
Abstract
While the impact of hemorrhagic and ischemic strokes on the blood–brain barrier has been extensively studied, the impact of these types of stroke on the choroid plexus, site of the blood-CSF barrier, has received much less attention. The purpose of this review is to examine evidence of choroid plexus injury in clinical and preclinical studies of intraventricular hemorrhage, subarachnoid hemorrhage, intracerebral hemorrhage and ischemic stroke. It then discusses evidence that the choroid plexuses are important in the response to brain injury, with potential roles in limiting damage. The overall aim of the review is to highlight deficiencies in our knowledge on the impact of hemorrhagic and ischemic strokes on the choroid plexus, particularly with reference to intraventricular hemorrhage, and to suggest that a greater understanding of the response of the choroid plexus to stroke may open new avenues for brain protection.
Similar content being viewed by others

Background
The choroid plexuses (CPs) are present in the lateral, third and fourth ventricles of the brain (LVCP, 3rd CP, 4th CP). Each CP is comprised of epithelial cells surrounding a richly vascularized core [1]. Compared to the cerebral capillaries that form the blood–brain barrier (BBB), CP capillaries are leakier with the endothelial cells having fenestrations, reflecting the role of the CPs in CSF secretion [2–6]. The CPs are the site of the blood-CSF barrier, with the CP epithelial cells being linked by tight junctions that limit paracellular diffusion [1, 7].
Many neurological conditions result in BBB dysfunction, including ischemic and hemorrhagic strokes [8–11]. That dysfunction participates in brain injury by, for example, causing vasogenic brain edema, allowing the entry of potentially neurotoxic compounds from blood to brain, and promoting leukocyte infiltration and neuroinflammation. In contrast to the wealth of studies on BBB injury, much less is known about whether CP injury occurs after stroke [12, 13]. One purpose of the present review is to discuss what is currently known about such injury. It addresses hemorrhagic and ischemic stroke, with a particular focus on the former including intraventricular hemorrhage (IVH), subarachnoid hemorrhage (SAH) and intracerebral hemorrhage (ICH).
Another purpose is to review a body of evidence that the CPs respond to injury (that may be distant from the CP) by releasing factors that protect the brain [14–16]. This has led to experimental studies with CP transplantation for a variety of conditions [17–19]. That section also addresses a related topic, namely the role of the CP in leukocyte entry into the brain after injury. Results on the protective effects of the CP in brain injury/disease may help to inform studies on the role of cerebral endothelial cells.
Choroid plexus as a site of injury
This section discusses evidence of CP injury in different forms of stroke in humans and animals. In human studies, it discusses morphological (post-mortem), imaging and permeability data to assess injury. One advantage of studies on the CP compared to the BBB is that the CSF (unlike the brain) can be sampled in patients, although it should be noted that CSF composition can reflect changes at both the blood-CSF and blood–brain barriers. In animal studies, the CP itself, as well as the CSF, can be sampled. Inflammatory changes will be discussed in the next section (CP as a responder to injury). Additionally, this section examines whether hemorrhage (particularly bleeding within the ventricles) can cause CP injury. It does not discuss the CP being the site of initial bleeding, although some evidence indicates CP hemorrhage is a major cause of IVH in babies born at full term [20] and can occur at other ages [21, 22].
Intraventricular hemorrhage
IVH commonly occurs in premature infants as a result of germinal matrix hemorrhage. It is a major cause of cerebral palsy in such infants and is often associated with later development of hydrocephalus [23]. In adults, intraventricular extension of bleeding occurs in ~50% of patients with ICH and ~45% of patients with SAH [24, 25]. Such ventricular extension is a risk factor for poor outcome after both ICH and SAH [25–27]. ICH is known to cause perihematomal tissue damage [28] and BBB dysfunction [10]. Clot-derived factors, including hemoglobin, iron and thrombin, play a major role in ICH-induced injury [28]. Despite this evidence in ICH and the proximity of intraventricular blood to the CPs, we did not find clinical studies examining the effects of IVH on the CP epithelial morphology in the literature. Clinically, IVH results in changes in CSF protein concentrations (e.g. [29–33]). While changes in blood-CSF barrier function are one parameter that can affect those concentrations, assessing barrier function from such data is very difficult, particularly in the setting of IVH. Thus, for example, total CSF protein is elevated after IVH [29], but this may reflect protein originating from the initial hemorrhage, protein influx at the BBB as well as the blood-CSF barrier and production from brain parenchyma. Examining the CSF/plasma ratio of specific proteins, such as albumin [30, 32], helps address the latter point, but albumin will still enter CSF from the initial IVH as well as any potential BBB disruption.
In animals, Simard et al. [34] examined a rat IVH model at 2 days and found induction of inflammatory signaling (nuclear factor κB activation) within the CP along with a significant intracellular uptake of IgG. Furthermore, Gram et al. [35] found a marked inflammatory response in a rabbit preterm model of IVH at 1 and 3 days. This was accompanied by CP cell death, caspase activation and pronounced ultrastructural changes. Similar inductions in inflammatory mediators and cell death were obtained in vitro when CP epithelial cells were exposed to met-hemoglobin, heme or CSF from preterm babies with IVH [35]. The hemoglobin scavenger, haptoglobin, prevented the adverse effects of IVH in vivo and hemoglobin or IVH CSF in vitro [35].
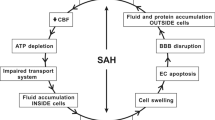
In contrast, Pang et al. [36] found only subtle changes in CP morphology at 3 months in a dog IVH model. In comparison, the ependymal lining of the lateral ventricles was destroyed or interrupted in many areas. Interestingly, they also examined the effects of urokinase in their model to lyse the IVH and found that this protected the ependyma but also resulted in CP atrophy in some animals for unknown reasons. While it is possible that there may be species differences in the effects of IVH on the CP, a more likely reason for the differences between the studies are the time points examined. As described below, in SAH and ischemia there is evidence of acute injury followed by a recovery in CP structure, although the latter may be accompanied by a reduction in CP size. Acute studies (1–3 days) in larger species and chronic studies in rodents would be informative (Fig. 1).

In general, there is a surprising lack of data on the effects of intraventricular and subarachnoid hemorrhage on choroid plexus (CP) injury, particularly in humans. In intraventricular hemorrhage, there are studies indicating acute CP damage but this may be absent in the long-term. This could reflect repair mechanisms (see Fig. 2 for cerebral ischemia) and there is a need for longitudinal studies in the same model. For subarachnoid hemorrhage, there is evidence for both acute and chronic CP injury
As opposed to the paucity of data on CP injury in human IVH or animal models, there is considerable data on damage to the ependymal cells that line the cerebral ventricles. Thus, Fukumizu et al. [37, 38] found marked ependymal damage and loss in human neonatal IVH. Mayfrank et al. [39] used a pig IVH model with and without tissue plasminogen activator-induced fibrinolysis and, similar to Pang et al. [36] in dog, they found marked IVH-induced ependymal damage at 1 and 7 weeks and that was reduced by fibrinolysis. In rat, Simard et al. [34] found that ependymal cells took up IgG at 48 h post IVH and Gao et al. [40] found ependymal damage 1 day after intracerebroventricular (ICV) injection of Fe3+, a degradation product of hemoglobin. These results indicate that there can be acute and chronic ependymal damage after IVH. In the CP after ischemic injury, there is evidence of tissue repair with time (see below). Why this does not happen in the ependyma is uncertain but it might relate to the ability of the CP to shrink in size when shedding dead cells. Because of the physical contact between the ependyma and underlying brain, such shrinkage is not possible for the ependyma.
Subarachnoid hemorrhage
SAH is often associated with extension of blood into the ventricular system (30–50%; [25, 27]). IVH is a risk factor for poor prognosis in SAH and for the development of hydrocephalus and vasospasm [25, 27, 41]. Rosen et al. [25] found that hydrocephalus developed in 62% of SAH patients with IVH compared to 26% of patients without IVH. Post-hemorrhagic hydrocephalus is generally assumed to result from a block in the CSF flow pathways or fibrosis at CSF outflow sites [42, 43]. Whether SAH (with or without associated IVH) causes CP injury has not to our knowledge been examined in patients. Given the preclinical data indicating that SAH causes CP damage in animals, this gap in knowledge needs to be corrected.
Several studies using cisterna magna blood injection in rabbits have reported effects on the CP (Fig. 1). Liszczak et al. [44] reported CP changes including electron-dense cytoplasmic inclusions and expansion of the lateral and subcellular spaces. Yilmaz et al. [45] examined both choroidal artery vasospasm and CP injury (using an injury scoring system) at 20 days and found evidence of a correlation between choroidal artery constriction and CP damage after SAH. In another study, the presence of water filled spaces within the CP was examined at 2 and 14 days after SAH [46]. There was a marked increase in the numbers of such spaces, particularly at 2 days and the authors hypothesized that these changes may be related to altered CSF secretion. Furthermore, changes in CP morphology, with reduced epithelial cell and microvillus height, and many TUNEL-positive cells have been reported 10 days after SAH (24% of cells in animals surviving SAH and 42% in animals that died) [47].
As in humans, animal models of SAH can also cause hydrocephalus. Thus, using a endovascular perforation model of SAH (perforation at the internal carotid artery bifurcation), Okobu et al. [48] found evidence of acute hydrocephalus in about 40% in rats. A question that has not been totally resolved is whether there has to be blood in the ventricles to induce CP injury and hydrocephalus, although Okobu et al. [48] found that hydrocephalus was always associated with blood in the ventricular system.
Intracerebral hemorrhage
Intraventricular extension of bleeding is a common finding in ICH and a risk factor for poor outcome [24, 26]. Whether ICH without such IVH can impact CP function has not been examined. Even though there is no extension of blood into the ventricular system, products from clot degradation may move into the CSF [49].
Ischemic stroke
In humans, the blood supply to the LVCP comes from the anterior, lateral posterior and medial posterior choroidal arteries [50]. The blood supply to the third ventricle CP comes from the bilateral medial posterior choroidal arteries [51], while that to the fourth ventricle CP is from the anterior and posterior inferior cerebellar arteries and the superior cerebellar artery [52]. The rich collateral circulation for each of the CPs may help protect them from focal ischemic events. Thus, using magnetic resonance imaging (MRI), only three CP infarcts have been described in the literature. Two related to choroidal artery occlusion [53, 54] and one due to a basilar artery occlusion [55]. Brain infarcts from anterior choroid artery (AChA) occlusion have been described by multiple groups (e.g. [56–59]). The AChA supplies multiple tissues (e.g. the internal capsule, optic tract and the cerebral peduncle; [58]) as well as the LVCPs. Whether changes in LVCP function occur and contribute to the neurological symptoms with AChA occlusion is uncertain. Infarcts related to the medial posterior choroidal arteries have rarely been described [54, 60] (Fig. 2). There have been few studies on the impact of transient global cerebral ischemia (e.g. cardiac arrest) on CP injury/function. However, in patients resuscitated after cardiac arrest, an increased expression of advanced glycation end-product receptors (RAGE) in CP epithelial cells and amyloid beta in both CP blood vessels and on the epithelial basement membrane have been reported [61, 62].

In animal studies, there is evidence that both transient global cerebral ischemia and focal cerebral ischemia cause choroid plexus (CP) injury and blood-CSF barrier (BSCFB) disruption. There is evidence that the damage after global ischemia is quickly repaired and this probably involves shedding of damaged cells resulting in CP atrophy. This has not been examined in focal cerebral ischemia. In humans, the effects of transient global cerebral ischemia (e.g. cardiac arrest) on the CP have not been examined. There are very rare reports of CP infarcts after focal cerebral ischemia in patients, but the occurrence of CP damage after focal cerebral ischemia is almost certainly limited by the rich collateral circulation to the CP
In animals, a number of studies using transient global ischemia models have shown CP epithelial cell death (Fig. 2). Thus, Pulsinelli et al. [63], using permanent bilateral basilar artery occlusion with 30 min transient common carotid artery occlusion (4VO model) in rats, found ischemic necrosis in the CP at 6 h and this is earlier than found in hippocampus suggesting selective vulnerability. Also, using the rat 4VO model, LVCP atrophy (~35%) [64] and histological damage [65] have been reported. Other studies have reported widespread epithelial cell death and CP edema early during reperfusion (0–12 h) after bilateral common carotid artery occlusion (10 min) with hypotension (2VO + hypotension) [66, 67]. Furthermore, CP DNA fragmentation occurred after 2VO + hypotension in rats at 18–24 h [68, 69]. In gerbils with 5 min bilateral common carotid artery occlusion with reperfusion, CP DNA fragmentation was found at ~1 week [70, 71].
In neonatal rats, Rothstein and Levinson [72] found that perinatal hypoxia/ischemia caused cell death in the CP and they concluded that the CP is selectively vulnerable. Likewise, Towfighi et al. [73] found pyknotic nuclei within the rat CP a few hours after hypoxia/ischemia and this progressed to necrosis by 24 h. Interestingly, similar to the adult, those necrotic cells disappeared within 4 days of the CP becoming atrophied. Sivakumar et al. [74] examined the effects of 2 h of hypoxia alone in neonatal rats and found CP morphological signs of injury at 3 and 24 h. These began to resolve at 3 days with the CPs having a normal appearance at 14 days. It should be noted that one study in neonatal mice reported no damage to the CP after hypoxia/ischemia injury [75]. It is interesting, however, that many of these studies report a recovery in CP structure and loss of damaged cells with time [63, 64, 66–69, 73]. This recovery may reflect the loss of damaged cells, the recruitment of new cells or both. The CP atrophy reported by Dienel [64] and Towfighi et al. [73] suggests the former, but there is some evidence for cell recruitment (see below). In terms of blood-CSF barrier function, barrier disruption was reported by Ikeda et al. [70] after bilateral common carotid occlusion in the gerbil. Similarly, Ennis and Keep [76] reported blood-CSF barrier disruption at 6 h after 2VO + hypotension in the rat.
There have been few studies on CP injury after focal ischemia [76–79] (Fig. 2). It should be noted that the commonly used suture model of middle cerebral artery (MCA) occlusion can result in reduced blood supply to the LVCP [76] as the branch point for the AChA from the internal carotid artery is close to the bifurcation of the MCA [80]. In the rat, Ennis and Keep [76] found a 38% decrease in LVCP blood flow after MCA occlusion and there was a 53% reduction if the animals underwent a tandem common carotid artery occlusion. Nagahiro et al. [79] examined the effects of different durations of tandem MCA and internal carotid occlusion in rats. They found evidence of blood-CSF barrier disruption with MRI using gadolinium-diethylene triamine pentaacetic acid (Gd-DTPA) in animals with 15 and 30 min of ischemia with 6 h of reperfusion, whereas BBB disruption was only found with longer (60 min) periods of ischemia. They did not find evidence of CP morphological changes after 6 h of reperfusion by H&E staining. Li et al. [78], studying 2 h transient MCA occlusion and reperfusion in rats, reported that CP cells became swollen (1–4 days) and that there was bromodeoxyuridine (BrdU) staining indicating cell proliferation, perhaps in compensation for CP cell death. No BrdU staining was observed in the CP in non-ischemic rats. Interestingly, the BrdU staining after MCA occlusion was not limited to the LVCP, but also occurred in the 3rd and 4th CPs indicating effects on proliferation distant to the ischemic parenchyma. Gillardon et al. [77] found evidence of LVCP epithelial cell death (TUNEL staining) after 6 h but not 1.5 h of permanent MCA occlusion in the rat and Ennis and Keep [76] found evidence of LVCP edema after MCA occlusion in rats with and without tandem common artery occlusion.
A number of co-morbidities increase the risk of ischemic stroke and result in more severe brain injury after stroke. These include aging, hypertension, hyperlipidemia and hyperglycemia and they are known to enhance BBB dysfunction after stroke [81]. The effects of such co-morbidities on CP injury after ischemic stroke are unknown. However, even in the absence of stroke, it is known that aging and Alzheimer’s disease result in CP dysfunction [5, 82–84].
Choroid plexus as a responder to injury
As well as being a site of injury, the CP may be involved in a neuroprotective response after brain injury. This includes CP secretion of protective factors, the CP as a site of neurogenesis/progenitor cell migration, the CP as a site of leukocyte infiltration (although this may have beneficial and detrimental effects) and potential changes in CSF production (Fig. 3). The evidence on such roles is limited and, therefore, this section discusses brain injury/disease in general and not just stroke. It should be noted that responses can be to brain injuries that are distant to the CP raising the question of how those injuries are ‘sensed’.

As well as being a potential site of injury after stroke, the choroid plexus (CP) also responds to stroke in multiple ways that may modify brain injury. It should be noted that the potential protective CP responses may be elicited by injury distant from the CP, indicating an unknown signaling mechanism. While most of the CP responses are thought to be protective, the effects of leukocyte diapedesis and changes in CSF production (if any) are uncertain. Elucidation of protective mechanisms involving the CP led to studies where CPs or CP epithelial cell transplants were used in animals with ischemic stroke with evidence of neuroprotection
Growth factor/neuropeptide release
The CPs produce a wide range of peptides/proteins: e.g. adrenomedullin, basic fibroblast growth factor (bFGF)-2, insulin-like growth factor (IGF)-2, transthyretin, transforming growth factor (TGF) β and vasopressin, many of which are secreted into the CSF [14–16]. There they can impact periventricular cells but they may also have an autocrine function since the CPs also possess an array of receptors: e.g. the FGFR1 and FGFR2 for fibroblast growth factor, IGF-1R and IGF-2R for insulin-like growth factor, TβRII for TGFβ, and the vasopressin receptors V1a, V1b and, during development, V2 [14]. There is evidence of CP-induced neuroprotection via growth factor release in a number of injury and disease states including stroke (see below) and traumatic brain injury [15, 85]. It should be noted, however, that some peptide release by the CP can have adverse effects. Thus, for example, there is evidence that vasopressin exacerbates brain injury after stroke [14].
With respect to animal models of stroke, studies have focused on cerebral ischemia and not the CP response to cerebral hemorrhage. Knuckey et al. [86] found that TGFβ-1, -2 and -3 mRNA expression in the CP increased at day 1 and 2 after transient global ischemia (2VO + hypotension), returning to normal at day 3. Those changes in mRNA expression were not mirrored in altered CP protein expression and it was suggested that increased mRNA levels might reflect secretion into CSF. Increases in CSF TGFβ-1 have been reported in ischemic stroke and SAH [87, 88]. In cerebral ischemia, TGFβ-1 appears to be neuroprotective [89, 90] but it may have an adverse effect in IVH-induced and other forms of hydrocephalus [42, 91].
Hayamizu et al. [92] used immunohistochemistry to examine the effects of transient global cerebral ischemia (2VO + hypotension) on bFGF-2 expression in the CP and found that ischemia caused a marked decrease in cytoplasmic (but not apical or lateral membrane associated) expression from 6 h to 14 days post-ischemia. The loss of cytoplasmic staining was interpreted as reflecting CP to CSF secretion. Since ICV injection of bFGF-2 is protective in animal models of global ischemia [93, 94], this suggests that CP secretion has a protective role. Beilharz et al. [95] examined the response of IGF-1 and -2 and different IGF binding proteins (IGFBP) to hypoxia/ischemia in 3 weeks-old rats. They found that IGFBP-6 mRNA and protein was induced in the CP (as well as the ependymal and reactive glial cells) and hypothesized that IGFBP-6 binds particularly to IGF-2 limiting adverse effects of IGF-2 after hypoxia/ischemia. Sivakumar et al. [74] found increased VEGF mRNA and protein levels in the CP at different times after 2 h of hypoxia in neonatal rats. VEGF expression is regulated by hypoxia-inducible factor α and VEGF concentrations in CSF are increased by conditions causing brain hypoxia [96]. Additionally, there are multiple papers indicating a protective effect of ICV administered VEGF in ischemic stroke [97–100] suggesting that CP production of VEGF could have protective effects in stroke. However, it should be noted that Liu et al. [101] recently found that ICV administration of an anti-VEGF receptor-2 antibody reduced early brain injury in a mouse SAH model.
A concern with such growth factor studies is that using data on CP mRNA or protein expression to imply secretion into CSF could be misleading. It should be noted, however, that there is also in vitro data indicating such secretion. For example, Borlongan et al. [18] found that CP explants secrete glial-derived neurotrophic factor, brain-derived neurotrophic factor and nerve growth factor. Another concern is the relative importance of the CPs in secreting such factors into CSF after stroke compared to other brain areas. Data using selective inhibition of CP growth factor production during stroke would be very informative (e.g. CP specific inducible KO mice; [102]).
With respect to human stroke, Flood et al. [87] examined CSF TGFβ1 in patients who had hydrocephalus after SAH and found markedly increased CSF levels early and late after SAH. While the initial rise was probably related to platelet-derived TGFβ1, the authors found that late increase in CSF levels correlated with increased CP expression. Adrenomedullin is a vasoactive peptide that is secreted by the CP and other non-BBB areas of the brain. After SAH, there is an early (day 3) and particularly a late (day 8) increase in CSF adrenomedullin that does not correlate with plasma levels. Higher CSF levels were correlated with an increased risk of delayed ischemic neurological deficits, occurrence of hyponatremia and reduced appetite [103, 104]. Whether the elevated CSF adrenomedullin levels are due to increased CP secretion has not been directly examined.
Apart from secreting proteins directly into CSF, recent evidence indicates that CP epithelial cells also secrete exosomes into the CSF [105–107]. These contain proteins, microRNAs and other components and form another type of communications between CP and the brain parenchyma [105–107]. The role of exosomes in the protective effects of the CP in brain injury is still unclear.
Choroid plexus enzymes and transporters
As well as secreting factors that may influence brain damage, the CPs also express many enzymes and transporters that may be important in the response to injury. Thus, unlike the CSF, the CP has high fibrinolytic activity [108, 109] and this may play a role in intraventricular hematoma resolution. In addition, considering the role that iron, as a degradation product of hemoglobin, has in brain injury after cerebral hemorrhage [28, 110, 111], it is noteworthy that the CP expresses a very wide array of iron-handling proteins including transporters: e.g. divalent metal transporter-1, ferroportin, transferrin and transferrin receptor-1 and -2, the iron storage protein ferritin, the heme metabolizing enzyme heme oxygenase-1, and proteins that change the oxidative sate of iron (ceruloplasmin and hephaestin) [112, 113]. The role of the CP in handling hemoglobin and iron after IVH merits further investigation. In relation to Alzheimer’s disease, the CP plays a role in regulating amyloid-β degradation and the levels of amyloid-β in the brain were reduced in a mouse model of Alzheimer’s disease by CP implants [17].
Choroid plexus transplantation
Because of the evidence that the CP can protect the brain from injury, there have been a number of studies examining the effects of CP transplants on different types of brain injury. Borlongan et al. found that transplanting rat [114] or pig [18] choroid plexus into the CNS protected against cerebral ischemia in rats. Matsumoto et al. [115] also showed similar protection with cultured choroid plexus epithelial cells in rat MCA occlusion. Outside cerebral ischemia, Bolos et al. [17] found that CP epithelial cell transplants reduced amyloid-β deposits, tau hyperphosphorylation and astrocytic reactivity in a mouse model of Alzheimer’s disease. They also improved behavioral outcomes including memory tests. Similarly, transplants of CP epithelial cells have enhanced axonal regeneration and locomotor improvement in rat spinal cord injury [19, 116]. Compared to approaches that focus on a single factor (e.g. a growth factor) to promote brain protection/recovery, CP epithelial cells transplants may have an advantage in producing multiple factors. It is also possible that the CP epithelial cells may have a modified response dependent upon the type of injury. Thus, for example, will CP epithelial cells exposed to an ischemic brain produce the same growth factor and express the same enzymes as those exposed to a brain with Alzheimer’s disease?
Choroid plexus proliferation and progenitor cells
In the adult CP, there is only a slow turnover of epithelial cells in rodents and primates [117, 118]. However, there is in vitro evidence that injury and growth factors (IGF and epidermal growth factor) can cause CP epithelial cell proliferation [119]. In vivo, Li et al. [78] found increased CP cell proliferation (BrdU positive cells) after MCA occlusion in the rat. The overall impact of such proliferation on CP injury in vivo is as yet uncertain (i.e. can it replace the cell loss caused by stroke?) and some of the BrdU cells were neuronal nuclear antigen (NeuN) and glial fibrillary acidic protein positive leading Li et al. to suggest they were neural precursor cells [78].
There has been considerable interest in the use of exogenous stem cells, either administered systemically or into the brain, to treat different neurological conditions. Endothelial progenitor cells administered intravenously can integrate into the cerebral endothelium after stroke and may be a source of growth factors [120]. Whether that is the case for the CP is unclear. Zhang et al. [121] found that bone marrow-derived endothelial progenitor cells migrated to the CP endothelium after intravenous injection in non-ischemic mice and Beck et al. [122] also found that bone marrow-derived cells given intravenously migrated to the CP in ischemic (MCA occlusion) and non-ischemic mice. They were uncertain whether those cells were integrating into the CP endothelium or migrating through the CP. However, mesenchymal stem cells given ICV can integrate into the CP epithelium [123, 124].
Leukocyte migration at the choroid plexus
In stroke, neuroinflammation may have both detrimental and beneficial effects. Thus, an early influx of leukocytes (particularly neutrophils) into the brain from the blood may exacerbate ischemic and hemorrhagic brain injury [125, 126], but a later inflammation may be beneficial in brain repair [127–129]. In the case of cerebral hemorrhage, infiltrating macrophages/resident microglia have an important role in hematoma resolution via phagocytosis [130, 131]. Infiltrating leukocytes may enter the brain across the BBB or the blood-CSF barrier after a stroke. While there is a wealth of knowledge about leukocyte entry at the BBB in stroke, studies on the role of the CP are very limited. Kowarik et al. [132] compared CSF levels of leukocytes in patients across multiple neurological conditions. For stroke (a mixture of ischemic and hemorrhagic), they examined CSF within 4 weeks of onset. Unlike some conditions (e.g. bacterial or viral meningitis), stroke was not associated with high CSF leukocyte counts nor did it alter the distribution of leukocyte subsets. The majority of CSF leukocytes after stroke were CD4+ T cells (71%) and CD8+ T cells (17%). A fuller time course of such changes in CSF, particularly soon after stroke, would be informative.
The CP is thought to be involved in immune surveillance by trafficking T-lymphocytes [133, 134]. In a model of hypoxia/ischemia with systemic inflammation (lipopolysaccharide; to mimic intrauterine infection) in rats, Yang et al. [135] found an early (4 h) infiltration of T-lymphocytes (CD43+) into the CP. This was blocked by fingolimod (FTY720; a sphingosine-1-phosphate receptor modulator) treatment which also markedly reduced the brain injury in lipopolysaccharide-enhanced hypoxia/ischemia injury. In other neurological conditions, such as multiple sclerosis, traumatic brain injury, spinal cord injury and meningitis, as well as peripheral inflammation, there has been interest in the role of the CP in leukocyte entry into CSF and brain [3, 136, 137]. For example, Szmydynger-Chodobska et al. [138, 139] found that the CP is a site of neutrophil and monocyte entry into the brain after traumatic brain injury (controlled cortical impact). It is interesting that in those studies the CPs are distant from the site of initial injury raising the question as to what signaling mechanisms trigger the diapedesis of leukocytes at the CP.
CSF production
Alterations in CSF production could impact brain injury. Increases could aid in the clearance of neurotoxic factors from the brain (CSF sink effect) but might also exacerbate hydrocephalus. With regards to the latter, choroid plexus hyperplasia and choroid plexus papillomas are associated with CSF hypersecretion and hydrocephalus [140]. Decreases in CSF production might serve to decrease the incidence of hydrocephalus and reduce intracranial pressure, and CP cauterization has been used as a treatment for hydrocephalus [141, 142]. After stroke, there is a lack of data on whether CSF production is changed. However, Holloway and Cassin [143] did report a 33% decline in CSF production with hypoxia in neonatal dogs.
The water channel, aquaporin 1 (Aqp1) is highly expressed in the CP epithelium and Aqp1 knockout mice have reduced CSF secretion [144]. There has, therefore, been an interest in examining the effects of stroke on Aqp1 expression. In the 4VO model of transient global cerebral ischemia, Akdemir et al. [65] reported an increase AQP1 protein expression in the CP epithelium between 24 and 48 h of reperfusion. Similarly, Sveinsdottir et al. [145] found an increase in CP Aqp1 protein expression in a preterm rabbit IVH model. However, at the mRNA level, there was a decrease in expression, an effect replicated in vitro by exposing cultured CP epithelial cells to the CSF of preterm infants with IVH or hemin. The reason for the different protein and mRNA responses are uncertain. Interestingly, Sveinsdottir et al. [145] also examined the expression of another aquaporin, Aqp5. They found a marked increase in both protein and mRNA expression in the preterm rabbit IVH model for that aquaporin and they also found increased Aqp5 mRNA after exposure of CP epithelial cells to hemin in vitro. This merits further investigation.
Injury sensing by the choroid plexus
In the earlier descriptions of stroke-induced CP injury, this review has focused on types of injury that directly impact the CP, including reductions in CP blood flow or blood in the ventricular system adjacent to the CPs. However, there is evidence that events distant from the CPs may trigger protective responses in the CP. For example, traumatic brain injury that does not directly impact the CP results in growth factor production [15, 85], alters CP protein expression (e.g. [146]), and infiltration of leukocytes across the CP [138], and Li et al. [78] found increased cell proliferation in CPs distant to the parenchymal damage induced by a MCA occlusion. How does the CP sense that distant injury and what pathways in the CP are triggered to elicit the protective response? As noted above, the CP expresses a wide range of peptide/protein receptors [14] and these may be involved, but these are as yet largely unaddressed questions.
Choroid plexus and cerebral endothelial responses to stroke
Our knowledge of the effects of stroke on cerebral endothelial cells, the site of the BBB, is much greater than at the CP epithelium. There are also differences in the types of experiment that can be performed in relation to each tissue. For example, with the cerebral endothelium, two-photon microscopy using fluorescently-tagged permeability markers and endothelial proteins (e.g. cells expressing GFP-tagged claudin-5) can be used to study stroke-induced BBB disruption in real time [147]. Such techniques can not currently be used to study the deeply-situated CPs. However, CP studies have an advantage in that CSF can be sampled (in animals and humans) in contrast to the cerebral endothelium studies where adjacent brain interstitial fluid is inaccessible. Thus, for example, the sampling of exosomes released from cerebral endothelial cells is currently limited to cultured cells [148]. While acknowledging that other sites (e.g. ependyma), as well as the CP, may contribute to CSF exosomes, analysis of such exosomes has given insight into exosome cargo in animals and humans in vivo (e.g. proteins, mRNAs and microRNAs [149–151]) as well as the impact of disease on exosomes [152].
Access to CSF has aided examination of the potential roles of the CP in protecting the brain from injury/disease. The results of such CP studies may aid in formulating brain endothelial studies. For example, to what extent will brain endothelial cells fulfil similar protective roles to the CP? While there has been considerable interest in the production of growth factors by neural and endothelial progenitor cells (endogenous and exogenous) after injury [153, 154], the role of the normal cerebral endothelium has received less attention.
Conclusions
There have been surprisingly few studies examining the degree to which hemorrhagic stroke causes CP injury. This is particularly noticeable in the setting of IVH, where the CP is in close proximity to the hemorrhage. In ICH, there is considerable evidence that clot derived factors (e.g. hemoglobin and iron) cause perihematomal injury [28] and are a therapeutic target (intracerebral hemorrhage deferoxamine trial; iDEF—NCT02175225). Further clinical and preclinical studies on the extent to which IVH and SAH cause CP injury are needed as they may identify a therapeutic target.
Because of its rich collateral circulation, focal cerebral ischemia probably rarely causes CP infarction. However, there is evidence suggesting that the CP is selectively vulnerable to ischemia. Further studies on the impact of transient global ischemia (e.g. cardiac arrest) on CP injury in patients are warranted. The finding that the CPs can produce factors that can protect the brain from injury and disease raises many opportunities. The factors involved are probably numerous, making the effects difficult to reproduce pharmacologically (especially within the brain). Hence, there has been an interest in using CP transplants. However, there is a question as to what can trigger the release of protective factors from the CP? Determining the signaling pathway(s) may be a promising therapeutic avenue. It should also be noted that a role of the CP in neuroprotection after IVH or SAH has not really been examined and this may be an important area of research.
References
Redzic ZB, Segal MB. The structure of the choroid plexus and the physiology of the choroid plexus epithelium. Adv Drug Deliv Rev. 2004;56(12):1695–716.
Damkier HH, Brown PD, Praetorius J. Cerebrospinal fluid secretion by the choroid plexus. Physiol Rev. 2013;93(4):1847–92.
Demeestere D, Libert C, Vandenbroucke RE. Clinical implications of leukocyte infiltration at the choroid plexus in (neuro)inflammatory disorders. Drug Discov Today. 2015;20(8):928–41.
Hladky SB, Barrand MA. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers CNS. 2014;11(1):26.
Marques F, Sousa JC, Brito MA, Pahnke J, Santos C, Correia-Neves M, Palha JA. The choroid plexus in health and in disease: dialogues into and out of the brain. Neurobiol Dis. 2016. doi:10.1016/j.nbd.2016.08.011.
Spector R, Johanson CE. Sustained choroid plexus function in human elderly and Alzheimer’s disease patients. Fluids Barriers CNS. 2013;10(1):28.
Tietz S, Engelhardt B. Brain barriers: crosstalk between complex tight junctions and adherens junctions. J Cell Biol. 2015;209(4):493–506.
Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, Sharp FR. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34(2):185–99.
Jin R, Yang G, Li G. Molecular insights and therapeutic targets for blood-brain barrier disruption in ischemic stroke: critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol Dis. 2010;38(3):376–85.
Keep RF, Zhou N, Xiang J, Andjelkovic AV, Hua Y, Xi G. Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS. 2014;11:18.
Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42(11):3323–8.
Ennis SR, Keep RF. Forebrain ischemia and the blood-cerebrospinal fluid barrier. Acta Neurochir Suppl. 2006;96:276–8.
Keep RF, Ennis SR, Xiang J. The blood-CSF barrier and cerebral ischemia. In: Zheng W, Chodobski A, editors. The blood-cerebrospinal fluid barrier. Boca Raton: Taylor & Francis; 2005. p. 345–60.
Chodobski A, Szmydynger-Chodobska J. Choroid plexus: target for polypeptides and site of their synthesis. Microsc Res Tech. 2001;52(1):65–82.
Johanson C, Stopa E, Baird A, Sharma H. Traumatic brain injury and recovery mechanisms: peptide modulation of periventricular neurogenic regions by the choroid plexus-CSF nexus. J Neural Transm. 2011;118(1):115–33.
Redzic ZB, Preston JE, Duncan JA, Chodobski A, Szmydynger-Chodobska J. The choroid plexus-cerebrospinal fluid system: from development to aging. Curr Top Dev Biol. 2005;71:1–52.
Bolos M, Antequera D, Aldudo J, Kristen H, Bullido MJ, Carro E. Choroid plexus implants rescue Alzheimer’s disease-like pathologies by modulating amyloid-beta degradation. Cell Mol Life Sci. 2014;71(15):2947–55.
Borlongan CV, Skinner SJ, Geaney M, Vasconcellos AV, Elliott RB, Emerich DF. Intracerebral transplantation of porcine choroid plexus provides structural and functional neuroprotection in a rodent model of stroke. Stroke. 2004;35(9):2206–10.
Ide C, Nakano N, Kanekiyo K. Cell transplantation for the treatment of spinal cord injury—bone marrow stromal cells and choroid plexus epithelial cells. Neural Regen Res. 2016;11(9):1385–8.
Lacey DJ, Terplan K. Intraventricular hemorrhage in full-term neonates. Dev Med Child Neurol. 1982;24(3):332–7.
Miyasaka Y, Yada K, Ohwada T, Morii S, Kitahara T, Kurata A, Tanaka R. Choroid plexus arteriovenous malformations. Neurol Med Chir. 1992;32(4):201–6.
Reeder JD, Kaude JV, Setzer ES. Choroid plexus hemorrhage in premature neonates: recognition by sonography. AJNR Am J Neuroradiol. 1982;3(6):619–22.
Ballabh P. Intraventricular hemorrhage in premature infants: mechanism of disease. Pediatr Res. 2010;67(1):1–8.
Bhattathiri PS, Gregson B, Prasad KS, Mendelow AD, Investigators S. Intraventricular hemorrhage and hydrocephalus after spontaneous intracerebral hemorrhage: results from the STICH trial. Acta Neurochir Suppl. 2006;96:65–8.
Rosen DS, Macdonald RL, Huo D, Goldenberg FD, Novakovic RL, Frank JI, Rosengart AJ. Intraventricular hemorrhage from ruptured aneurysm: clinical characteristics, complications, and outcomes in a large, prospective, multicenter study population. J Neurosurg. 2007;107(2):261–5.
Hanley DF. Intraventricular hemorrhage: severity factor and treatment target in spontaneous intracerebral hemorrhage. Stroke. 2009;40(4):1533–8.
Jabbarli R, Reinhard M, Roelz R, Shah M, Niesen WD, Kaier K, Taschner C, Weyerbrock A, Velthoven VV. The predictors and clinical impact of intraventricular hemorrhage in patients with aneurysmal subarachnoid hemorrhage. Int J Stroke. 2016;11(1):68–76.
Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11(8):720–31.
Hallevi H, Walker KC, Kasam M, Bornstein N, Grotta JC, Savitz SI. Inflammatory response to intraventricular hemorrhage: time course, magnitude and effect of t-PA. J Neurol Sci. 2012;315(1–2):93–5.
Kaestner S, Dimitriou I. TGF beta1 and TGF beta2 and their role in posthemorrhagic hydrocephalus following SAH and IVH. J Neurol Surg Part A Cent Eur Neurosurg. 2013;74(5):279–84.
Kramer AH, Jenne CN, Zygun DA, Roberts DJ, Hill MD, Holodinsky JK, Todd S, Kubes P, Wong JH. Intraventricular fibrinolysis with tissue plasminogen activator is associated with transient cerebrospinal fluid inflammation: a randomized controlled trial. J Cereb Blood Flow Metab. 2015;35(8):1241–8.
Milman N, Graudal NA, Olsen TS, Wandall JH, Pedersen NS. Cerebrospinal fluid ferritin in patients with meningitis and cerebral infarction or bleeding. Dan Med Bull. 1993;40(4):490–2.
Morales DM, Silver SA, Morgan CD, Mercer D, Inder TE, Holtzman DM, Wallendorf MJ, Rao R, McAllister JP, Limbrick DD Jr. Lumbar cerebrospinal fluid biomarkers of posthemorrhagic hydrocephalus of prematurity: amyloid precursor protein, soluble amyloid precursor protein alpha, and L1 cell adhesion molecule. Neurosurgery. 2017;80(1):82–90.
Simard PF, Tosun C, Melnichenko L, Ivanova S, Gerzanich V, Simard JM. Inflammation of the choroid plexus and ependymal layer of the ventricle following intraventricular hemorrhage. Transl Stroke Res. 2011;2(2):227–31.
Gram M, Sveinsdottir S, Cinthio M, Sveinsdottir K, Hansson SR, Morgelin M, Akerstrom B, Ley D. Extracellular hemoglobin—mediator of inflammation and cell death in the choroid plexus following preterm intraventricular hemorrhage. J Neuroinflammation. 2014;11:200.
Pang D, Sclabassi RJ, Horton JA. Lysis of intraventricular blood clot with urokinase in a canine model: Part 3. Effects of intraventricular urokinase on clot lysis and posthemorrhagic hydrocephalus. Neurosurgery. 1986;19(4):553–72.
Fukumizu M, Takashima S, Becker LE. Neonatal posthemorrhagic hydrocephalus: neuropathologic and immunohistochemical studies. Pediatr Neurol. 1995;13(3):230–4.
Fukumizu M, Takashima S, Becker LE. Glial reaction in periventricular areas of the brainstem in fetal and neonatal posthemorrhagic hydrocephalus and congenital hydrocephalus. Brain Develop. 1996;18(1):40–5.
Mayfrank L, Kim Y, Kissler J, Delsing P, Gilsbach JM, Schroder JM, Weis J. Morphological changes following experimental intraventricular haemorrhage and intraventricular fibrinolytic treatment with recombinant tissue plasminogen activator. Acta Neuropathol. 2000;100(5):561–7.
Gao F, Liu F, Chen Z, Hua Y, Keep RF, Xi G. Hydrocephalus after intraventricular hemorrhage: the role of thrombin. J Cereb Blood Flow Metab. 2014;34(3):489–94.
Wilson TJ, Stetler WR Jr, Davis MC, Giles DA, Khan A, Chaudhary N, Gemmete JJ, Xi G, Thompson BG, Pandey AS. Intraventricular hemorrhage is associated with early hydrocephalus, symptomatic vasospasm, and poor outcome in aneurysmal subarachnoid hemorrhage. J Neurol Surg. 2015;76(2):126–32.
Cherian S, Whitelaw A, Thoresen M, Love S. The pathogenesis of neonatal post-hemorrhagic hydrocephalus. Brain Pathol. 2004;14(3):305–11.
Whitelaw A, Thoresen M, Pople I. Posthaemorrhagic ventricular dilatation. Arch Dis Child Fetal Neonatal Ed. 2002;86(2):F72–4.
Liszczak TM, Black PM, Tzouras A, Foley L, Zervas NT. Morphological changes of the basilar artery, ventricles, and choroid plexus after experimental SAH. J Neurosurg. 1984;61(3):486–93.
Yilmaz A, Aydin MD, Kanat A, Musluman AM, Altas S, Aydin Y, Calik M, Gursan N. The effect of choroidal artery vasospasm on choroid plexus injury in subarachnoid hemorrhage: experimental study. Turk Neurosurg. 2011;21(4):477–82.
Kanat A, Turkmenoglu O, Aydin MD, Yolas C, Aydin N, Gursan N, Tumkaya L, Demir R. Toward changing of the pathophysiologic basis of acute hydrocephalus after subarachnoid hemorrhage: a preliminary experimental study. World Neurosurg. 2013;80(3–4):390–5.
Kotan D, Aydin MD, Gundogdu C, Aygul R, Aydin N, Ulvi H. Parallel development of choroid plexus degeneration and meningeal inflammation in subarachnoid hemorrhage—experimental study. Adv Clin Exp Med. 2014;23(5):699–704.
Okubo S, Strahle J, Keep RF, Hua Y, Xi G. Subarachnoid hemorrhage-induced hydrocephalus in rats. Stroke. 2013;44(2):547–50.
Norrving B, Olsson JE. The diagnostic value of spectrophotometric analysis of the cerebrospinal fluid in cerebral hematomas. J Neurol Sci. 1979;44(1):105–14.
Marinkovic S, Gibo H, Milisavljevic M, Djulejic V, Jovanovic VT. Microanatomy of the intrachoroidal vasculature of the lateral ventricle. Neurosurgery. 2005;57(1 Suppl):22–36 (discussion 22–36).
Wolfram-Gabel R, Maillot C, Koritke JG, Laude M. Vascularization of the tela choroidea of the 3rd ventricle in man. Archives d’Anatomie, d’Histologie, et d’Embryologie Normales et Experimentales. 1984;67:3–42.
Sharifi M, Ciolkowski M, Krajewski P, Ciszek B. The choroid plexus of the fourth ventricle and its arteries. Folia Morphol. 2005;64(3):194–8.
Koral K, Dowling MM, Rollins NK. Choroid plexus infarction in a child. Pediatr Neurol. 2007;37(6):452–3.
Liebeskind DS, Hurst RW. Infarction of the choroid plexus. AJNR Am J Neuroradiol. 2004;25(2):289–90.
Nabavizadeh SA, Tangestanipoor A, Mowla A, Hurst R, Mamourian AC. Infarction of the choroid plexus in basilar artery occlusion. J Neurol Sci. 2015;358(1–2):467–8.
Helgason CM. A new view of anterior choroidal artery territory infarction. J Neurol. 1988;235(7):387–91.
Hupperts RM, Lodder J, Heuts-van Raak EP, Kessels F. Infarcts in the anterior choroidal artery territory. Anatomical distribution, clinical syndromes, presumed pathogenesis and early outcome. Brain. 1994;117(Pt 4):825–34.
Pezzella FR, Vadala R. Anterior choroidal artery territory infarction. Front Neurol Neurosci. 2012;30:123–7.
Takahashi S, Ishii K, Matsumoto K, Higano S, Ishibashi T, Suzuki M, Sakamoto K. The anterior choroidal artery syndrome. II. CT and/or MR in angiographically verified cases. Neuroradiology. 1994;36(5):340–5.
Neau JP, Bogousslavsky J. The syndrome of posterior choroidal artery territory infarction. Ann Neurol. 1996;39(6):779–88.
Maslinska D, Laure-Kamionowska M, Taraszewska A, Deregowski K, Maslinski S. Immunodistribution of amyloid beta protein (Abeta) and advanced glycation end-product receptors (RAGE) in choroid plexus and ependyma of resuscitated patients. Folia Neuropathol. 2011;49(4):295–300.
Wisniewski HM, Maslinska D. Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 1996;34(2):65–71.
Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11(5):491–8.
Dienel GA. Regional accumulation of calcium in postischemic rat brain. J Neurochem. 1984;43(4):913–25.
Akdemir G, Kaymaz F, Gursoy-Ozdemir Y, Akalan N, Akdemir ES. The time course changes in expression of aquaporin 4 and aquaporin 1 following global cerebral ischemic edema in rat. Surg Neurol Int. 2016;7:4.
Johanson CE, Palm DE, Primiano MJ, McMillan PN, Chan P, Knuckey NW, Stopa EG. Choroid plexus recovery after transient forebrain ischemia: role of growth factors and other repair mechanisms. Cell Mol Neurobiol. 2000;20(2):197–216.
Palm D, Knuckey N, Guglielmo M, Watson P, Primiano M, Johanson C. Choroid plexus electrolytes and ultrastructure following transient forebrain ischemia. Am J Physiol. 1995;269(1 Pt 2):R73–9.
Ferrand-Drake M. Cell death in the choroid plexus following transient forebrain global ischemia in the rat. Microsc Res Tech. 2001;52(1):130–6.
Ferrand-Drake M, Wieloch T. The time-course of DNA fragmentation in the choroid plexus and the CA1 region following transient global ischemia in the rat brain. The effect of intra-ischemic hypothermia. Neuroscience. 1999;93(2):537–49.
Ikeda J, Mies G, Nowak TS Jr, Joo F, Klatzo I. Evidence for increased calcium influx across the choroid plexus following brief ischemia of gerbil brain. Neurosci Lett. 1992;142(2):257–9.
Kitagawa H, Setoguchi Y, Fukuchi Y, Mitsumoto Y, Koga N, Mori T, Abe K. DNA fragmentation and HSP72 gene expression by adenovirus-mediated gene transfer in postischemic gerbil hippocampus and ventricle. Metab Brain Dis. 1998;13(3):211–23.
Rothstein RP, Levison SW. Damage to the choroid plexus, ependyma and subependyma as a consequence of perinatal hypoxia/ischemia. Dev Neurosci. 2002;24(5):426–36.
Towfighi J, Zec N, Yager J, Housman C, Vannucci RC. Temporal evolution of neuropathologic changes in an immature rat model of cerebral hypoxia: a light microscopic study. Acta Neuropathol. 1995;90(4):375–86.
Sivakumar V, Lu J, Ling EA, Kaur C. Vascular endothelial growth factor and nitric oxide production in response to hypoxia in the choroid plexus in neonatal brain. Brain Pathol. 2008;18(1):71–85.
D’Angelo B, Ek CJ, Sandberg M, Mallard C. Expression of the Nrf2-system at the blood-CSF barrier is modulated by neonatal inflammation and hypoxia-ischemia. J Inherit Metab Dis. 2013;36(3):479–90.
Ennis SR, Keep RF. The effects of cerebral ischemia on the rat choroid plexus. J Cereb Blood Flow Metab. 2006;26(5):675–83.
Gillardon F, Lenz C, Kuschinsky W, Zimmermann M. Evidence for apoptotic cell death in the choroid plexus following focal cerebral ischemia. Neurosci Lett. 1996;207(2):113–6.
Li Y, Chen J, Chopp M. Cell proliferation and differentiation from ependymal, subependymal and choroid plexus cells in response to stroke in rats. J Neurol Sci. 2002;193(2):137–46.
Nagahiro S, Goto S, Korematsu K, Sumi M, Takahashi M, Ushio Y. Disruption of the blood-cerebrospinal fluid barrier by transient cerebral ischemia. Brain Res. 1994;633(1–2):305–11.
He Z, Yang SH, Naritomi H, Yamawaki T, Liu Q, King MA, Day AL, Simpkins JW. Definition of the anterior choroidal artery territory in rats using intraluminal occluding technique. J Neurol Sci. 2000;182(1):16–28.
Lucke-Wold BP, Logsdon AF, Turner RC, Rosen CL, Huber JD. Aging, the metabolic syndrome, and ischemic stroke: redefining the approach for studying the blood–brain barrier in a complex neurological disease. Adv Pharmacol. 2014;71:411–49.
Serot JM, Bene MC, Faure GC. Comparative immunohistochemical characteristics of human choroid plexus in vascular and Alzheimer’s dementia. Hum Pathol. 1994;25(11):1185–90.
Serot JM, Zmudka J, Jouanny P. A possible role for CSF turnover and choroid plexus in the pathogenesis of late onset Alzheimer’s disease. J Alzheimer’s Dis. 2012;30(1):17–26.
Vandenbroucke RE. A hidden epithelial barrier in the brain with a central role in regulating brain homeostasis. Implications for aging. Ann Am Thorac Soc. 2016;13(Supplement_5):S407–10.
Walter HJ, Berry M, Hill DJ, Cwyfan-Hughes S, Holly JM, Logan A. Distinct sites of insulin-like growth factor (IGF)-II expression and localization in lesioned rat brain: possible roles of IGF binding proteins (IGFBPs) in the mediation of IGF-II activity. Endocrinology. 1999;140(1):520–32.
Knuckey NW, Finch P, Palm DE, Primiano MJ, Johanson CE, Flanders KC, Thompson NL. Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat hippocampus following transient forebrain ischemia. Brain Res Mol Brain Res. 1996;40(1):1–14.
Flood C, Akinwunmi J, Lagord C, Daniel M, Berry M, Jackowski A, Logan A. Transforming growth factor-beta1 in the cerebrospinal fluid of patients with subarachnoid hemorrhage: titers derived from exogenous and endogenous sources. J Cereb Blood Flow Metab. 2001;21(2):157–62.
Krupinski J, Vodovotz Y, Li C, Slowik A, Beevers D, Flanders KC, Lip G, Kumar P, Szczudlik A. Inducible nitric oxide production and expression of transforming growth factor-beta1 in serum and CSF after cerebral ischaemic stroke in man. Nitric Oxide. 1998;2(6):442–53.
Buisson A, Lesne S, Docagne F, Ali C, Nicole O, MacKenzie ET, Vivien D. Transforming growth factor-beta and ischemic brain injury. Cell Mol Neurobiol. 2003;23(4–5):539–50.
Dobolyi A, Vincze C, Pal G, Lovas G. The neuroprotective functions of transforming growth factor beta proteins. Int J Mol Sci. 2012;13(7):8219–58.
Aojula A, Botfield H, McAllister JP 2nd, Gonzalez AM, Abdullah O, Logan A, Sinclair A. Diffusion tensor imaging with direct cytopathological validation: characterisation of decorin treatment in experimental juvenile communicating hydrocephalus. Fluids Barriers CNS. 2016;13(1):9.
Hayamizu TF, Chan PT, Johanson CE. FGF-2 immunoreactivity in adult rat ependyma and choroid plexus: responses to global forebrain ischemia and intraventricular FGF-2. Neurol Res. 2001;23(4):353–8.
Koketsu N, Berlove DJ, Moskowitz MA, Kowall NW, Caday CG, Finklestein SP. Pretreatment with intraventricular basic fibroblast growth factor decreases infarct size following focal cerebral ischemia in rats. Ann Neurol. 1994;35(4):451–7.
Nakata N, Kato H, Kogure K. Protective effects of basic fibroblast growth factor against hippocampal neuronal damage following cerebral ischemia in the gerbil. Brain Res. 1993;605(2):354–6.
Beilharz EJ, Russo VC, Butler G, Baker NL, Connor B, Sirimanne ES, Dragunow M, Werther GA, Gluckman PD, Williams CE, et al. Co-ordinated and cellular specific induction of the components of the IGF/IGFBP axis in the rat brain following hypoxic-ischemic injury. Brain Res Mol Brain Res. 1998;59(2):119–34.
Scheufler KM, Drevs J, van Velthoven V, Reusch P, Klisch J, Augustin HG, Zentner J, Marme D. Implications of vascular endothelial growth factor, sFlt-1, and sTie-2 in plasma, serum and cerebrospinal fluid during cerebral ischemia in man. J Cereb Blood Flow Metab. 2003;23(1):99–110.
Harrigan MR, Ennis SR, Sullivan SE, Keep RF. Effects of intraventricular infusion of vascular endothelial growth factor on cerebral blood flow, edema, and infarct volume. Acta Neurochir (Wien). 2003;145(1):49–53.
Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci USA. 2002;99(18):11946–50.
Kaya D, Gursoy-Ozdemir Y, Yemisci M, Tuncer N, Aktan S, Dalkara T. VEGF protects brain against focal ischemia without increasing blood–brain permeability when administered intracerebroventricularly. J Cereb Blood Flow Metab. 2005;25(9):1111–8.
Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Investig. 2003;111(12):1843–51.
Liu L, Fujimoto M, Kawakita F, Nakano F, Imanaka-Yoshida K, Yoshida T, Suzuki H. Anti-vascular endothelial growth factor treatment suppresses early brain injury after subarachnoid hemorrhage in mice. Mol Neurobiol. 2016;53(7):4529–38.
Crouthamel MH, Kelly EJ, Ho RJ. Development and characterization of transgenic mouse models for conditional gene knockout in the blood-brain and blood-CSF barriers. Transgenic Res. 2012;21(1):113–30.
Kubo Y, Koji T, Kashimura H, Otawara Y, Ogawa A, Ogasawara K. Adrenomedullin concentration in the cerebrospinal fluid is related to appetite loss and delayed ischemic neurological deficits after subarachnoid hemorrhage. Neurol Res. 2013;35(7):713–8.
Kubo Y, Ogasawara K, Kakino S, Kashimura H, Yoshida K, Ogawa A. Cerebrospinal fluid adrenomedullin concentration correlates with hyponatremia and delayed ischemic neurological deficits after subarachnoid hemorrhage. Cerebrovasc Dis. 2008;25(1–2):164–9.
Balusu S, Van Wonterghem E, De Rycke R, Raemdonck K, Stremersch S, Gevaert K, Brkic M, Demeestere D, Vanhooren V, Hendrix A, et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol Med. 2016;8(10):1162–83.
Grapp M, Wrede A, Schweizer M, Huwel S, Galla HJ, Snaidero N, Simons M, Buckers J, Low PS, Urlaub H, et al. Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat Commun. 2013;4:2123.
Tietje A, Maron KN, Wei Y, Feliciano DM. Cerebrospinal fluid extracellular vesicles undergo age dependent declines and contain known and novel non-coding RNAs. PLoS ONE. 2014;9(11):e113116.
Fodstad H, Kok P, Algers G. Fibrinolytic activity of cerebral tissue after experimental subarachnoid haemorrhage: inhibitory effect of tranexamic acid (AMCA). Acta Neurol Scand. 1981;64(1):29–46.
Masuda T, Dohrmann GJ, Kwaan HC, Erickson RK, Wollman RL. Fibrinolytic activity in experimental intracerebral hematoma. J Neurosurg. 1988;68(2):274–8.
Gao C, Du H, Hua Y, Keep RF, Strahle J, Xi G. Role of red blood cell lysis and iron in hydrocephalus after intraventricular hemorrhage. J Cereb Blood Flow Metab. 2014;34(6):1070–5.
Strahle JM, Garton T, Bazzi AA, Kilaru H, Garton HJ, Maher CO, Muraszko KM, Keep RF, Xi G. Role of hemoglobin and iron in hydrocephalus after neonatal intraventricular hemorrhage. Neurosurgery. 2014;75(6):696–705 (discussion 706).
Mesquita SD, Ferreira AC, Sousa JC, Santos NC, Correia-Neves M, Sousa N, Palha JA, Marques F. Modulation of iron metabolism in aging and in Alzheimer’s disease: relevance of the choroid plexus. Front Cell Neurosci. 2012;6:25.
Rouault TA, Zhang DL, Jeong SY. Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab Brain Dis. 2009;24(4):673–84.
Borlongan CV, Skinner SJ, Geaney M, Vasconcellos AV, Elliott RB, Emerich DF. CNS grafts of rat choroid plexus protect against cerebral ischemia in adult rats. NeuroReport. 2004;15(10):1543–7.
Matsumoto N, Taguchi A, Kitayama H, Watanabe Y, Ohta M, Yoshihara T, Itokazu Y, Dezawa M, Suzuki Y, Sugimoto H, et al. Transplantation of cultured choroid plexus epithelial cells via cerebrospinal fluid shows prominent neuroprotective effects against acute ischemic brain injury in the rat. Neurosci Lett. 2010;469(3):283–8.
Kanekiyo K, Nakano N, Noda T, Yamada Y, Suzuki Y, Ohta M, Yokota A, Fukushima M, Ide C. Transplantation of choroid plexus epithelial cells into contusion-injured spinal cord of rats. Restor Neurol Neurosci. 2016;34(3):347–66.
Chauhan AN, Lewis PD. A quantitative study of cell proliferation in ependyma and choroid plexus in the postnatal rat brain. Neuropathol Appl Neurobiol. 1979;5(4):303–9.
Kaplan MS. Proliferation of epithelial cells in the adult primate choroid plexus. Anat Rec. 1980;197(4):495–502.
Barkho BZ, Monuki ES. Proliferation of cultured mouse choroid plexus epithelial cells. PLoS ONE. 2015;10(3):e0121738.
Malinovskaya NA, Komleva YK, Salmin VV, Morgun AV, Shuvaev AN, Panina YA, Boitsova EB, Salmina AB. Endothelial progenitor cells physiology and metabolic plasticity in brain angiogenesis and blood–brain barrier modeling. Front Physiols. 2016;7:599.
Zhang ZG, Zhang L, Jiang Q, Chopp M. Bone marrow-derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ Res. 2002;90(3):284–8.
Beck H, Voswinckel R, Wagner S, Ziegelhoeffer T, Heil M, Helisch A, Schaper W, Acker T, Hatzopoulos AK, Plate KH. Participation of bone marrow-derived cells in long-term repair processes after experimental stroke. J Cereb Blood Flow Metab. 2003;23(6):709–17.
Hamisha KN, Tfilin M, Yanai J, Turgeman G. Mesenchymal stem cells can prevent alterations in behavior and neurogenesis induced by Ass25-35 administration. J Mol Neurosci. 2015;55(4):1006–13.
Violatto MB, Santangelo C, Capelli C, Frapolli R, Ferrari R, Sitia L, Tortarolo M, Talamini L, Previdi S, Moscatelli D, et al. Longitudinal tracking of triple labeled umbilical cord derived mesenchymal stromal cells in a mouse model of amyotrophic lateral sclerosis. Stem Cell Res. 2015;15(1):243–53.
Petrovic-Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory Disequilibrium in Stroke. Circ Res. 2016;119(1):142–58.
Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92(4):463–77.
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808.
Kim JY, Park J, Chang JY, Kim SH, Lee JE. Inflammation after ischemic stroke: the role of leukocytes and glial cells. Exp Neurobiol. 2016;25(5):241–51.
Vexler ZS, Yenari MA. Does inflammation after stroke affect the developing brain differently than adult brain? Dev Neurosci. 2009;31(5):378–93.
Schallner N, Pandit R, LeBlanc R 3rd, Thomas AJ, Ogilvy CS, Zuckerbraun BS, Gallo D, Otterbein LE, Hanafy KA. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J Clin Investig. 2015;125(7):2609–25.
Zhao X, Sun G, Ting SM, Song S, Zhang J, Edwards NJ, Aronowski J. Cleaning up after ICH: the role of Nrf2 in modulating microglia function and hematoma clearance. J Neurochem. 2015;133(1):144–52.
Kowarik MC, Grummel V, Wemlinger S, Buck D, Weber MS, Berthele A, Hemmer B. Immune cell subtyping in the cerebrospinal fluid of patients with neurological diseases. J Neurol. 2014;261(1):130–43.
Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, Mirlas-Neisberg N, Cardon M, Vaknin I, Cahalon L, et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci USA. 2013;110(6):2264–9.
Ritzel RM, Crapser J, Patel AR, Verma R, Grenier JM, Chauhan A, Jellison ER, McCullough LD. Age-associated resident memory CD8 T cells in the central nervous system are primed to potentiate inflammation after ischemic brain injury. J Immunol. 2016;196(8):3318–30.
Yang D, Sun YY, Bhaumik SK, Li Y, Baumann JM, Lin X, Zhang Y, Lin SH, Dunn RS, Liu CY, et al. Blocking lymphocyte trafficking with FTY720 prevents inflammation-sensitized hypoxic-ischemic brain injury in newborns. J Neurosci. 2014;34(49):16467–81.
Coisne C, Engelhardt B. Tight junctions in brain barriers during central nervous system inflammation. Antioxid Redox Signal. 2011;15(5):1285–303.
Shrestha B, Paul D, Pachter JS. Alterations in tight junction protein and IgG permeability accompany leukocyte extravasation across the choroid plexus during neuroinflammation. J Neuropathol Exp Neurol. 2014;73(11):1047–61.
Szmydynger-Chodobska J, Strazielle N, Gandy JR, Keefe TH, Zink BJ, Ghersi-Egea JF, Chodobski A. Posttraumatic invasion of monocytes across the blood-cerebrospinal fluid barrier. J Cereb Blood Flow Metab. 2012;32(1):93–104.
Szmydynger-Chodobska J, Strazielle N, Zink BJ, Ghersi-Egea JF, Chodobski A. The role of the choroid plexus in neutrophil invasion after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29(9):1503–16.
Karimy JK, Duran D, Hu JK, Gavankar C, Gaillard JR, Bayri Y, Rice H, DiLuna ML, Gerzanich V, Marc Simard J, et al. Cerebrospinal fluid hypersecretion in pediatric hydrocephalus. Neurosurg Focus. 2016;41(5):E10.
Scarff JE. The treatment of nonobstructive (communicating) hydrocephalus by endoscopic cauterization of the choroid plexuses. J Neurosurg. 1970;33(1):1–18.
Weil AG, Westwick H, Wang S, Alotaibi NM, Elkaim L, Ibrahim GM, Wang AC, Ariani RT, Crevier L, Myers B, et al. Efficacy and safety of endoscopic third ventriculostomy and choroid plexus cauterization for infantile hydrocephalus: a systematic review and meta-analysis. Child’s Nerv Syst. 2016;32(11):2119–31.
Holloway LS Jr, Cassin S. Cerebrospinal fluid dynamics in the newborn dog during normoxia and hypoxia. Am J Physiol. 1972;223(3):499–502.
Oshio K, Watanabe H, Song Y, Verkman AS, Manley GT. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel Aquaporin-1. FASEB J. 2005;19(1):76–8.
Sveinsdottir S, Gram M, Cinthio M, Sveinsdottir K, Morgelin M, Ley D. Altered expression of aquaporin 1 and 5 in the choroid plexus following preterm intraventricular hemorrhage. Dev Neurosci. 2014;36(6):542–51.
Podvin S, Gonzalez AM, Miller MC, Dang X, Botfield H, Donahue JE, Kurabi A, Boissaud-Cooke M, Rossi R, Leadbeater WE, et al. Esophageal cancer related gene-4 is a choroid plexus-derived injury response gene: evidence for a biphasic response in early and late brain injury. PLoS ONE. 2011;6(9):e24609.
Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, Steinberg GK, Barres BA, Nimmerjahn A, Agalliu D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014;82(3):603–17.
Andras IE, Toborek M. Extracellular vesicles of the blood-brain barrier. Tissue Barriers. 2016;4(1):e1131804.
Chiasserini D, van Weering JR, Piersma SR, Pham TV, Malekzadeh A, Teunissen CE, de Wit H, Jimenez CR. Proteomic analysis of cerebrospinal fluid extracellular vesicles: a comprehensive dataset. J Proteom. 2014;106:191–204.
Street JM, Barran PE, Mackay CL, Weidt S, Balmforth C, Walsh TS, Chalmers RT, Webb DJ, Dear JW. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J Transl Med. 2012;10:5.
Yagi Y, Ohkubo T, Kawaji H, Machida A, Miyata H, Goda S, Roy S, Hayashizaki Y, Suzuki H, Yokota T. Next-generation sequencing-based small RNA profiling of cerebrospinal fluid exosomes. Neurosci Lett. 2017;636:48–57.
Yang Y, Keene CD, Peskind ER, Galasko DR, Hu SC, Cudaback E, Wilson AM, Li G, Yu CE, Montine KS, et al. Cerebrospinal fluid particles in Alzheimer disease and Parkinson disease. J Neuropathol Exp Neurol. 2015;74(7):672–87.
Chen J, Venkat P, Zacharek A, Chopp M. Neurorestorative therapy for stroke. Front Hum Neurosci. 2014;8:382.
Zhang ZG, Chopp M. Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol. 2009;8(5):491–500.
Authors’ contributions
All the authors contributed to the writing and editing of this manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors have read the final version of the manuscript and consented to publication.
Funding
This work was supported by the National Institutes of Health Grants NS073595, NS079157, NS090925, NS091545, NS093399, NS096917 and NS007222. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Lisa Juul Routhe is recipient of a Ph.D-stipend obtained from the Lundbeck foundation (Grant No: R191-2015-1360).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xiang, J., Routhe, L.J., Wilkinson, D.A. et al. The choroid plexus as a site of damage in hemorrhagic and ischemic stroke and its role in responding to injury. Fluids Barriers CNS 14, 8 (2017). https://doi.org/10.1186/s12987-017-0056-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-017-0056-3