
Abstract
Nucleo-cytoplasmic large DNA viruses are doubled stranded DNA viruses capable of infecting eukaryotic cells. Since the discovery of Mimivirus and Pandoravirus, there has been no doubt about their extraordinary features compared to “classic” viruses. Recently, we reported the expansion of the proposed family Pithoviridae, with the description of Cedratvirus and Orpheovirus, two new viruses related to Pithoviruses. Studying the major capsid protein of Orpheovirus, we detected a homologous sequence in a mine drainage metagenome. The in-depth exploration of this metagenome, using the MG-Digger program, enabled us to retrieve up to 10 contigs with clear evidence of viral sequences. Moreover, phylogenetic analyses further extended our screening with the discovery in another marine metagenome of a second virus closely related to Orpheovirus IHUMI-LCC2. This virus is a misidentified virus confused with and annotated as a Rickettsiales bacterium. It presents a partial genome size of about 170 kbp.
Similar content being viewed by others


Main text
Metagenomic analysis is a powerful method to detect micro-organisms in their ecosystems. These micro-organisms belong to all the components of the tree of life but also to those of the viral world. The commitment to be totally independent from the culture process constitutes an extremely important part of its success [1, 2]. These techniques have pushed researchers to explore different environments, or microbiota as in the Human Microbiome Project [3], and sometimes multiplying samples collection in various environments such as the international space station or as the Permafrost in “omics” study [4, 5]. Nevertheless, a wide part of these metagenomic results are still unknown in database, and is referred to as “dark matter” [6]. On the opposite side, the culture tools and notably some culturomics studies allows us to describe and characterize isolates, which is impossible by the use of metagenomic analysis alone [7]. This confirms the extreme complementary of both methods.
On the other hand, giant viruses are double stranded DNA viruses described as potentially comparable to certain bacteria by their genomic length and their particle size [8, 9]. Based on the reconstructions of ancestral sequences of viral RNA polymerase II subunits as baits, successful studies were carried out to identify individual sequences as well as partial or near complete genomes [10,11,12,13]. Finally, each new viral description which came from culture isolation allows us to retrospectively better understand unknown old or new metagenomic reports and finally permits to better decipher the “dark matter”. Recently, we characterised a new giant virus named Orpheovirus and its growth on the amoeba Vermamoeba vermiformis [14]. The gene ORPV_1034 was predicted to encode a 587 amino-acid viral major capsid protein (MCP). With the goal of investigating homologies in database to increase efficiency of our annotation we used, with standard parameter (i.e. e-value inclusion threshold 1e-3), an online software (HHblits) able to detect homologies and structure. The result showed 65 hits but only 2 proteins matched with a special interest (Additional file 1). The probability obtained was at 97,72% with a length of 302 amino-acids between Orpheovirus and Pithovirus sibericum and it was at 97,04% with alignments of 237 amino-acids between Orpheovirus and unknown sequence present in a metagenome. Then, using PSI-BLAST against metagenomes database, we also detected homologies between Orpheovirus and Cedratvirus A11 (55% coverage, 29%identity) as well as with Pithovirus sibericum (41% coverage, 21% identity). PSI-BLAST against metagenomes database of the Orpheovirus’s MCP detected an homology with the same protein detected by HHblits (47% coverage, 24%identity) with the sequence EQD26795.1 and also, with 2 additional proteins issued from different marine metagenomes [15, 16]. We observed that the best hit with this viral capsid came from an acid mine drainage metagenome located in Spain.
We decided to investigate this pyrite mine using MG-Digger program [17] with updated giant viruses database (e.g Pithovirus massiliensis LC8, Pacmanvirus A23, Cedratvirus A11, Cedratvirus lausannensis, Orpheovirus IHUMI-LCC2, Kaumoebavirus and Klosneuvirinae). Projects have been previously registered [18] under the IDs PRJNA193663 sample (B1A), PRJNA193664 (sample B1B) and PRJNA193665 (sample B2A) at NCBI. This Whole Genome Shotgun project has been deposited under the accession AUZX00000000- AUZY00000000-AUZZ00000000. The parameters used for MG-Digger were fixed with an e-value cut-off of 10− 3 and no limit of both coverage and identity percent are defined to detect giant viruses-like sequences in the alignment of the BLAST options. All contigs available in the mine drainage metagenome represent 41,233 contigs. The MG-Digger program accepts protein or nucleic acid sequences in input [17] and in this case, we used nucleic acid contigs in input against updated database.
We detected and revealed 10 contigs from the series AUZX and AUZY as showing best hit informing the viral presence (samples B1A and B1B) (Table 1, Additional file 2) with no doubt that their origins are viral. Indeed, they presented a score higher than 60 for all queries with an average coverage of 79,2% (ranging from 53 to 99%), e-value inferior at 10− 7 and identity with an average of about 38% (ranging from 27% to 48%). Each contig detected encode for one putative protein. To complete the annotation, all proteins corresponding to each viral contig were downloaded and all annotations were confirmed by DELTA-BLASTP program [19]. Six viral contigs retrieved as best hit proteins from the members of the proposed family Pithoviridae (Pithoviruses and Cedratviruses) and Orpheovirus. Detailed annotation of these 6 proteins encoded by these contigs revealed 1 DNA polymerase B family, 1 DNA topoisomerase IIA, 1 mRNA capping enzyme, 1, ribonucleoside-diphosphate reductase, 1 Very Early Transcription Factor (VETF) and the previously found MCP. Moreover, we found 2 putative Ankyrin-repeat proteins matching with Mimiviruses, and one hypothetical protein and one protein coding for D5 helicase primase protein present best hits with Iridoviruses (Table 1, Additional file 2).
Subsequently, phylogenetic analyses were performed at first glance on these predicted proteins (data not shown). Indeed, we retrieved homologous sequences by Blast protein strategy with Orpheovirus, Cedratviruses, Pithoviruses and some of the 9 contigs and also with a Rickettsiales bacterium present in the NCBI database (belonging to another marine metagenome from Atlantic North under the accession number: NVVL00000000.1).
Then, this marine metagenome for which the annotation was performed by automatic pipeline, was further investigated. Amongst 111 contigs, all genes and proteins were predicted de novo using GenemarkS [20], with a blastp analysis of the 1947 predicted proteins followed by GC% assignment of putative viral contigs (Additional files 3 and 4). Indeed, we used an e-value cut-off at 10− 2 and our results highlight that some proteins have a best hit (Additional file 3) with viral strains notably with proteins previously described as fundamental proteins in Nucleo-Cytoplasmic-Large DNA viruses (e.g DNA-directed RNA polymerase subunit RPB2, Flap endonuclease 1, transcription initiation factor TFIIB, DNA polymerase delta catalytic subunit). Finally, we identified 15 viral contigs totalizing 177,601 base pairs with a GC % close to 33%. Initially annotated as a Rickettsiales hit, we propose to re-annotate these viral sequences like a Misidentifiedvirus. On the other hand, true Rickettsiales contigs present a GC% more variable around 40% and we identified 11 contigs that could potentially be a ciliate protist close to Oxytricha spp. (Additional file 5). This ciliate could be the host of the Misidentifiedvirus as previously described for others protists [12].
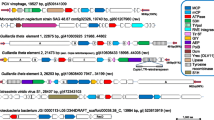
For all phylogenetic analyses we used Muscle algorithm [21] to perform alignment. Alignments were curated manually and finally, FastTree program was used to build maximum-likelihood trees with standard program (Jones-Taylor-Thornton model). Then, ITolV3 online was used to visualized trees [22]. Phylogenetic analysis based on the MCP anchors these 2 different viruses from marine and mine metagenomes closely-related to Orpheovirus (Fig. 1, Additional file 6). DNA topoisomerase II, VETF, and mRNA capping enzyme trees confirmed these results (Additional file 7). Nevertheless, due to the partial sequences for the mine drainage, their exact position could not definitively be determined.

Phylogenetic tree based on the viral major capsid protein. Alignment was performed using Muscle in the MEGA6.0 software. MVIEW [26] was used to visualized the alignment, and after we performed a maximum likelihood tree on the 423 positions with 1000 bootstrap replication with FastTree program (JTT model) with standard parameter. ITolV3 online [22] was used to visualized the tree. Branches under 0.5 as bootstrap value were deleted
Retrieving best hit virus constitute an unambiguous evidence for viral presence, especially when we found structural gene described like hallmark genes in Nucleo-cytoplasmic Large DNA viruses [23, 24]. Altogether, these data confirm a viral presence in this mine pyrite and a Misidentifiedvirus in the marine metagenome as two putative viruses possessing a close relationship with the proposed Pithoviridae and Orpheoviridae families. The rapid expansion of gigantic dsDNA reports and their genomic descriptions permit to evaluate and re-evaluate published and novel metagenomes. MG-digger program is a functional and appropriate tool to investigate them. Nevertheless, it is limited by the detection of best hit that it confers its confidence. Finally, a complementary method is emerging promoted by the next-generation sequencing. Indeed, the metagenomic binning approach represent a major step in the genome reconstruction from different metagenomes [12]. Such additional approaches could be implemented in future automatic pipelines. There is no doubt that the development of new programs [25] associated with efficient new viral isolation would enable further discoveries and start filling the knowledge gap in the current dark matter content.
Abbreviations
- MCP:
-
Major capsid protein
- VETF:
-
Very Early Transcription Factor
References
Schloss PD, Handelsman J. Metagenomics for studying unculturable microorganisms: cutting the Gordian knot. Genome Biol. 2005;6:229.
Taylor-Brown A, Spang L, Borel N, Polkinghorne A. Culture-independent metagenomics supports discovery of uncultivable bacteria within the genus chlamydia. Sci Rep. 2017;7:10661.
Consortium THMP. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14.
Hultman J, Waldrop MP, Mackelprang R, David MM, McFarland J, Blazewicz SJ, Harden J, Turetsky MR, McGuire AD, Shah MB, VerBerkmoes NC, Lee LH, Mavrommatis K, Jansson JK. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature. 2015;521:208–12.
Be NA, Avila-Herrera A, Allen JE, Singh N, Checinska Sielaff A, Jaing C, Venkateswaran K. Whole metagenome profiles of particulates collected from the international Space Station. Microbiome. 2017;5:81.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J-F, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu W-T, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–7.
Lagier J-C, Khelaifia S, Alou MT, Ndongo S, Dione N, Hugon P, Caputo A, Cadoret F, Traore SI, Seck EH, Dubourg G, Durand G, Mourembou G, Guilhot E, Togo A, Bellali S, Bachar D, Cassir N, Bittar F, Delerce J, Mailhe M, Ricaboni D, Bilen M, Dangui Nieko NPM, Dia Badiane NM, Valles C, Mouelhi D, Diop K, Million M, Musso D, et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nature Microb. 2016;1:16203.
La Scola B. A Giant Virus in Amoebae. Science. 2003;299:2033.
Philippe N, Legendre M, Doutre G, Couté Y, Poirot O, Lescot M, Arslan D, Seltzer V, Bertaux L, Bruley C, Garin J, Claverie J-M, Abergel C. Pandoraviruses: amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science. 2013;341:281–6.
Sharma V, Colson P, Giorgi R, Pontarotti P, Raoult D. DNA-dependent RNA polymerase detects hidden giant viruses in published databanks. Genome Biol Evol. 2014;6:1603–10.
Bekliz M, Verneau J, Benamar S, Raoult D, La Scola B, Colson P. A new Zamilon-like Virophage partial genome assembled from a bioreactor metagenome. Front Microbiol. 2015;6:1308.
Schulz F, Yutin N, Ivanova NN, Ortega DR, Lee TK, Vierheilig J, Daims H, Horn M, Wagner M, Jensen GJ, Kyrpides NC, Koonin EV, Woyke T. Giant viruses with an expanded complement of translation system components. Science. 2017;356:82–5.
Roux S, Chan L-K, Egan R, Malmstrom RR, McMahon KD, Sullivan MB. Ecogenomics of virophages and their giant virus hosts assessed through time series metagenomics. Nat Commun. 2017;8:858.
Andreani J, Khalil JYB, Baptiste E, Hasni I, Michelle C, Raoult D, Levasseur A, La Scola B. Orpheovirus IHUMI-LCC2: a new virus among the giant viruses. Front Microbiol. 2018;8:779.
Kawai M, Futagami T, Toyoda A, Takaki Y, Nishi S, Hori S, Arai W, Tsubouchi T, Morono Y, Uchiyama I, Ito T, Fujiyama A, Inagaki F, Takami H. High frequency of phylogenetically diverse reductive dehalogenase-homologous genes in deep subseafloor sedimentary metagenomes. Front Microbiol. 2014;5:80.
Spang A, Saw JH, Jørgensen SL, Zaremba-Niedzwiedzka K, Martijn J, Lind AE, van Eijk R, Schleper C, Guy L, Ettema TJG. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature. 2015;521:173–9.
Verneau J, Levasseur A, Raoult D, La Scola B, Colson P. MG-digger: an automated pipeline to search for Giant virus-related sequences in metagenomes. Front Microbiol. 2016;7:428.
Méndez-García C, Mesa V, Sprenger RR, Richter M, Diez MS, Solano J, Bargiela R, Golyshina OV, Manteca Á, Ramos JL, Gallego JR, Llorente I, Martins dos Santos VAP, Jensen ON, Peláez AI, Sánchez J, Ferrer M. Microbial stratification in low pH oxic and suboxic macroscopic growths along an acid mine drainage. ISME J. 2014;8:1259–74.
Boratyn GM, Schäffer AA, Agarwala R, Altschul SF. Domain enhanced lookup time accelerated BLAST. Biol Direct. 2012;7:12. https://doi.org/10.1186/1745-6150-7-12. https://www.ncbi.nlm.nih.gov/pubmed/22510480.
Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucl Acids Res. 2001;29:2607–18.
Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucl Acids Res. 2016;44:W242–5.
Iyer LM, Aravind L, Koonin EV. Common origin of four diverse families of large eukaryotic DNA viruses. J Virol. 2001;75:11720–34.
Colson P, De Lamballerie X, Yutin N, Asgari S, Bigot Y, Bideshi DK, Cheng X-W, Federici BA, Van Etten JL, Koonin EV, La Scola B, Raoult D. “Megavirales,” a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch Virol. 2013;158:2517–21.
Lin H-H, Liao Y-C. Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci Rep. 2016;6:24175.
EMBL-EBI 2018. https://www.ebi.ac.uk/Tools/msa/mview/ (2018). Accessed 20 Feb 2018.
Acknowledgements
The authors would particularly like to thank Claire Andréani for her help in the English correction.
Funding
This work was supported by the French Government under the « Investissements d’avenir » (Investments for the Future) program managed by the Agence Nationale de la Recherche (ANR, fr: National Agency for Research), (reference: Méditerranée Infection 10-IAHU-03).
Availability of data and materials
Authors can confirm that all relevant data obtained are included in the article and/or its supplementary information files.
Author information
Authors and Affiliations
Contributions
JA, JV and AL performed bio-informatic analyses. JA, DR and BL conceived the study. All authors wrote the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Data sheet about HHblits results obtained for the predicted protein ORPV_1034. 65 hits are mentioned with their probabilities, alignments, e-value and target length. (XLSX 24 kb)
Additional file 2:
MG-digger results. The NCBI databases available are up to October 2017. Data sheet contains all queries matching with each identified subjects. We added annotation in the column F. BLAST scores are indicate in columns from G to S. (XLSX 13 kb)
Additional file 3:
Blastp result obtained using de novo predicted proteins for the marine metagenome NVVL00000000.1. Data sheet represent all blastp results obtained and extracted specific blast for each contig. We added the annotation of the Misidentifiedvirus in the column H of the datasheet named “Genermarks-111contig”. (XLSX 14868 kb)
Additional file 4:
Marine metagenome proteins predicted. GenemarkS was used to predict de novo proteins from 111 “Rickettsiales Bacterium” contigs. (PDF 2040 kb)
Additional file 5:
List of NVVL00000000.1 contigs. We identified 15 and 11 contigs among 111 “Rickettsiales” contigs as viruses and as ciliate protist. (PDF 60 kb)
Additional file 6:
Protein alignment of Major capsid protein. This alignment was done MUSCLE with standard parameter and was used to build the tree in the Fig. 1. (PDF 104 kb)
Additional file 7:
Additional phylogenetic trees. Maximum likelihood trees were added based on VETF, mRNA capping enzyme and DNA topoisomerase II proteins. (PDF 744 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Andreani, J., Verneau, J., Raoult, D. et al. Deciphering viral presences: two novel partial giant viruses detected in marine metagenome and in a mine drainage metagenome. Virol J 15, 66 (2018). https://doi.org/10.1186/s12985-018-0976-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-018-0976-9