
Abstract
Background
During the last two decades, structural biology analyses have shown that viruses infecting hosts far apart in evolution share similar architectural features, prompting a new virus classification based on structural lineages. Until recently, only a few prokaryotic viruses had been described for one of the lineages, whose main characteristic is a capsid protein with a perpendicular double jelly roll.
Main body
Metagenomics analyses are showing that the variety of prokaryotic viruses encoding double jelly roll capsid proteins is much larger than previously thought. The newly discovered viruses have novel genome organisations with interesting implications for virus structure, function and evolution. There are also indications of their having a significant ecological impact.
Conclusion
Viruses with double jelly roll capsid proteins that infect prokaryotic hosts form a large part of the virosphere that had so far gone unnoticed. Their discovery by metagenomics is only a first step towards many more exciting findings. Work needs to be invested in isolating these viruses and their hosts, characterizing the structure and function of the proteins their genomes encode, and eventually access the wealth of biological information they may hold.
Similar content being viewed by others

Structural biology and the first glimpses of the double jelly roll reach
Towards the end of last century, many virus structures had been determined by protein crystallography, showing that the β-barrel fold (consisting of eight antiparallel β-strands organized in two sheets that form the opposite sides of the barrel) was a common feature in the organization of icosahedral virus capsids [1]. ssDNA viruses infecting bacteria (Microviridae such as ΦX174), as well as ssRNA viruses infecting plants (e.g. tombusviruses), insects (tetra-, noda-, dicistroviruses), cattle (foot-and-mouth disease virus) and humans (rhinovirus, poliovirus) all were found to build their capsids using proteins that fold as a “jelly roll” β-barrel. Back then, only one dsDNA virus, human adenovirus, was known to utilize the β-barrel fold in its capsid, albeit in an odd way. The adenovirus major coat protein contains two β-barrels instead of one, an arrangement also referred to as double jelly roll [2] (Fig. 1). The adenovirus β-barrels are not parallel, but perpendicular to the capsid surface, and form pseudo-hexagonal capsomers, allowing trimeric proteins to fill in the six-fold coordinated positions of the icosahedral capsid [3].

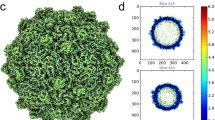
From the simplest to the most complex double jelly roll virus structures solved so far. The structures of the major capsid protein monomer (top row) and trimer (middle row) are shown, together the complete capsid (bottom row) of bacteriophage PM2, human adenovirus, and Faustovirus. These viruses represent the simplest and most complex examples for which both the high resolution structure of the major coat protein and at least the general capsid organization have been solved. While the PM2 major coat protein is formed by the double jelly roll motif with no more elaborations, the adenovirus and Faustovirus proteins have extensive tower domains which establish intricate interlacing in the trimer. Database identifiers and triangulation numbers are shown by each structure. The colour bar indicates capsid radii, in nm
Then, in 1999, the major coat protein structure of a peculiar, tail-less, membrane-containing dsDNA bacteriophage called PRD1 was solved, and unexpectedly proved that the human adenovirus structural solution was present also in viruses with prokaryotic hosts [4]. This finding raised questions on virus evolution, hinting at a possible common ancestor of viruses infecting prokaryotic and eukaryotic organisms [5]. At about the same time, it was also realized that herpesviruses share architectural characteristics with tailed phages, and that reoviruses have structural similarity with the bacterial cystoviruses [6, 7]. All these observations evolved into the proposal that a classification on structural lineages, based on major coat protein folds, might be more useful, and better reflect the evolutionary history of viruses, than previous classifications based on genome type or host [8,9,10].
Four icosahedral virus structural lineages are recognized at present [10], with indications that other lineages may exist, for example one encompassing positive and negative ssRNA viruses [11]. The dsDNA herpesviruses, which infect animals, form one structural lineage with tailed bacteriophages: they share many structural characteristics, including their assembly pathway and major coat protein fold. A second structural lineage includes the dsRNA cystoviruses (bacteriophages like Φ6) together with eukaryotic dsRNA viruses such as reo- or totiviruses. A third lineage encompasses picorna-like viruses, with coat proteins folding as a β-barrel lying parallel to the capsid surface. Adenoviruses, which infect vertebrates, and the tectivirus PRD1 were the founding members of the fourth icosahedral structural lineage, which encompasses dsDNA viruses infecting organisms across the evolutionary tree (Table 1): bacteria (tectiviruses, corticoviruses), archaea (turriviruses), unicellular animals (giant viruses like mimivirus and their relatives, also their virophages) and algae (phycodnaviruses), insects, fish, amphibians and reptiles (iridoviruses), pigs (asfarviruses), and vertebrates in general including humans (adenoviruses) [10, 12]. The infectious particles of all these viruses are built from trimeric double jelly roll capsomers (Fig. 1), arranged with triangulation numbers ranging between T = 21 [13] and 499 [14]. The triangulation number of the giant mimivirus capsid, which has not been unequivocally determined yet, is estimated to be in the 972–1200 range [15]. Members of the double jelly roll lineage have also a single perpendicular jelly roll protein forming the pentameric vertex capsomers. Remarkably, a scaffold protein of the non-icosahedral poxviruses involved in the initial stages of assembly also folds as a double β-barrel pseudo-hexamer [16].
How did the double jelly roll fold jump from prokaryotic to eukaryotic hosts?
The fact that viruses with different hosts share a common structural solution suggests that the architecture was established in the early stages of evolution, before the branches of the evolutionary tree diverged into the three kingdoms known today (archaea, bacteria and eukarya). Intriguingly, an evolutionary connection has been found between viruses in the double jelly roll lineage and large (15–20 kbp) eukaryotic double-stranded DNA transposons called Polintons [17]. Polintons are so named because they all encode a protein-primed DNA polymerase (to sustain self-replication, POL) and a retroviral-like integrase (INT). Most of them also include genes for a DNA-packaging ATPase and a maturation protease like those found in double jelly roll lineage viruses. Exhaustive sequence analyses revealed that these transposable elements also encode genes that could translate into double or single jelly roll proteins, suggesting that at some point in time, or in certain conditions, they could form icosahedral capsids.
In the light of all these findings, an evolutionary model was proposed in which a primordial, PRD1-like double jelly roll phage (encoding a double jelly roll capsid protein, a protein-primed DNA polymerase and a packaging ATPase) would have invaded a proto-eukaryotic host with a bacterial endosymbiont (mitochondria), somehow reached the nucleus, and recombined with a eukaryotic transposable DNA element carrying the integrase and maturation protease. This “polintovirus” element would have then evolved in separate ways to produce the polintons (transposable, capsid-less integrating elements), and a variety of eukaryotic “free-standing” viruses, all the way from adenovirus to mimiviruses [18].
New findings from metagenomics extend the double jelly roll reach
The great majority of known dsDNA viruses belong to either the tailed phage/herpes lineage or to the double jelly roll lineage. The tailed phage/herpes lineage is massively dominated by the tailed phages, with herpesviruses the only eukaryotic members. Conversely, there is a large variety of double jelly roll viruses infecting eukaryotic hosts, from algae to humans, while only a few lineage members with prokaryotic hosts (bacteria and archaea) have been isolated (Table 1). Even within this paucity, some discoveries hinted at variant uses of the double jelly roll architecture, and its possible widespread use in the prokaryotic world. On the one hand, the Flavobacterium-infecting, lipid containing phage FLiP, has a double jelly roll architecture but a circular ssDNA instead of a dsDNA genome [19], demonstrating the use of similar architectural solutions irrespective of genome nature. On the other, some viruses infecting archaea or extremophile bacteria encode two major coat proteins, each folding as a single β-barrel, that combine in hetero-multimers to produce capsids with the single jelly rolls perpendicular to the surface [20,21,22]. The existence of these later viruses supports the hypothesis that double jelly roll coat proteins may have evolved from single jelly rolls by gene duplication [23].
Progress in structural biology technologies facilitated the studies on large, complex coat proteins and virus particles that were instrumental in revealing the structural lineages. In parallel, highly advanced DNA sequencing methods became common, paving the way for environmental metagenomics projects that are nowadays the main source of virus discovery [24, 25]. Metagenomics allows virus discovery even if the host is not known or cannot be cultured in laboratory conditions. By providing previously inaccessible, large amounts of sequence data, metagenomics has also facilitated the analysis of virus evolution trends. Marine metagenome analyses have recently revealed a new group of putative polinton-like viruses in algae [26]. Polinton-like virus genomes contain genes for single and double jelly roll proteins and a packaging ATPase, but lack the protease and integrase genes. Therefore, polinton-like viruses could represent a minimal version of the double jelly roll lineage in eukaryotic hosts, or perhaps the first eukaryotic dsDNA viruses to evolve from bacterial ancestors [26].
Morphological surveys on marine samples suggested that non-tailed phages might even be more abundant than the tailed ones, despite their scarcity in culture and sequence collections [27]. More recently, examination of agents infecting marine Vibrionaceae bacteria has revealed that a new group of double jelly roll viruses, the autolykiviruses, has a very broad host range, and may be responsible for a large part of deaths in marine bacteria, indicating the ecological relevance of double jelly roll tail-less phages [28, 29]. With 10 kbp long genomes and 49 nm diameter capsids, the autolykiviruses would be the smallest members of the double jelly roll lineage found so far.
A more recent study used the previously identified prokaryotic double jelly roll major coat protein sequences as bait for mining the GenBank and metagenomics databases [30]. Some of the hits found were flanked by typical bacterial genes, reminding us that analyses limited to genomic sequences might identify non-functional prophages as well as actual viruses. But once this was taken into account, the authors found indications that many more double jelly roll virus families may exist in the prokaryotic landscape, including a completely new group of viruses (termed Odin), which has no characterized members. It was remarkable that, when the database search was carried out with just the presence of the double jelly roll major coat protein as a common trait, a large variety of genome organizations was found. It was observed that two genes previously thought to be fundamental lineage traits can be absent: the protein-primed replication polymerase, and the packaging ATPase. These were considered part of the “primordial” double jelly roll virus in bacteria that recombined with transposons in eukaryotic cells [18]. The finding that double jelly roll prokaryotic viruses may exist without these two genes raises questions about their mode of assembly and replication, and their place in the evolutionary landscape.
The role of the packaging ATPase is still a mystery for many double jelly roll viruses. While it seems to function as a bona fide portal for genome translocation into a preformed capsid in bacteriophage PRD1 [31, 32], such a function does not appear so obvious for members of the lineage where topological constraints are at odds with genome translocation. For example, it is not clear how the corticovirus PM2, with its circular, supercoiled dsDNA genome, or adenovirus, with a linear dsDNA genome heavily covered by protein, would use a portal with a packaging ATPase for genome translocation [13, 33, 34]. Until recently, only FLiP, the single lineage member with a circular ssDNA genome, had been found to lack the ATPase gene [19]. Now it is found that viruses in the Odin group also lack it, and have instead an open reading frame coding for a small protein preceding the major coat protein gene. This small protein has no detected similarity to any known proteins, but is conserved throughout the group.
Conclusions
Prokaryotic double jelly roll viruses are much more abundant and hold much more genomic variability than previously thought. These realizations open the way to exciting future findings: more new viruses, new modes of genome replication and particle assembly, new host-pathogen interactions, and ecological relevance. To achieve all this new knowledge, several steps need to be addressed first, such as identifying the virus hosts, isolating the virus particles themselves, solving the structure of the capsid and determining the folds of other virus protein structures.
References
Chapman MS, Liljas L. Structural folds of viral proteins. Adv Prot Chem. 2003;64:125–96.
Roberts MM, White JL, Grutter MG, Burnett RM. 3-dimensional structure of the adenovirus major coat protein hexon. Science. 1986;232:1148–51.
San Martín C. Latest insights on adenovirus structure and assembly. Viruses. 2012;4:847–77.
Benson SD, Bamford JKH, Bamford DH, Burnett RM. Viral evolution revealed by bacteriophage PRD1 and human adenovirus coat protein structures. Cell. 1999;98:825–33.
Hendrix RW. Evolution: the long evolutionary reach of viruses. Curr Biol. 1999;9:R914–7.
Bamford DH, Burnett RM, Stuart DI. Evolution of viral structure. Theor Popul Biol. 2002;61:461–70.
Baker ML, Jiang W, Rixon FJ, Chiu W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J Virol. 2005;79:14967–70.
Bamford DH. Do viruses form lineages across different domains of life? Res Microbiol. 2003;154:231–6.
Bamford DH, Grimes JM, Stuart DI. What does structure tell us about virus evolution? Curr Opin Struct Biol. 2005;15:655–63.
Abrescia NG, Bamford DH, Grimes JM, Stuart DI. Structure unifies the viral universe. Annu Rev Biochem. 2012;81:795–822.
Valle M. Structural homology between nucleoproteins of ssRNA viruses. In: Harris J, Bhella D, editors. Virus protein and nucleoprotein complexes. Subcellular biochemistry, vol. 88. Singapore: Springer; 2018. p. 129–45.
Krupovic M, Bamford DH. Virus evolution: how far does the double beta-barrel viral lineage extend? Nat Rev Microbiol. 2008;6:941–8.
Huiskonen JT, Kivela HM, Bamford DH, Butcher SJ. The PM2 virion has a novel organization with an internal membrane and pentameric receptor binding spikes. Nat Struct Mol Biol. 2004;11:850–6.
Xiao C, Fischer MG, Bolotaulo DM, Ulloa-Rondeau N, Avila GA, Suttle CA. Cryo-EM reconstruction of the Cafeteria roenbergensis virus capsid suggests novel assembly pathway for giant viruses. Sci Rep. 2017;7:5484.
Xiao C, Kuznetsov YG, Sun S, Hafenstein SL, Kostyuchenko VA, Chipman PR, et al. Structural studies of the giant mimivirus. PLoS Biol. 2009;7:e92.
Bahar MW, Graham SC, Stuart DI, Grimes JM. Insights into the evolution of a complex virus from the crystal structure of vaccinia virus D13. Structure. 2011;19:1011–20.
Fischer MG, Suttle CA. A virophage at the origin of large DNA transposons. Science. 2011;332:231–4.
Krupovic M, Koonin EV. Polintons: a hotbed of eukaryotic virus, transposon and plasmid evolution. Nat Rev Microbiol. 2015;13:105–15.
Laanto E, Mantynen S, De Colibus L, Marjakangas J, Gillum A, Stuart DI, et al. Virus found in a boreal lake links ssDNA and dsDNA viruses. Proc Natl Acad Sci U S A. 2017;114:8378–83.
Jaalinoja HT, Roine E, Laurinmaki P, Kivela HM, Bamford DH, Butcher SJ. Structure and host-cell interaction of SH1, a membrane-containing, halophilic euryarchaeal virus. Proc Natl Acad Sci USA. 2008;105:8008–13.
Rissanen I, Grimes JM, Pawlowski A, Mantynen S, Harlos K, Bamford JK, et al. Bacteriophage P23-77 capsid protein structures reveal the archetype of an ancient branch from a major virus lineage. Structure. 2013;21:718–6.
Gil-Carton D, Jaakkola ST, Charro D, Peralta B, Castano-Diez D, Oksanen HM, et al. Insight into the assembly of viruses with vertical single beta-barrel major capsid proteins. Structure. 2015;23:1866–77.
Krupovic M, Koonin EV. Multiple origins of viral capsid proteins from cellular ancestors. Proc Natl Acad Sci U S A. 2017;114:E2401–10.
Mardis ER. A decade’s perspective on DNA sequencing technology. Nature. 2011;470:198–203.
Simmonds P, Adams MJ, Benko M, Breitbart M, Brister JR, Carstens EB, et al. Consensus statement: virus taxonomy in the age of metagenomics. Nat Rev Microbiol. 2017;15:161–8.
Yutin N, Shevchenko S, Kapitonov V, Krupovic M, Koonin EV. A novel group of diverse Polinton-like viruses discovered by metagenome analysis. BMC Biol. 2015;13:95.
Brum JR, Schenck RO, Sullivan MB. Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 2013;7:1738–51.
Kauffman KM, Hussain FA, Yang J, Arevalo P, Brown JM, Chang WK, et al. A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria. Nature. 2018;554:118–22.
Ignacio-Espinoza JC, Fuhrman JA. A non-tailed twist in the viral tale. Nature. 2018;554:38–9.
Yutin N, Backstrom D, Ettema TJG, Krupovic M, Koonin EV. Vast diversity of prokaryotic virus genomes encoding double jelly-roll major capsid proteins uncovered by genomic and metagenomic sequence analysis. Virol J. 2018;15:67.
Hong C, Oksanen HM, Liu X, Jakana J, Bamford DH, Chiu W. A structural model of the genome packaging process in a membrane-containing double stranded DNA virus. PLoS Bio. 2014;12:e1002024.
Stromsten NJ, Bamford DH, Bamford JK. In vitro DNA packaging of PRD1: a common mechanism for internal-membrane viruses. J Mol Biol. 2005;348:617–29.
Abrescia NG, Grimes JM, Kivela HM, Assenberg R, Sutton GC, Butcher SJ, et al. Insights into virus evolution and membrane biogenesis from the structure of the marine lipid-containing bacteriophage PM2. Mol Cell. 2008;31:749–61.
Condezo GN, San Martín C. Localization of adenovirus morphogenesis players, together with visualization of assembly intermediates and failed products, favor a model where assembly and packaging occur concurrently at the periphery of the replication center. PLoS Pathog. 2017;13:e1006320.
ICTV Taxonomy. https://talk.ictvonline.org/taxonomy/. Accessed 30 Oct 2018.
Abrescia NGA, Cockburn JJB, Grimes JM, Sutton GC, Diprose JM, Butcher SJ, et al. Insights into assembly from structural analysis of bacteriophage PRD1. Nature. 2004;432:68–74.
Aalto AP, Bitto D, Ravantti JJ, Bamford DH, Huiskonen JT, Oksanen HM. Snapshot of virus evolution in hypersaline environments from the characterization of a membrane-containing Salisaeta icosahedral phage 1. Proc Natl Acad Sci U S A. 2012;109:7079–84.
Veesler D, Ng TS, Sendamarai AK, Eilers BJ, Lawrence CM, Lok SM, Younge MJ, et al. Atomic structure of the 75 MDa extremophile Sulfolobus turreted icosahedral virus determined by CryoEM and X-ray crystallography. Proc Natl Acad Sci U S A. 2013;110:5504–9.
Nandhagopal N, Simpson AA, Gurnon JR, Yan X, Baker TS, Graves MV, et al. The structure and evolution of the major capsid protein of a large, lipid-containing DNA virus. Proc Natl Acad Sci U S A. 2002;99:14758–63.
Wilson WH, Van Etten JL, Schroeder DC, Nagasaki K, Brussaard C, Bratbak G, et al. Phycodnaviridae. In: ICTV 9th report; 2011. https://talk.ictvonline.org/ictv-reports/ictv_9th_report/dsdna-viruses-2011/w/dsdna_viruses/123/phycodnaviridae. Accessed 30 Oct 2018.
Yan X, Chipman PR, Castberg T, Bratbak G, Baker TS. The marine algal virus PpV01 has an icosahedral capsid with T=219 quasisymmetry. J Virol. 2005;79:9236–43.
Fischer MG, Allen MJ, Wilson WH, Suttle CA. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc Natl Acad Sci U S A. 2010;107:19508–13.
Klose K, Kuznetsov YG, Xiao C, Sun S, McPherson A, Rossmann MG. The three-dimensional structure of Mimivirus. Intervirology. 2010;53:268–73.
Zhang X, Sun S, Xiang Y, Wong J, Klose T, Raoult D, et al. Structure of sputnik, a virophage, at 3.5-Å resolution. Proc Natl Acad Sci U S A. 2012;109:18431–6.
Okamoto K, Miyazaki N, Reddy HKN, Hantke MF, Maia FRNC, Larsson DSD, et al. Cryo-EM structure of a Marseilleviridae virus particle reveals a large internal microassembly. Virology. 2018;516:239–45.
Doutre G, Philippe N, Abergel C, Claverie JM. Genome analysis of the first Marseilleviridae representative from Australia indicates that most of its genes contribute to virus fitness. J Virol. 2014;88:14340–9.
Klose T, Reteno DG, Benamar S, Hollerbach A, Colson P, La Scola B, et al. Structure of faustovirus, a large dsDNA virus. Proc Natl Acad Sci USA. 2016;113:6206–11.
Unclassified viruses. In: ICTV report. https://talk2x.ictvonline.2xorg/ictv-reports/ictv_online_report/unclassified-viruses/w/unclassified-viruses Accessed 30 Oct 2018.
Reteno DG, Benamar S, Khalil JB, Andreani J, Armstrong N, Klose T, et al. Faustovirus, an asfarvirus-related new lineage of giant viruses infecting amoebae. J Virol. 2015;89:6585–94.
Andreani J, Khalil JYB, Sevvana M, Benamar S, Di Pinto F, Bitam I, et al. Pacmanvirus, a new giant icosahedral virus at the crossroads between Asfarviridae and Faustoviruses. J Virol. 2017;91:e00212–7.
Yan X, Olson NH, Van Etten JL, Bergoin M, Rossmann MG, Baker TS. Structure and assembly of large lipid-containing dsDNA viruses. Nat Struct Biol 2000;7:101–103.
Yan X, Yu Z, Zhang P, Battisti AJ, Holdaway HA, Chipman PR, et al. The capsid proteins of a large, icosahedral dsDNA virus. J Mol Biol. 2009;385:1287–99.
Claverie JM, Abergel C. Mimiviridae: an expanding family of highly diverse large dsDNA viruses infecting a wide phylogenetic range of aquatic eukaryotes. Viruses. 2018;10:e506.
Acknowledgements
We apologize to those colleagues whose research has not been mentioned in this mini-review, and acknowledge them all for theirwork.
Funding
Work was funded by grants BFU2014–53425-P and BFU2016–74868-P, co-funded by the Spanish State Research Agency and the European Regional Development Fund, as well as BIO2015–68990-REDT (the Spanish Adenovirus Network, AdenoNet), from the Spanish Ministry of Economy, Industry and Competitiveness. The funding bodies did not play any role in the writing of the manuscript.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
CSM and MJvR wrote the manuscript. Both authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they do not have any competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
San Martín, C., van Raaij, M.J. The so far farthest reaches of the double jelly roll capsid protein fold. Virol J 15, 181 (2018). https://doi.org/10.1186/s12985-018-1097-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-018-1097-1