
Abstract
Background
Infectious bronchitis is a severe disease caused by infectious bronchitis virus (IBV) that affects fowl flocks worldwide. The understanding of the mechanisms involved in IBV evolution and variation would provide important theoretical basis for prevention and control of the disease in the future.
Methods
IBV strain GD was isolated from southern China in 2013 and the complete genome sequencing and phylogenetic analysis were performed.
Results
The genome of approximately 27,680 nt comprised six genes, with insertions and mutations in most of the structural genes. The S1 gene showed the highest identity to strain TW2575/98 isolated in Taiwan, and was distantly related to the H120 vaccine strain. Phylogenetic analysis showed that the S1 gene of strain GD was also related to that of TW-type strains. Recombination analysis indicated that strain GD was a chimera whose putative parental strains belonged to the QX- and TW-type subgroups.
Conclusions
An increasing number of TW-type strains have been isolated from China in recent years, which is in agreement with our findings, suggesting the emergence and increased prevalence of new TW-type strains in southern China.
Similar content being viewed by others


Background
Infectious bronchitis virus (IBV), a member of genus Gammacoronavirus, subfamily Coronavirinae, family Coronaviridae, affects the performance of both meat-type and egg-laying birds, causing tremendous economic losses in the poultry industry worldwide [1, 2].
IBV is an enveloped, positive-sense, non-segmented, single-stranded RNA virion,approximately 27.6 kb in length that contains 5ʹ and 3ʹ untranslated regions [3, 4]. The genome of IBV contains at least 10 open reading frames (ORFs) as follows: 5ʹ-1a-1b-S (S1, S2)-3a-3b-3c (E)-M-5a-5b-N-Poly (A)-3ʹ [5]. The two overlapping ORFs of 1a and 1b at the 5ʹ end are encoded by Gene 1, which constitutes approximately two-thirds of the genome, and are translated as the large polyprotein 1ab that is associated with RNA replication and transcription. The other regions of the genome encode four main structural proteins (the glycosylation spike glycoprotein (S), the envelope protein (E), the membrane protein (M), and the nucleocapsid protein (N)), as well as two accessory genes, 3 and 5, which express accessory proteins 3a and 3b, and 5a and 5b, respectively [6]. The spike protein of IBV is cleaved into S1 and S2 glycoproteins. The S1 subunit is the major target of neutralizing antibodies and carries serotype-specific antigenic determinants. The C-terminal portion of the S2 protein intercalates in the virus envelope and assists S1 protein anchoring in the membrane [7]. Both M and E proteins are membrane-associated proteins required for the formation of virus-like particles and virus budding [8]. N protein is associated with the RNA genome and forms the ribonucleoprotein [2].
An increasing number of new serotypes or variants of IBV caused by frequent gene mutation have become the major challenge of the prevention and control of IB [9]. Multiple IBV serotypes have been identified worldwide and the number of emergent IBV antigenic variants is increasing [7, 10, 11]. Meanwhile, the little cross-protection between different variants and vaccine has led to the outbreaks of infectious bronchitis in vaccinated chicken flocks [11–13]. Genomic recombination has been shown to occur at high frequency in coronaviruses, particularly IBV [14–17], also potentially leading to outbreaks of disease. All of these findings suggest the importance in the investigation of new and emerging IBV isolates.
In China, the outbreaks of infectious bronchitis in vaccinated and non-vaccinated flocks has led to severe economic losses in recent years, highlighting the need for a comprehensive study in the epidemic status of IBV serotypes. Therefore, we monitored the clinical manifestations of infectious bronchitis among Chinese chicken flocks and isolated a number of strains affecting flocks from different regions. One such strain, strain GD, was isolated from the flock in Guangdong Province and the complete genome was sequenced. Phylogenetic analysis was performed by comparing the genome sequence of strain GD with that of other IBV strains, including reference strains, vaccine strains, and other Chinese isolates reported recently. Recombination sites were analyzed to determine whether strain GD was a chimera. Our results provide insight into the current distribution of IBV genotypes across China and the molecular characteristics of prevalent strains. These findings may provide important theoretical basis and practical guidance for the effective vaccine strategies in prevention and control of IBV.
Results
Sequence analysis of the strain GD genome
The full-length genome sequence of IBV isolate GD was obtained and was found to be about 27,680 nucleotides (nt) in length. The genome comprised six genes and 10 ORFs in the following organization: 5ʹ-1ab-S-3a-3b-E-M-5a-5b-N-3ʹ. Gene 1 was 19,904 nt in length and consisted of two overlapping ORFs: 1a and 1b. Gene 2 encodes the S protein and contained 3, 498 nt. S gene was found to be cleaved into two subunits, S1 and S2, which were 1, 620 and 1, 878 nt in length, respectively, encoding the S1 protein of 540 aa and the S2 protein of 626 aa. The cleavage site on the spike protein was R-R-F-R-R. Gene 3 encoded non-structural proteins 3a and 3b, and structural protein E, which were 174, 189, and 327 nt in length, respectively, and the size of animo acid were 58, 63 and 109 aa. Gene 4 encoded the M protein of 226 aa and contained 678 nt. Gene 5 encoded two proteins, 5a and 5b, which were 66 aa and 83 aa, translated by 198 and 249 nt, respectively. Gene 6 was 1230 nt and encoded the N protein of 410 aa.
Sequence comparisons with other IBV genomes
Comparison of the full-length genome sequences showed that the nt identities between strain GD and other IBV strains were ranged from 84.6 to 94.4 %. Strain GD had the highest nt sequence identity (94.4 %) to strain YX10 (GenBank accession number JX840411) isolated from southern China and the lowest nt sequence identity (84.6 %) to strain Georgia 1998, a vaccine strain. The nt identity between strain GD and the H120 vaccine strain was 86.5 % (Table 1).
Sequence comparisons between gene segments
For the S1 gene, the nt and aa sequence identities between strain GD and other IBV strains were 61.6–98.3 and 49.8–96.8 %, respectively. Isolate GD showed the highest identity to strain TW2575/98 (98.3 % nt and 96.8 % aa sequence identity), which is much higher than other strains. The nt and aa identities of S1 gene of strain GD and H120 vaccine strain did not exceed 82 % (Table 1). There were three insertions in the deduced aa sequence of S1 when compared with strain H120. One was between positions 87 and 88, the other two were between 119 and 120 (H120 aa numbering). The insertion positions were located in or near hypervariable region 1 (HVR1) and HVR2, the regions that had a close relationship with a neutralizing antibody. The insertion of glycine between positions 87 and 88 was also detected in strain TW2575/98 and TW1171/92 which were classified in TW-type but was not detected in other reference strains. We also found that the second insertion of lysine between 119 and 120 was different from the other reference strains except TW-type IBVs (Fig. 1). Furthermore, there were 98 aa mutations and no deletion in the S1 protein of strain GD. Of these 98 mutations, 68 were located in the N-terminal 300 aa that contained HVR1, HVR2, and HVR3.

Sequence alignment of S1 amino acids sequence of GD strain and major reference IBV strains with H120. A dot indicates an amino acid identical to that of the H120 strain. A dash indicates an amino acid deletion in comparison with the H120 strain. The red box indicate the insertions between the position of 87 and 88 and the position of 119 to 120 (numbered by H120)
The nt and aa sequence identity among the other structural and nonstructural genes of S2, 3a, 3b, E, M, 5a, 5b, and N of strain GD and other strains were 74.7–98.0 and 58.1–97.8 %, respectively (Table 1).
Phylogenetic analysis of IBV sequences
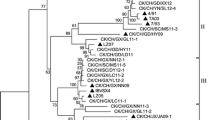
The phylogenetic tree in Fig. 1 showed the genetic relationships of the full-length sequences of strain GD with a range of other strains representing the majority of known IBV genotypes clustered into two groups (groups I and II). Strain GD belonged to group I and was closely related to most of the prevalent isolates, but distantly related to the Massachusetts-type strains comprising the most common vaccine strains in China (Fig. 2a).

Phylogenetic tree of IBV strains showing the evolutionary relatedness of genomes and individual gene sequences. Complete genome (a), S1 gene (b), S2gene (c), E gene (d), M gene (e), and N gene (f). The neighbor-joining method was used with a bootstrap of 1000 replicates. A black dot (●) indicates strain GD
Phylogenetic trees were constructed from the S1 genes of 90 IBV strains prevalent over the last 5 years. The findings indicated that IBVs could be divided into six main clades. Strain GD was located in group IV, also called the TW-type group as it contained the Taiwanese strains, and this group showed low similarity with strains from mainland China. The Massachusetts-type vaccines, such as strain H120, was clustered into group V (Fig. 2b).
Phylogenetic analysis based on the S2, E, M, and N gene sequences suggested that strain GD was closely related phylogenetically to the QX-type or YN-type IBV isolates, which were currently prevalent in China, and distantly related to the Massachusetts-type vaccine strains. So the results showed that GD strain was clustered in different classification based on the phylogenetic analysis of different genes, indicating that recombination events might occur between the S1 gene and other genes (Fig. 2c–f).
Recombination in the genome of strain GD
As supported by the data from all of the employed recombination detection methods, strain GD was found to undergo potential recombination events in four areas of the genome containing all of the structural genes (Table 2). Both 1ab gene and the S gene showed a high frequency of recombination events. The recombination events mainly occurred between QX- and YN-type IBVs, between QX- and TW-type IBVs, and between QX- and QX-type IBVs.
Analysis of TW-type strains isolated in China
There is no TW-type strains isolated from 2006 to 2008 and few sequence data of IBV S1 gene released from 2013 to 2015. Therefore, the data of TW-type strains available from year 2009 to 2012 were used in our analysis. Based on sequence and phylogenetic analysis, 61 TW-type strains were identified, comprising 20 strains from Taiwan and 41 from mainland China. Interestingly, there were two TW-type strains, strains GX-G and GX-XD, isolated in Guangxi Province in 1988. Another strain, CK/CH/LSD/05I, classified as a TW-type strain, was isolated in Shandong Province in 2005 and first reported in 2008. These results demonstrated an increasing ratio of TW-type strains in mainland China in the period of 2009–2012 (Fig. 3).

Proportion of TW-type domestic strains in recent years in China. Each denominator (224, 146, 251, 59) represent the numbers of IBV strains during different years, while the numerator (4, 10, 12, 8) represent the numbers of TW-type strains. All the data used were from NCBI
Discussion
Infectious bronchitis has had a devastating effect on the domestic poultry industry since the emergence and increased prevalence of IBV in China. Because of the high rate of mutation and recombination in coronaviruses, new serotypes and genotypes of IBV have been frequently reported in recent years around the world [2]. These new isolates were generated by insertions, point mutations, and recombination, highlighting the importance of genomic sequence analysis of new isolates of IBV [18]. This study was undertaken to investigate the genetic characteristics of a new IBV strain isolated in Guangdong Province of China in 2013. Sequence, phylogenetic, and recombination analysis were performed to explore the genetic features of this new epidemic isolate, alongside analysis of TW-type strains isolated in mainland China.
The S gene of IBV was expressed as precursor protein (S0), then cleaved into two subunits S1 and S2. The S1 protein determines the genotype, serotype, and phenotype of IBVs [19]. In this study, the S1 cleavage site of strain GD was R-R-F-R-R (Arg- Arg- Phe- Arg- Arg), which was commonly observed in recent Chinese isolates and was identical to the most of TW-type IBVs isolated in China mainland. However, it has been reported that the cleavage recognition site in the S1 protein was not associated with the pathogenicity of IBV [20, 21]. The S1 gene of strain GD was most closely related to TW-type strains due to the highest identity identified between strain GD and TW2575/98 (isolated in Taiwan). Previous studies suggested that only a few changes in the aa sequence of the S1 protein of IBV could lead to the emergence of a new serotype [22], suggesting that amino acid insertions and mutations in the S1 protein might lead to the changes of immunogenicity in strain GD, which make it possible to escape the immunization of current vaccine. Further investigations are still required.
Phylogenetic analysis of the S1 gene in this study suggested that all strains were clustered into six distinct genetic branches. Group I strains were proposed to be QX-like strains as the Chinese QXIBV strain was involved. QX-like IBV strains have become the predominant genotype in China and other countries in Asia and Europe [23]. Group III strains were designated as YN-type strains due to the YN strain isolated in southern China in 2005 [16]. The group IV strains, known as the TW-type strains because of inclusion of the Taiwanese reference strains, formed a unique genotype that was separated from the Chinese genotypes [24, 25].
An increasing number of recombination events have been reported in IBVs, which occurred between different IBV strains, not only between field isolates but also between field and vaccine viruses [26], causing the emergence and evolution of new IBV genotypes [27, 28]. Previous studies have suggested that recombination within the 1ab, S1, S2, 3a,3b, E, M, and N structural proteins had likely occurred in most parts of the world [1, 15, 16, 29, 30]. In this study, convincing evidence was obtained to suggest that four independent recombination events occurred in strain GD affecting all of the structural genes. The major parent of strain GD was identified as a QX-type IBV, while the minor parents were TW-type and YN-type IBVs, confirming that RNA recombination of IBV included more than two strains and occurred between multiple genes.
Sequence, phylogenetic, and recombination analysis inferred that isolate GD was closely related to TW-type IBV strains. In addition, there was similar background between GD strain and a majority of the TW-type domestic IBVs, such as the similar days of the affected broilers, the morbidity and mortality rate. These results were in agreement with those of previous studies that reported an increasing number of TW-type strains isolated in southern China in recent years [24, 25, 31]. Taiwan is a location-independent island separated by a strait from mainland China, and no livestock trading occurred between these countries. The main reason for the emergence of IBV variants in Taiwan has been proposed to be the migration of birds [32]. Since the first TW-type domestic strain, CK/CH/LSD/05I, was isolated in Shandong Province, an increasing number of TW-type strains have been isolated in mainland China [33], as confirmed by our findings. Most of the TW-type strains were isolated from about 30-days-old broilers while few were from more than 200-days-old layer hens. Moreover, the majority of TW-type strains (thirty-one over forty-one) have been isolated in southern China, with only seven strains isolated in northeast China. Geographical factor is one of the factors affecting the evolution of IBVs. Northeast China is located further from Taiwan island than the southern. That may be the reason we can explain why most of the isolates are from in Southern China. Our findings indicated that mutations and/or recombination events were common among IBVs isolated in China. The increasing ratio of TW-type strains suggested that conventional vaccines such as H120 might not be effective in protecting poultry.
When live-attenuated and inactivated vaccines of IBV were first introduced they were effective at controlling infectious bronchitis among poultry flocks [34]. However, serious production losses in vaccinated flocks have been experienced in many areas of China in recent years [22, 35, 36]. The isolation of IBV strains from vaccinated flocks suggested that IBV strains had evolved to evade the immune defenses induced by current vaccines, highlighting the importance of close monitoring and analysis of emerging strains.
Conclusion
In summary, our data suggested that both inter-strain recombination and mutation were contributing to the generation of IBV variants in the field. We showed here that genomic recombination between IBV strains might lead to the replacements of large RNA fragments in multiple genes. Sequence and phylogenetic analysis of strain GD revealed that this isolate was likely a recombinant of QX- and TW-type strains. The emergence of new TW-type strains has been increasing in southern China in recent years, and TW-type isolates are genetically distantly related to the predominant vaccine strain H120. Our research therefore emphasizes the significance of continuous monitoring and new vaccine strategy in view of the current circulating strains, which are the fundamental of the prevention and control infectious bronchitis among poultry.
Methods
Virus isolation and amplification
A strain of IBV was isolated from 32-day-old broiler chicks from a flock which were vaccinated with an IBV vaccine containing the ‘Massachusetts-type’ serotypes in Guangdong Province of China in 2013. The flock exhibited IB features described as decrease in feed consumption, respiratory symptoms and had a morbidity of 60 % and a mortality of 5 ~ 6 %. The isolate was propagated in the allantoic cavity of 10-day-old embryonated specific-pathogen-free eggs and the embryos were incubated at 37 °C for 40 h. The allantoic fluid was harvested sterilely from the embryos after been passaged three times, and was frozen at −80 °C prior to use.
PCR primer design
Twenty-eight pairs of primers were designed based on the complete genome sequence of IBV strain TW2575/98 from the GenBank database (GenBank accession number DQ646405) and these primers were used for the amplification of the complete genome of strain GD (Table 3).
Viral RNA extraction, reverse transcription (RT)-polymerase chain reaction (PCR), and DNA sequencing
Viral RNA was extracted from the virus-infected allantoic fluid using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. RT was carried out using 4 μL of total RNA, 2 μL of dNTPs (2.5 mM), 1 μL of RNasin (50 U/μL), and M-MLV (10 U/μL, Promega, Madison, WI, USA) at 37 °C for 1 h, and 95 °C for 5 min. For PCR, 25 μL of 2 × PCRmix (TransGen Biotech, Beijing, China) and 20 pmol of each primer were added to 100 ng of template cDNA in a total reaction volume of 50 μL. The reaction conditions were as follows: 94 °C for 5 min followed by 30 cycles of 94 °C for 45 s, annealing for 45 s (temperature depended on the primers), 72 °C for 2 min, with a final extension step of 72 °C for 10 min. The PCR products were analyzed by 1 % agarose gel electrophoresis. Amplified sequences were purified using the AxyPrep DNA Gel Extraction kit (Axygen, Union City, CA, USA), and the purified products were inserted into the pMD18-T vector (Promega). Nucleotide sequencing of the recombinant plasmids was performed by TsingKe Biological Technology (Beijing, China).
Every passages of the virus-infected allantoic fluid were sequenced according to the method above and the last passage was sequenced for three times to ensure the accuracy of the sequence of GD strain.
Sequence and phylogenetic analysis
Assembly of the sequence of the IBV GD isolate was conducted using the SeqMan program of the DNASTAR software suite version 7.1 (DNASTAR, Madison, WI, USA). The sequence of the full-length genome and the individual structural genes were compared with other IBVs using the MegAlign program of the DNASTAR software. Deduced amino acid (aa) sequences were aligned and phylogenetic trees were mapped with the full-length genomes and structural genes using MEGA 5.0 software (www.megasoftware.net) and the Kimura 2-parameter nucleotide substitution model. The maximum likelihood method was used with 1000 bootstrap replicates.
Recombination detection
The complete genome sequence of IBV strain GD and other reference strains were aligned using the ClustaW program of the MEGA 5.0 software and were analyzed using the Recombination Detection Program version 4.0 (RDP4.0, Simmonics, University of Warwick, Coventry, UK) and the algorithms of RDP, Bootscan, GENECONV, MaxChi, Chimaera, SiScan, and 3Seq. Default settings were used for all programs. More than one method was used to analyze the data because results from only a single method were not reliable and could have resulted in misleading results.
Analysis of TW-type strains isolated in China
Using the data released from NCBI, all S1 gene sequences from IBVs deposited up until November 2015 were collected and phylogenetic analysis was performed. The proportion of TW-type strains among the isolates were computed for each year.
References
He K, Li M, Wei P, Mo ML, Wei TC, Li KR. Complete genome sequence of an infectious bronchitis virus chimera between cocirculating heterotypic strains. J Virol. 2012;86:13887–8.
Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet Res. 2007;38:281–97.
Abro SH, Renstrom LH, Ullman K, Belak S, Baule C. Characterization and analysis of the full-length genome of a strain of the European QX-like genotype of infectious bronchitis virus. Arch Virol. 2012;157:1211–5.
Fellahi S, El Harrak M, Ducatez M, Loutfi C, Koraichi SI, Kuhn JH, et al. Phylogenetic analysis of avian infectious bronchitis virus S1 glycoprotein regions reveals emergence of a new genotype in Moroccan broiler chicken flocks. Virol J. 2015;12, e116.
Liu XL, Su JL, Zhao JX, Zhang GZ. Complete genome sequence analysis of a predominant infectious bronchitis virus (IBV) strain in China. Virus Genes. 2009;38:56–65.
Cook JK, Jackwood M, Jones R. The long view: 40 years of infectious bronchitis research. Avian Pathol. 2012;41:239–50.
Li M, Wang XY, Wei P, Chen QY, Wei ZJ, Mo ML. Serotype and genotype diversity of infectious bronchitis viruses isolated during 1985–2008 in Guangxi, China. Arch Virol. 2012;157:467–74.
Zulperi ZM, Omar AR, Arshad SS. Sequence and phylogenetic analysis of S1, S2, M, and N genes of infectious bronchitis virus isolates from Malaysia. Virus Genes. 2009;38:383–91.
Jackwood MW. Review of infectious bronchitis virus around the world. Avian Dis. 2012;56:634–41.
Abro SH, Renstrom LH, Ullman K, Isaksson M, Zohari S, Jansson DS, et al. Emergence of novel strains of avian infectious bronchitis virus in Sweden. Vet Microbiol. 2012;155:237–46.
Gelb Jr J, Weisman Y, Ladman BS, Meir R. S1 gene characteristics and efficacy of vaccination against infectious bronchitis virus field isolates from the United States and Israel (1996 to 2000). Avian Pathol. 2005;34:194–203.
Xu C, Zhao J, Hu X, Zhang G. Isolation and identification of four infectious bronchitis virus strains in China and analyses of their S1 glycoprotein gene. Vet Microbiol. 2007;122:61–71.
Lim TH, Lee HJ, Lee DH, Lee YN, Park JK, Youn HN, et al. An emerging recombinant cluster of nephropathogenic strains of avian infectious bronchitis virus in Korea. Infect Genet Evol. 2011;11:678–85.
Jia W, Karaca K, Parrish C, Naqi S. A novel variant of avian infectious bronchitis virus resulting from recombination among three different strains. Arch Virol. 1995;140:259–71.
Mo ML, Hong SM, Kwon HJ, Kim IH, Song CS, Kim JH. Genetic diversity of spike, 3a, 3b and E genes of infectious bronchitis viruses and emergence of new recombinants in Korea. Viruses. 2013;5:550–67.
Zhao Y, Liu XY, Cheng JL, Wu YP, Zhang GZ. Molecular characterization of an infectious bronchitis virus strain isolated from northern China in 2012. Arch Virol. 2014;159:3457–61.
Cavanagh D, Davis P, Cook J. Infectious bronchitis virus: evidence for recombination within the Massachusetts serotype. Avian Pathol. 1992;21:401–8.
Awad F, Chhabra R, Baylis M, Ganapathy K. An overview of infectious bronchitis virus in chickens. Worlds Poult Sci J. 2014;70:375–84.
Liu SW, Zhang QX, Chen JD, Han ZX, Liu X, Feng L, et al. Genetic diversity of avian infectious bronchitis coronavirus strains isolated in China between 1995 and 2004. Arch Virol. 2006;151:1133–48.
Jackwood MW, Hilt DA, Callison SA, Lee CW, Plaza H, Wade E. Spike glycoprotein cleavage recognition site analysis of infectious bronchitis virus. Avian Dis. 2001;45:366–72.
Yan F, Zhao Y, Yue W, Lv L, Ji W, Li X, et al. Phylogenetic analysis of S1 gene of infectious bronchitis virus isolates from China. Avian Dis. 2011;55:451–8.
Liu S, Kong X. A new genotype of nephropathogenic infectious bronchitis virus circulating in vaccinated and non-vaccinated flocks in China. Avian Pathol. 2004;33:321–7.
Sun C, Han Z, Ma H, Zhang Q, Yan B, Shao Y, et al. Phylogenetic analysis of infectious bronchitis coronaviruses newly isolated in China, and pathogenicity and evaluation of protection induced by Massachusetts serotype H120 vaccine against QX-like strains. Avian Pathol. 2011;40:43–54.
Feng K, Xue Y, Wang F, Chen F, Shu D, Xie Q. Analysis of S1 gene of avian infectious bronchitis virus isolated in southern China during 2011-2012. Virus Genes. 2014;49:292–303.
Luo H, Qin J, Chen F, Xie Q, Bi Y, Cao Y, et al. Phylogenetic analysis of the S1 glycoprotein gene of infectious bronchitis viruses isolated in China during 2009–2010. Virus Genes. 2012;44:19–23.
Zhang Y, Wang HN, Wang T, Fan WQ, Zhang AY, Wei K, et al. Complete genome sequence and recombination analysis of infectious bronchitis virus attenuated vaccine strain H120. Virus Genes. 2010;41:377–88.
Jackwood MW, Boynton TO, Hilt DA, McKinley ET, Kissinger JC, Paterson AH, et al. Emergence of a group 3 coronavirus through recombination. Virology. 2010;398:98–108.
Thor SW, Hilt DA, Kissinger JC, Paterson AH, Jackwood MW. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses. 2011;3:1777–99.
Ovchinnikova EV, Bochkov YA, Shcherbakova LO, Nikonova ZB, Zinyakov NG, Elatkin NP, et al. Molecular characterization of infectious bronchitis virus isolates from Russia and neighbouring countries: identification of intertypic recombination in the S1 gene. Avian Pathol. 2011;40:507–14.
Kuo SM, Wang CH, Hou MH, Huang YP, Kao HW, Su HL. Evolution of infectious bronchitis virus in Taiwan: characterisation of RNA recombination in the nucleocapsid gene. Vet Microbiol. 2010;144:293–302.
Zou NL, Zhao FF, Wang YP, Liu P, Cao SJ, Wen XT, et al. Genetic analysis revealed LX4 genotype strains of avian infectious bronchitis virus became predominant in recent years in Sichuan area, China. Virus Genes. 2010;41:202–9.
Chen HW, Huang YP, Wang CH. Identification of Taiwan and China-like recombinant avian infectious bronchitis viruses in Taiwan. Virus Res. 2009;140:121–9.
Liu S, Wang Y, Ma Y, Han Z, Zhang Q, Shao Y, et al. Identification of a newly isolated avian infectious bronchitis coronavirus variant in China exhibiting affinity for the respiratory tract. Avian Dis. 2008;52:306–14.
Cook JK, Chesher J, Baxendale W, Greenwood N, Huggins MB, Orbell SJ. Protection of chickens against renal damage caused by a nephropathogenic infectious bronchitis virus. Avian Pathol. 2001;30:423–6.
Nix WA, Troeber DS, Kingham BF, Keeler CL, Gelb J. Emergence of subtype strains of the Arkansas serotype of infectious bronchitis virus in Delmarva broiler chickens. Avian Dis. 2000;44:568–81.
Farsang A, Ros C, Renström LH, Baule C, Soos T, Belak S. Molecular epizootiology of infectious bronchitis virus in Sweden indicating the involvement of a vaccine strain. Avian Pathol. 2002;31:229–36.
Funding
This study was supported by the China Agriculture Research System Poultry-related Science and Technology Innovation Team of Peking.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: GX XYL GZZ. Performed the experiments: XYL GX YC. Analyzed the data: GX JZ YZ. Contributed reagents/materials/analysis tools: JZ GZZ. Wrote the paper: GX JZ YZ. Checked and revised the manuscript: GX GZZ. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, G., Liu, Xy., Zhao, Y. et al. Characterization and analysis of an infectious bronchitis virus strain isolated from southern China in 2013. Virol J 13, 40 (2016). https://doi.org/10.1186/s12985-016-0497-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-016-0497-3