Is Associated With Endothelial Injury and Systemic Inflammation. Circ Res. 2016;119(11):1204–14. https://doi.org/10.1161/circresaha.116.309279.
Hajat A, Allison M, Diez-Roux AV, Jenny NS, Jorgensen NW, Szpiro AA, et al. Long-term exposure to air pollution and markers of inflammation, coagulation, and endothelial activation: a repeat-measures analysis in the Multi-Ethnic Study of Atherosclerosis (MESA). Epidemiology. 2015;26(3):310–20. https://doi.org/10.1097/ede.0000000000000267.
Wu CF, Shen FH, Li YR, Tsao TM, Tsai MJ, Chen CC, et al. Association of short-term exposure to fine particulate matter and nitrogen dioxide with acute cardiovascular effects. Sci Total Environ. 2016;569-570:300–5. https://doi.org/10.1016/j.scitotenv.2016.06.084.
Shen C, Liu J, Zhu F, Lei R, Cheng H, Zhang C, et al. The effects of cooking oil fumes-derived PM2.5 on blood vessel formation through ROS-mediated NLRP3 inflammasome pathway in human umbilical vein endothelial cells. Ecotoxicol Environ Saf. 2019;174:690–8. https://doi.org/10.1016/j.ecoenv.2019.03.028.
Soppa VJ, Shinnawi S, Hennig F, Sasse B, Hellack B, Kaminski H, et al. Effects of short-term exposure to fine and ultrafine particles from indoor sources on arterial stiffness - A randomized sham-controlled exposure study. Int J Hyg Environ Health. 2019;222(8):1115–32. https://doi.org/10.1016/j.ijheh.2019.08.002.
Pate M, Damarla V, Chi DS, Negi S, Krishnaswamy G. Endothelial cell biology: role in the inflammatory response. Adv Cin Chem. 2010;52:109–30.
Brook RD, Brook JR, Urch B, Vincent R, Rajagopalan S, Silverman F. Inhalation of fine particulate air pollution and ozone causes acute arterial vasoconstriction in healthy adults. Circulation. 2002;105(13):1534–6. https://doi.org/10.1161/01.cir.0000013838.94747.64.
Daiber A, Steven S, Weber A, Shuvaev VV, Muzykantov VR, Laher I, et al. Targeting vascular (endothelial) dysfunction. Br J Pharmacol. 2017;174(12):1591–619. https://doi.org/10.1111/bph.13517.
Lawal AO, Davids LM, Marnewick JL. Diesel exhaust particles and endothelial cells dysfunction: An update. Toxicol in Vitro. 2016;32:92–104. https://doi.org/10.1016/j.tiv.2015.12.015.
Fang X, Li R, Xu Q, Bottai M, Fang F, Cao Y. A Two-Stage Method to Estimate the Contribution of Road Traffic to PM(2).(5) Concentrations in Beijing, China. Int J Environ Res Public Health. 2016;13:1. https://doi.org/10.3390/ijerph13010124.
Ito K, Xue N, Thurston GJAE. Spatial variation of PM2.5 chemical species and source-apportioned mass concentrations in New York City. Atmos Environ. 2004;38(31):5269–82. https://doi.org/10.1016/j.atmosenv.2004.02.063.
Chuersuwan N, Nimrat S, Lekphet S, Kerdkumrai T. Levels and major sources of PM2.5 and PM10 in Bangkok Metropolitan Region. Environ Int. 2008;34(5):671–7. https://doi.org/10.1016/j.envint.2007.12.018.
Miller MR, Newby DE. Air pollution and cardiovascular disease: car sick. Cardiovasc Res. 2020;116(2):279–94. https://doi.org/10.1093/cvr/cvz228.
Choi SH, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, et al. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ Res. 2009;104(12):1355–63. https://doi.org/10.1161/circresaha.108.192880.
Geng J, Liu H, Ge P, Hu T, Zhang Y, Zhang X, et al. PM2.5 promotes plaque vulnerability at different stages of atherosclerosis and the formation of foam cells via TLR4/MyD88/NFkappaB pathway. Ecotoxicol Environ Saf. 2019;176:76–84. https://doi.org/10.1016/j.ecoenv.2019.03.068.
Liu J, Liang S, Du Z, Zhang J, Sun B, Zhao T, et al. PM(2.5) aggravates the lipid accumulation, mitochondrial damage and apoptosis in macrophage foam cells. Environ Pollut. 2019;249:482–90. https://doi.org/10.1016/j.envpol.2019.03.045.
Holme SAN, Sigsgaard T. Effects of particulate matter on atherosclerosis: a link via high-density lipoprotein (HDL) functionality? Part Fibre Toxicol. 2020;17(1):36. https://doi.org/10.1186/s12989-020-00367-x.
Doran AC, Yurdagul A Jr, Tabas I. Efferocytosis in health and disease. Nat Rev Immunol. 2020;20(4):254–67. https://doi.org/10.1038/s41577-019-0240-6.
Wan Q, Liu Z, Yang Y. Puerarin inhibits vascular smooth muscle cells proliferation induced by fine particulate matter via suppressing of the p38 MAPK signaling pathway. BMC Complement Altern Med. 2018;18(1):146. https://doi.org/10.1186/s12906-018-2206-9.
Cao Y, Long J, Ji Y, Chen G, Shen Y, Gong Y, et al. Foam cell formation by particulate matter (PM) exposure: a review. Inhal Toxicol. 2016;28(13):583–90. https://doi.org/10.1080/08958378.2016.1236157.
Perez L, Wolf K, Hennig F, Penell J, Basagana X, Foraster M, et al. Air pollution and atherosclerosis: a cross-sectional analysis of four European cohort studies in the ESCAPE study. Environ Health Perspect. 2015;123(6):597–605. https://doi.org/10.1289/ehp.1307711.
Hartiala J, Breton CV, Tang WH, Lurmann F, Hazen SL, Gilliland FD, et al. Ambient Air Pollution Is Associated With the Severity of Coronary Atherosclerosis and Incident Myocardial Infarction in Patients Undergoing Elective Cardiac Evaluation. J Am Heart Assoc. 2016;5:8. https://doi.org/10.1161/jaha.116.003947.
Kalsch H, Hennig F, Moebus S, Mohlenkamp S, Dragano N, Jakobs H, et al. Are air pollution and traffic noise independently associated with atherosclerosis: the Heinz Nixdorf Recall Study. Eur Heart J. 2014;35(13):853–60. https://doi.org/10.1093/eurheartj/eht426.
Kaufman JD, Adar SD, Barr RG, Budoff M, Burke GL, Curl CL, et al. Association between air pollution and coronary artery calcification within six metropolitan areas in the USA (the Multi-Ethnic Study of Atherosclerosis and Air Pollution): a longitudinal cohort study. Lancet. 2016;388(10045):696–704. https://doi.org/10.1016/s0140-6736(16)00378-0.
Ranzani OT, Mila C, Sanchez M, Bhogadi S, Kulkarni B, Balakrishnan K, et al. Association between ambient and household air pollution with carotid intima-media thickness in peri-urban South India: CHAI-Project. Int J Epidemiol. 2020;49(1):69–79. https://doi.org/10.1093/ije/dyz208.
Akintoye E, Shi L, Obaitan I, Olusunmade M, Wang Y, Newman JD, et al. Association between fine particulate matter exposure and subclinical atherosclerosis: A meta-analysis. Eur J Prev Cardiol. 2016;23(6):602–12. https://doi.org/10.1177/2047487315588758.
Bell G, Mora S, Greenland P, Tsai M, Gill E, Kaufman JD. Association of Air Pollution Exposures With High-Density Lipoprotein Cholesterol and Particle Number: The Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37(5):976–82. https://doi.org/10.1161/atvbaha.116.308193.
Wu S, Yang D, Wei H, Wang B, Huang J, Li H, et al. Association of chemical constituents and pollution sources of ambient fine particulate air pollution and biomarkers of oxidative stress associated with atherosclerosis: A panel study among young adults in Beijing, China. Chemosphere. 2015;135:347–53. https://doi.org/10.1016/j.chemosphere.2015.04.096.
Wang M, Hou ZH, Xu H, Liu Y, Budoff MJ, Szpiro AA, et al. Association of Estimated Long-term Exposure to Air Pollution and Traffic Proximity With a Marker for Coronary Atherosclerosis in a Nationwide Study in China. JAMA Net Open. 2019;2(6):e196553. https://doi.org/10.1001/jamanetworkopen.2019.6553.
Su TC, Hwang JJ, Shen YC, Chan CC. Carotid Intima-Media Thickness and Long-Term Exposure to Traffic-Related Air Pollution in Middle-Aged Residents of Taiwan: A Cross-Sectional Study. Environ Health Perspect. 2015;123(8):773–8. https://doi.org/10.1289/ehp.1408553.
Ramanathan G, Yin F, Speck M, Tseng CH, Brook JR, Silverman F, et al. Effects of urban fine particulate matter and ozone on HDL functionality. Part Fibre Toxicol. 2016;13(1):26. https://doi.org/10.1186/s12989-016-0139-3.
Adar SD, Sheppard L, Vedal S, Polak JF, Sampson PD, Diez Roux AV, et al. Fine particulate air pollution and the progression of carotid intima-medial thickness: a prospective cohort study from the multi-ethnic study of atherosclerosis and air pollution. PLoS Med. 2013;10(4):e1001430. https://doi.org/10.1371/journal.pmed.1001430.
Kim SY, Sheppard L, Kaufman JD, Bergen S, Szpiro AA, Larson TV, et al. Individual-level concentrations of fine particulate matter chemical components and subclinical atherosclerosis: a cross-sectional analysis based on 2 advanced exposure prediction models in the multi-ethnic study of atherosclerosis. Am J Epidemiol. 2014;180(7):718–28. https://doi.org/10.1093/aje/kwu186.
Tonne C, Salmon M, Sanchez M, Sreekanth V, Bhogadi S, Sambandam S, et al. Integrated assessment of exposure to PM2.5 in South India and its relation with cardiovascular risk: Design of the CHAI observational cohort study. Int J Hyg Environ Health. 2017;220(6):1081–8. https://doi.org/10.1016/j.ijheh.2017.05.005.
Hennig F, Moebus S, Reinsch N, Budde T, Erbel R, Jockel KH, et al. Investigation of air pollution and noise on progression of thoracic aortic calcification: results of the Heinz Nixdorf Recall Study. Eur J Prev Cardiol. 2019:2047487319854818. https://doi.org/10.1177/2047487319854818.
Gondalia R, Holliday KM, Baldassari A, Justice AE, Stewart JD, Liao D, et al. Leukocyte Traits and Exposure to Ambient Particulate Matter Air Pollution in the Women's Health Initiative and Atherosclerosis Risk in Communities Study. Environ Health Perspect. 2020;128(1):17004. https://doi.org/10.1289/ehp5360.
Lenters V, Uiterwaal CS, Beelen R, Bots ML, Fischer P, Brunekreef B, et al. Long-term exposure to air pollution and vascular damage in young adults. Epidemiology. 2010;21(4):512–20. https://doi.org/10.1097/EDE.0b013e3181dec3a7.
O'Neill MS, Diez-Roux AV, Auchincloss AH, Shen M, Lima JA, Polak JF, et al. Long-term exposure to airborne particles and arterial stiffness: the Multi-Ethnic Study of Atherosclerosis (MESA). Environ Health Perspect. 2011;119(6):844–51. https://doi.org/10.1289/ehp.0901524.
Huynh Q, Marwick TH, Venkataraman P, Knibbs LD, Johnston FH, Negishi K. Long-term exposure to ambient air pollution is associated with coronary artery calcification among asymptomatic adults. Eur Heart J Cardiovasc Imaging. 2020. https://doi.org/10.1093/ehjci/jeaa073.
Fuks K, Moebus S, Hertel S, Viehmann A, Nonnemacher M, Dragano N, et al. Long-term urban particulate air pollution, traffic noise, and arterial blood pressure. Environ Health Perspect. 2011;119(12):1706–11. https://doi.org/10.1289/ehp.1103564.
Aguilera I, Dratva J, Caviezel S, Burdet L, de Groot E, Ducret-Stich RE, et al. Particulate Matter and Subclinical Atherosclerosis: Associations between Different Particle Sizes and Sources with Carotid Intima-Media Thickness in the SAPALDIA Study. Environ Halth Perspect. 2016;124(11):1700–6. https://doi.org/10.1289/ehp161.
Sun M, Kaufman JD, Kim SY, Larson TV, Gould TR, Polak JF, et al. Particulate matter components and subclinical atherosclerosis: common approaches to estimating exposure in a Multi-Ethnic Study of Atherosclerosis cross-sectional study. Environ Health. 2013;12:39. https://doi.org/10.1186/1476-069x-12-39.
Yang S, Lee SP, Park JB, Lee H, Kang SH, Lee SE, et al. PM2.5 concentration in the ambient air is a risk factor for the development of high-risk coronary plaques. Eur Heart J Cardiovasc Imaging. 2019;20(12):1355–64. https://doi.org/10.1093/ehjci/jez209.
Duan C, Talbott EO, Broadwin R, Brooks M, Matthews K, Barinas-Mitchell E. Residential Exposure to PM2.5 and Ozone and Progression of Subclinical Atherosclerosis Among Women Transitioning Through Menopause: The Study of Women's Health Across the Nation. J Women's Health. 2019;28(6):802–11. https://doi.org/10.1089/jwh.2018.7182.
Hoffmann B, Moebus S, Mohlenkamp S, Stang A, Lehmann N, Dragano N, et al. Residential exposure to traffic is associated with coronary atherosclerosis. Circulation. 2007;116(5):489–96. https://doi.org/10.1161/circulationaha.107.693622.
Dorans KS, Wilker EH, Li W, Rice MB, Ljungman PL, Schwartz J, et al. Residential proximity to major roads, exposure to fine particulate matter and aortic calcium: the Framingham Heart Study, a cohort study. BMJ Open. 2017;7(3):e013455. https://doi.org/10.1136/bmjopen-2016-013455.
Zhang Z, Chang LY, Lau AKH, Chan TC, Chieh Chuang Y, Chan J, et al. Satellite-based estimates of long-term exposure to fine particulate matter are associated with C-reactive protein in 30 034 Taiwanese adults. Int J Epidemiol. 2017;46(4):1126–36. https://doi.org/10.1093/ije/dyx069.
Breton CV, Wang X, Mack WJ, Berhane K, Lopez M, Islam TS, et al. Childhood air pollutant exposure and carotid artery intima-media thickness in young adults. Circulation. 2012;126(13):1614–20. https://doi.org/10.1161/circulationaha.112.096164.
Duan C, Talbott E, Brooks M, Park SK, Broadwin R, Matthews K, et al. Five-year exposure to PM(2.5) and ozone and subclinical atherosclerosis in late midlife women: The Study of Women's Health Across the Nation. Int J Hyg Environ Health. 2019;222(2):168–76. https://doi.org/10.1016/j.ijheh.2018.09.001.
Bauer M, Moebus S, Möhlenkamp S, Dragano N, Nonnemacher M, Fuchsluger M, et al. Urban particulate matter air pollution is associated with subclinical atherosclerosis: results from the HNR (Heinz Nixdorf Recall) study. J Am Coll Cardiol. 2010;56(22):1803–8. https://doi.org/10.1016/j.jacc.2010.04.065.
Baumgartner J, Carter E, Schauer JJ, Ezzati M, Daskalopoulou SS, Valois MF, et al. Household air pollution and measures of blood pressure, arterial stiffness and central haemodynamics. Heart. 2018;104(18):1515–21. https://doi.org/10.1136/heartjnl-2017-312595.
Painschab MS, Davila-Roman VG, Gilman RH, Vasquez-Villar AD, Pollard SL, Wise RA, et al. Chronic exposure to biomass fuel is associated with increased carotid artery intima-media thickness and a higher prevalence of atherosclerotic plaque. Heart. 2013;99(14):984–91. https://doi.org/10.1136/heartjnl-2012-303440.
Künzli N, Jerrett M, Mack WJ, Beckerman B, LaBree L, Gilliland F, et al. Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect. 2005;113(2):201–6. https://doi.org/10.1289/ehp.7523.
Allen RW, Criqui MH, Diez Roux AV, Allison M, Shea S, Detrano R, et al. Fine particulate matter air pollution, proximity to traffic, and aortic atherosclerosis. Epidemiology. 2009;20(2):254–64. https://doi.org/10.1097/EDE.0b013e31819644cc.
Diez Roux AV, Auchincloss AH, Franklin TG, Raghunathan T, Barr RG, Kaufman J, et al. Long-term exposure to ambient particulate matter and prevalence of subclinical atherosclerosis in the Multi-Ethnic Study of Atherosclerosis. Am J Epidemiol. 2008;167(6):667–75. https://doi.org/10.1093/aje/kwm359.
Pang Y, Zhang B, Xing D, Shang J, Chen F, Kang H, et al. Increased risk of carotid atherosclerosis for long-term exposure to indoor coal-burning pollution in rural area, Hebei Province, China. Environ Pollut. 2019;255(Pt 2):113320. https://doi.org/10.1016/j.envpol.2019.113320.
Lee SH, Kim JH, Kang MJ, Lee YA, Won Yang S, Shin CH. Implications of nocturnal hypertension in children and adolescents with type 1 diabetes. Diabetes Care. 2011;34(10):2180–5. https://doi.org/10.2337/dc11-0830.
Du X, Jiang S, Zeng X, Zhang J, Pan K, Zhou J, et al. Air pollution is associated with the development of atherosclerosis via the cooperation of CD36 and NLRP3 inflammasome in ApoE(-/-) mice. Toxicol Lett. 2018;290:123–32. https://doi.org/10.1016/j.toxlet.2018.03.022.
Wan Q, Liu Z, Yang M, Wu J. Acceleratory effects of ambient fine particulate matter on the development and progression of atherosclerosis in apolipoprotein E knockout mice by down-regulating CD4(+)CD25(+)Foxp3(+) regulatory T cells. Toxicol Lett. 2019;316:27–34. https://doi.org/10.1016/j.toxlet.2019.09.005.
Wan Q, Cui X, Shao J, Zhou F, Jia Y, Sun X, et al. Beijing ambient particle exposure accelerates atherosclerosis in ApoE knockout mice by upregulating visfatin expression. Cell Stress Chaperones. 2014;19(5):715–24. https://doi.org/10.1007/s12192-014-0499-2.
Chen T, Jia G, Wei Y, Li J. Beijing ambient particle exposure accelerates atherosclerosis in ApoE knockout mice. Toxicol Lett. 2013;223(2):146–53. https://doi.org/10.1016/j.toxlet.2013.09.004.
Zhao J, Mi X, Zhao L, Midgley AC, Tang H, Tian M, et al. Validation of PM(2.5) model particle through physicochemical evaluation and atherosclerotic plaque formation in ApoE(-/-) mice. Ecotoxicol Environ Saf. 2020;192:110308. https://doi.org/10.1016/j.ecoenv.2020.110308.
Quan C, Sun Q, Lippmann M, Chen LC. Comparative effects of inhaled diesel exhaust and ambient fine particles on inflammation, atherosclerosis, and vascular dysfunction. Inhal Toxicol. 2010;22(9):738–53. https://doi.org/10.3109/08958371003728057.
Wang S, Wang F, Yang L, Li Q, Huang Y, Cheng Z, et al. Effects of coal-fired PM(2.5) on the expression levels of atherosclerosis-related proteins and the phosphorylation level of MAPK in ApoE(-/-) mice. BMC Pharmacol Toxicol. 2020;21(1):34. https://doi.org/10.1186/s40360-020-00411-8.
Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, et al. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. Jama. 2005;294(23):3003–10. https://doi.org/10.1001/jama.294.23.3003.
Araujo JA, Barajas B, Kleinman M, Wang X, Bennett BJ, Gong KW, et al. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res. 2008;102(5):589–96. https://doi.org/10.1161/circresaha.107.164970.
Chen LC, Nadziejko C. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. V. CAPs exacerbate aortic plaque development in hyperlipidemic mice. Inhal Toxicol. 2005;17(4-5):217–24. https://doi.org/10.1080/08958370590912815.
Zhu X, Zhao P, Lu Y, Huo L, Bai M, Yu F, et al. Potential injurious effects of the fine particulate PM2.5 on the progression of atherosclerosis in apoE-deficient mice by activating platelets and leukocytes. Arch Med Sci. 2019;15(1):250–61. https://doi.org/10.5114/aoms.2018.81039.
Bai Y, Sun Q. Fine particulate matter air pollution and atherosclerosis: Mechanistic insights. Biochim Biophys Acta. 2016;1860(12):2863–8. https://doi.org/10.1016/j.bbagen.2016.04.030.
Grahame TJ, Schlesinger RB. Oxidative stress-induced telomeric erosion as a mechanism underlying airborne particulate matter-related cardiovascular disease. Part Fibre Toxicol. 2012;9:21. https://doi.org/10.1186/1743-8977-9-21.
Bourdrel T, Bind MA, Béjot Y, Morel O, Argacha JF. Cardiovascular effects of air pollution. Arch Cardiovasc Dis. 2017;110(11):634–42. https://doi.org/10.1016/j.acvd.2017.05.003.
Dai J, Chen W, Lin Y, Wang S, Guo X, Zhang QQ. Exposure to Concentrated Ambient Fine Particulate Matter Induces Vascular Endothelial Dysfunction via miR-21. Int J Biol Sci. 2017;13(7):868–77. https://doi.org/10.7150/ijbs.19868.
Guo S, Som AT, Arai K, Lo EH. Effects of angiotensin-II on brain endothelial cell permeability via PPARalpha regulation of para- and trans-cellular pathways. Brain Res. 2019;1722:146353. https://doi.org/10.1016/j.brainres.2019.146353.
Han W, Dan W, Shuo Y, Fang Z, Wenjun D. Oxidative stress induced by urban fine particles in cultured EA.hy926 cells. Hum Exp Toxicol. 2011;30(7):579–90. https://doi.org/10.1177/0960327110374207.
Wang Y, Tang M. PM2.5 induces autophagy and apoptosis through endoplasmic reticulum stress in human endothelial cells. Sci Total Environ. 2020;710:136397. https://doi.org/10.1016/j.scitotenv.2019.136397.
Wang Y, Tang M. PM2.5 induces ferroptosis in human endothelial cells through iron overload and redox imbalance. Environ Pollut. 2019;254(Pt A):112937. https://doi.org/10.1016/j.envpol.2019.07.105.
Sharma K, Lee HH, Gong DS, Park SH, Yi E, Schini-Kerth V, et al. Fine air pollution particles induce endothelial senescence via redox-sensitive activation of local angiotensin system. Environ Pollut. 2019;252(Pt A):317–29. https://doi.org/10.1016/j.envpol.2019.05.066.
Long YM, Yang XZ, Yang QQ, Clermont AC, Yin YG, Liu GL, et al. PM2.5 induces vascular permeability increase through activating MAPK/ERK signaling pathway and ROS generation. J Hazard Mater. 2020;386:121659. https://doi.org/10.1016/j.jhazmat.2019.121659.
Montiel-Dávalos A, Ibarra-Sánchez Mde J, Ventura-Gallegos JL, Alfaro-Moreno E, López-Marure R. Oxidative stress and apoptosis are induced in human endothelial cells exposed to urban particulate matter. Toxicol in Vitro. 2010;24(1):135–41. https://doi.org/10.1016/j.tiv.2009.08.004.
Xu X, Xu H, Qimuge A, Liu S, Wang H, Hu M, et al. MAPK/AP-1 pathway activation mediates AT1R upregulation and vascular endothelial cells dysfunction under PM2.5 exposure. Ecotoxicol Environ Saf. 2019;170:188–94. https://doi.org/10.1016/j.ecoenv.2018.11.124.
Chen S, Wu X, Hu J, Dai G, Rong A, Guo G. PM2.5 exposure decreases viability, migration and angiogenesis in human umbilical vein endothelial cells and human microvascular endothelial cells. Mol Med Rep. 2017;16 3:2425-2430; doi: 10.3892/mmr.2017.6877.
Rui W, Guan L, Zhang F, Zhang W, Ding W. PM2.5-induced oxidative stress increases adhesion molecules expression in human endothelial cells through the ERK/AKT/NF-kappaB-dependent pathway. J Appl Toxicol. 2016;36(1):48–59. https://doi.org/10.1002/jat.3143.
Zhu F, Cheng H, Lei R, Shen C, Liu J, Hou L, et al. Effects of cooking oil fume derived fine particulate matter on blood vessel formation through the VEGF/VEGFR2/MEK1/2/ERK1/2/mTOR pathway in human umbilical vein endothelial cells. Environ Toxicol Pharmacol. 2019;69:112–9. https://doi.org/10.1016/j.etap.2019.04.008.
Le Y, Hu X, Zhu J, Wang C, Yang Z, Lu D. Ambient fine particulate matter induces inflammatory responses of vascular endothelial cells through activating TLR-mediated pathway. Toxicol Ind Health. 2019;35(10):670–8. https://doi.org/10.1177/0748233719871778.
Yin J, Xia W, Li Y, Guo C, Zhang Y, Huang S, et al. COX-2 mediates PM2.5-induced apoptosis and inflammation in vascular endothelial cells. Am J Transl Res. 2017;9(9):3967–76.
Wang JS, Tseng CY, Chao MW. Diesel Exhaust Particles Contribute to Endothelia Apoptosis via Autophagy Pathway. Toxicol Sci. 2017;156(1):72–83. https://doi.org/10.1093/toxsci/kfw237.
Li R, Ning Z, Cui J, Yu F, Sioutas C, Hsiai T. Diesel exhaust particles modulate vascular endothelial cell permeability: implication of ZO-1 expression. Toxicol Lett. 2010;197(3):163–8. https://doi.org/10.1016/j.toxlet.2010.05.017.
Zhao J, Xie Y, Jiang R, Kan H, Song W. Effects of atorvastatin on fine particle-induced inflammatory response, oxidative stress and endothelial function in human umbilical vein endothelial cells. Hum Exp Toxicol. 2011;30(11):1828–39. https://doi.org/10.1177/0960327111401050.
Lai CY, Lai CH, Chuang HC, Pan CH, Yen CC, Lin WY, et al. Physicochemistry and cardiovascular toxicity of metal fume PM2.5: a study of human coronary artery endothelial cells and welding workers. Sci Rep. 2016;6:33515. https://doi.org/10.1038/srep33515.
Montiel-Dávalos A, Alfaro-Moreno E, López-Marure R. PM2.5 and PM10 induce the expression of adhesion molecules and the adhesion of monocytic cells to human umbilical vein endothelial cells. Inhal Toxicol. 2007;19(Suppl 1):91–8. https://doi.org/10.1080/08958370701495212.
Zhou Z, Shao T, Qin M, Miao X, Chang Y, Sheng W, et al. The effects of autophagy on vascular endothelial cells induced by airborne PM2.5. J Environ Sci. 2018;66:182–7. https://doi.org/10.1016/j.jes.2017.05.019.
Wang FF, Geng CM, Hao WD, Zhao YD, Li Q, Wang HM, et al. The Cellular Toxicity of PM2.5 Emitted from Coal Combustion in Human Umbilical Vein Endothelial Cells. Biomed Environ Sci. 2016;29(2):107–16. https://doi.org/10.3967/bes2016.012.
Wu S, Wang B, Yang D, Wei H, Li H, Pan L, et al. Ambient particulate air pollution and circulating antioxidant enzymes: A repeated-measure study in healthy adults in Beijing, China. Environ Pollut. 2016;208(Pt A):16–24. https://doi.org/10.1016/j.envpol.2015.06.002.
Feng B, Qi R, Gao J, Wang T, Xu H, Zhao Q, et al. Exercise training prevented endothelium dysfunction from particulate matter instillation in Wistar rats. Sci Total Environ. 2019;694:133674. https://doi.org/10.1016/j.scitotenv.2019.133674.
Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative Stress in Atherosclerosis. Curr Atheroscler Rep. 2017;19(11):42. https://doi.org/10.1007/s11883-017-0678-6.
Miao X, Li W, Niu B, Li J, Sun J, Qin M, et al. Mitochondrial dysfunction in endothelial cells induced by airborne fine particulate matter (<2.5 mum). J Appl Toxicol. 2019;39(10):1424–32. https://doi.org/10.1002/jat.3828.
Kim D, Mouritzen U, Larsen BD, Roy S. Inhibition of Cx43 gap junction uncoupling prevents high glucose-induced apoptosis and reduces excess cell monolayer permeability in retinal vascular endothelial cells. Exp Eye Res. 2018;173:85–90. https://doi.org/10.1016/j.exer.2018.05.003.
Hennig B, Chow CK. Lipid peroxidation and endothelial cell injury: implications in atherosclerosis. Free Radic Biol Med. 1988;4(2):99–106. https://doi.org/10.1016/0891-5849(88)90070-6.
Krouwer VJ, Hekking LH, Langelaar-Makkinje M, Regan-Klapisz E, Post JA. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc Cell. 2012;4(1):12. https://doi.org/10.1186/2045-824x-4-12.
Grimsley-Myers CM, Isaacson RH, Cadwell CM, Campos J, Hernandes MS, Myers KR, et al. VE-cadherin endocytosis controls vascular integrity and patterning during development. J Cell Biol. 2020;219:5. https://doi.org/10.1083/jcb.201909081.
Wang T, Shimizu Y, Wu X, Kelly GT, Xu X, Wang L, et al. Particulate matter disrupts human lung endothelial cell barrier integrity via Rho-dependent pathways. Pulm Circ. 2017;7(3):617–23. https://doi.org/10.1086/689906.
Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26(5):441–54. https://doi.org/10.1016/j.devcel.2013.08.020.
Marasciulo FL, Montagnani M, Potenza MA. Endothelin-1: the yin and yang on vascular function. Curr Med Chem. 2006;13(14):1655–65. https://doi.org/10.2174/092986706777441968.
Niu J, Wu J, Li X, Zhang F. Association between endothelin-1/endothelin receptor A and inflammation in mouse kidneys following acute ischemia/reperfusion. Mol Med Rep. 2015;11(5):3981–7. https://doi.org/10.3892/mmr.2014.3138.
Mazzuca MQ, Khalil RA. Vascular endothelin receptor type B: structure, function and dysregulation in vascular disease. Biochem Pharmacol. 2012;84(2):147–62. https://doi.org/10.1016/j.bcp.2012.03.020.
Gimbrone MA Jr, Garcia-Cardena G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. 2016;118(4):620–36. https://doi.org/10.1161/circresaha.115.306301.
Deweirdt J, Quignard JF, Crobeddu B, Baeza-Squiban A, Sciare J, Courtois A, et al. Involvement of oxidative stress and calcium signaling in airborne particulate matter - induced damages in human pulmonary artery endothelial cells. Toxicol in Vitro. 2017;45(Pt 3):340–50. https://doi.org/10.1016/j.tiv.2017.07.001.
Xiao X, Cao L, Wang R, Shen ZX, Cao YX. Airborne fine particulate matter alters the expression of endothelin receptors in rat coronary arteries. Environ Pollut. 2016;218:487–96. https://doi.org/10.1016/j.envpol.2016.07.028.
Chan EA, Buckley B, Farraj AK, Thompson LC. The heart as an extravascular target of endothelin-1 in particulate matter-induced cardiac dysfunction. Pharmacol Ther. 2016;165:63–78. https://doi.org/10.1016/j.pharmthera.2016.05.006.
Lin YJ, Kwok CF, Juan CC, Hsu YP, Shih KC, Chen CC, et al. Angiotensin II enhances endothelin-1-induced vasoconstriction through upregulating endothelin type A receptor. Biochem Biophys Res Commun. 2014;451(2):263–9. https://doi.org/10.1016/j.bbrc.2014.07.119.
Haberzettl P, O'Toole TE, Bhatnagar A, Conklin DJ. Exposure to Fine Particulate Air Pollution Causes Vascular Insulin Resistance by Inducing Pulmonary Oxidative Stress. Environ Health Perspect. 2016;124(12):1830–9. https://doi.org/10.1289/ehp212.
Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, et al. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28(11):1982–8. https://doi.org/10.1161/atvbaha.108.169722.
Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113(15):1888–904. https://doi.org/10.1161/circulationaha.105.563213.
Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation. 2004;109(21 Suppl 1):Ii27–33. https://doi.org/10.1161/01.CIR.0000129501.88485.1f.
Bayraktutan U. Endothelial progenitor cells: Potential novel therapeutics for ischaemic stroke. Pharmacol Res. 2019;144:181–91. https://doi.org/10.1016/j.phrs.2019.04.017.
Noor R, Shuaib U, Wang CX, Todd K, Ghani U, Schwindt B, et al. High-density lipoprotein cholesterol regulates endothelial progenitor cells by increasing eNOS and preventing apoptosis. Atherosclerosis. 2007;192(1):92–9. https://doi.org/10.1016/j.atherosclerosis.2006.06.023.
Haberzettl P, Conklin DJ, Abplanalp WT, Bhatnagar A, O'Toole TE. Inhalation of Fine Particulate Matter Impairs Endothelial Progenitor Cell Function Via Pulmonary Oxidative Stress. Arterioscler Thromb Vasc Biol. 2018;38(1):131–42. https://doi.org/10.1161/atvbaha.117.309971.
Haberzettl P, McCracken JP, Bhatnagar A, Conklin DJ. Insulin sensitizers prevent fine particulate matter-induced vascular insulin resistance and changes in endothelial progenitor cell homeostasis. Am J Physiol Heart Circ Physiol. 2016;310(11):H1423–38. https://doi.org/10.1152/ajpheart.00369.2015.
Cui Y, Sun Q, Liu Z. Ambient particulate matter exposure and cardiovascular diseases: a focus on progenitor and stem cells. J Cell Mol Med. 2016;20(5):782–93. https://doi.org/10.1111/jcmm.12822.
Jian D, Wang W, Zhou X, Jia Z, Wang J, Yang M, et al. Interferon-induced protein 35 inhibits endothelial cell proliferation, migration and re-endothelialization of injured arteries by inhibiting the nuclear factor-kappa B pathway. Acta Physiol (Ox). 2018;223(3):e13037. https://doi.org/10.1111/apha.13037.
Robertson S, Miller MR. Ambient air pollution and thrombosis. Part Fibre Toxicol. 2018;15(1):1. https://doi.org/10.1186/s12989-017-0237-x.
Esmon CT. Coagulation inhibitors in inflammation. Biochem Soc Trans. 2005;33(Pt 2):401–5. https://doi.org/10.1042/bst0330401.
Hu H, Wu J, Li Q, Asweto C, Feng L, Yang X, et al. Fine particulate matter induces vascular endothelial activation via IL-6 dependent JAK1/STAT3 signaling pathway. Toxicol Res. 2016;5(3):946–53. https://doi.org/10.1039/c5tx00351b.
Liang S, Zhao T, Hu H, Shi Y, Xu Q, Miller MR, et al. Repeat dose exposure of PM2.5 triggers the disseminated intravascular coagulation (DIC) in SD rats. Sci Total Environ. 2019;663:245–53. https://doi.org/10.1016/j.scitotenv.2019.01.346.
Kawecki C, Lenting PJ, Denis CV. von Willebrand factor and inflammation. J Thromb Haemost. 2017;15(7):1285–94. https://doi.org/10.1111/jth.13696.
Lordan R, Tsoupras A, Zabetakis I. Platelet activation and prothrombotic mediators at the nexus of inflammation and atherosclerosis: Potential role of antiplatelet agents. Blood Rev. 2020;100694. https://doi.org/10.1016/j.blre.2020.100694.
Ten Cate H, Hackeng TM, Garcia de Frutos P. Coagulation factor and protease pathways in thrombosis and cardiovascular disease. Thromb Haemost. 2017;117(7):1265–71. https://doi.org/10.1160/th17-02-0079.
Liu C, Cai J, Qiao L, Wang H, Xu W, Li H, et al. The Acute Effects of Fine Particulate Matter Constituents on Blood Inflammation and Coagulation. Environ Sci Technol. 2017;51(14):8128–37. https://doi.org/10.1021/acs.est.7b00312.
Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nature reviews Disease primers. 2019;5 1:56; doi: https://doi.org/10.1038/s41572-019-0106-z.
Tabas I, Bornfeldt KE. Intracellular and Intercellular Aspects of Macrophage Immunometabolism in Atherosclerosis. Circ Res. 2020;126(9):1209–27. https://doi.org/10.1161/circresaha.119.315939.
Goldberg AC, Leiter LA, Stroes ESG, Baum SJ, Hanselman JC, Bloedon LT, et al. Effect of Bempedoic Acid vs Placebo Added to Maximally Tolerated Statins on Low-Density Lipoprotein Cholesterol in Patients at High Risk for Cardiovascular Disease: The CLEAR Wisdom Randomized Clinical Trial. Jama. 2019;322(18):1780–8. https://doi.org/10.1001/jama.2019.16585.
Hansson GK, Jonasson L. The discovery of cellular immunity in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2009;29(11):1714–7. https://doi.org/10.1161/atvbaha.108.179713.
Huang L, Chambliss KL, Gao X, Yuhanna IS, Behling-Kelly E, Bergaya S, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019;569(7757):565–9. https://doi.org/10.1038/s41586-019-1140-4.
Mehta JL, Chen J, Hermonat PL, Romeo F, Novelli G. Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders. Cardiovasc Res. 2006;69(1):36–45. https://doi.org/10.1016/j.cardiores.2005.09.006.
Li D, Mehta JL. Intracellular signaling of LOX-1 in endothelial cell apoptosis. Circ Res. 2009;104(5):566–8. https://doi.org/10.1161/circresaha.109.194209.
Akhmedov A, Rozenberg I, Paneni F, Camici GG, Shi Y, Doerries C, et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur Heart J. 2014;35(40):2839–48. https://doi.org/10.1093/eurheartj/eht532.
Markstad H, Edsfeldt A, Yao Mattison I, Bengtsson E, Singh P, Cavalera M, et al. High Levels of Soluble Lectinlike Oxidized Low-Density Lipoprotein Receptor-1 Are Associated With Carotid Plaque Inflammation and Increased Risk of Ischemic Stroke. J Ame Heart Assoc. 2019;8(4):e009874. https://doi.org/10.1161/JAHA.118.009874.
Lund AK, Lucero J, Harman M, Madden MC, McDonald JD, Seagrave JC, et al. The oxidized low-density lipoprotein receptor mediates vascular effects of inhaled vehicle emissions. Am J Respir Crit Care Med. 2011;184(1):82–91. https://doi.org/10.1164/rccm.201012-1967OC.
Kodavanti UP, Thomas R, Ledbetter AD, Schladweiler MC, Shannahan JH, Wallenborn JG, et al. Vascular and cardiac impairments in rats inhaling ozone and diesel exhaust particles. Environ Health Perspect. 2011;119(3):312–8. https://doi.org/10.1289/ehp.1002386.
Lucero J, Suwannasual U, Herbert LM, McDonald JD, Lund AK. The role of the lectin-like oxLDL receptor (LOX-1) in traffic-generated air pollution exposure-mediated alteration of the brain microvasculature in Apolipoprotein (Apo) E knockout mice. Inhal Toxicol. 2017;29(6):266–81. https://doi.org/10.1080/08958378.2017.1357774.
Liu J, Liang S, Du Z, Zhang J, Sun B, Zhao T, et al. PM2.5 aggravates the lipid accumulation, mitochondrial damage and apoptosis in macrophage foam cells. Environ Pollut. 2019;249:482–90. https://doi.org/10.1016/j.envpol.2019.03.045.
WHO, Burden of disease from the joint effects of household and ambient air pollution. 2018, Available from: www.who.int/airpollution/data/.
Brauner EV, Forchhammer L, Moller P, Barregard L, Gunnarsen L, Afshari A, et al. Indoor particles affect vascular function in the aged: an air filtration-based intervention study. Am J Respir Crit Care Med. 2008;177(4):419–25. https://doi.org/10.1164/rccm.200704-632OC.
Ding R, Zhang C, Zhu X, Cheng H, Zhu F, Xu Y, et al. ROS-AKT-mTOR axis mediates autophagy of human umbilical vein endothelial cells induced by cooking oil fumes-derived fine particulate matters in vitro. Free Radic Biol Med. 2017;113:452–60. https://doi.org/10.1016/j.freeradbiomed.2017.10.386.
Acknowledgments
Not applicable.
Funding
This work was supported by National Key Research and Development Program of China (2017YFC0211600, 2017YFC0211602, 2017YFC0211606), National Natural Science Foundation of China (91943301), Beijing Nova Program (Z181100006218027) and Beijing Outstanding Talent Training Program, Beijing Natural Science Foundation Program and Scientific Research Key Program of Beijing Municipal Commission of Education (KZ202110025040).
Author information
Authors and Affiliations
Contributions
All authors contributed to the design, concept and grammar in this article. Junchao Duan and Zhiwei Sun revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declared they had no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, S., Zhang, J., Ning, R. et al. The critical role of endothelial function in fine particulate matter-induced atherosclerosis. Part Fibre Toxicol 17, 61 (2020). https://doi.org/10.1186/s12989-020-00391-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12989-020-00391-x