
Abstract
Infectious bronchitis (IB) is one of the major diseases in poultry flocks all over the world caused by infectious bronchitis virus (IBV). In the study, the complete genome sequence of strain A2 was sequenced and analyzed, which was a predominant IBV strain in China. The results indicated that there were mutations, insertions, and deletions distributed in the whole genome. The A2 virus had the highest identity to S14 and BJ in terms of full genome, whereas had a further distance to Massachusetts strains. Phylogenetic analysis showed that A2 isolate clustered together with most Chinese strains. The results of this study suggest that strain A2 may play an important role in IBV’s evolution and A2-like IBVs are predominant strains in China.
Similar content being viewed by others


Introduction
Infectious bronchitis virus (IBV) causes tremendous economic losses to the poultry industry in many countries. Domestic chicken is the most important but not the only host for IBV [1]. IB has been reported in peafowl, teal, partridge, turkey, pheasant, racing pigeon, and guinea fowl [2–5]. IBV is the prototype of Coronaviridae, which is a diverse family consists of three groups based on genetic and serologic properties [6, 7]. IBV belongs to Coronaviridae group 3, which differs extensively from group 1 and group 2 that are consisting of mammalian coronaviruses.
Infectious bronchitis virus is an enveloped positive-sense, single-strand RNA virus, with a large genome RNA about 27 kb in size [8]. The whole IBV genome has at least 10 open reading frames (ORF), from 5′ to 3′ are as follows: 5′-1a-1b-S(S1,S2)-3a,b,c(E)-M-5a,b-N-Poly(A)-3′, consisting of four main structural proteins (glycosylation spike glycoprotein (S), small envelope protein (E), membrane glycoprotein (M), and nucleocapsid protein (N)) and numerous non-structural proteins of known and unknown functions [1, 9–12]. S protein was cleavaged into N-terminal S1 and C-terminal S2 glycopolypeptides. The S1 spike glycoprotein carries virus-neutralizing and serotype-specific antigenic determinants, which can evolve rapidly especially within the three hypervariable regions (HVR). S2 protein anchors to viron and can affect S1 protein fusion to membrane. N protein plays important role in virus replication, assembly, and cell immunity. E and M proteins are required in virus assembly and budding. Gene 1 codes two polymerase proteins 1a and 1b, which are incised into 15 non-structural proteins (nsp) and are associated with RNA replication and transcription. NS proteins, encoded by gene 3 and gene 5, are not essential in virus replication, but they may play a role in antagonism of innate immune responses. They can also serve as targets for rational attenuation of IBV pathogenicity [1, 8, 13–16].
Protection for IBV infection is not satisfactory because of the numerous existed and newly emerged serotypes, poor cross-protection and the inefficient application of vaccine [17–24]. Currently, more than 60 serotypes have been reported as a consequence of mutation in its large genome. Choosing appropriate vaccine to control IB infection is very important due to the poor cross-protection among the different serotypes. In North America, the mainly used vaccines are Massachusetts, Connecticut, and Arkansas serotypes. California strains and other important region serotypes are also used. In Europe, various “Holland variants” are available. In Australia, native B and C serotypes are used to control IB.
In China, IB was first reported in 1972 and the virus was found nearly all over the country in the following years. The disease has occurred frequently in chicken flocks and caused severe economic losses in recent years, although vaccines based on Massachusetts strains, such as H120 and H52, have been broadly used for prevention and control of the infection. Our previous study revealed that there are at least four groups of IBVs circulating in China and the disease outbreaks might have been caused by infection of multiple strains of IBV, but the predominant strain was correlative tightly with 4/91 serotype [21].
In this study, we sequenced the complete genome of IBV strain A2, which was an early isolate and closely related to strain 4/91 in neutralization test and the immune protection test. Sequence and phylogenetic analysis were carried out to compare A2 and other IBVs, which included the reference strains, the vaccine strains or the isolates from China reported in recent years. The results from this study will be useful to better understand the evolution of IBV in China and improve the efficacy of the vaccines for IBV infection in poultry industry.
Materials and methods
Virus amplification
Strain A2 was inoculated into the allantoic cavity of 10-day-old embryonated specific pathogen free (SPF) eggs. The embryos were incubated at 37°C and examined twice daily for their viability. The allantoic fluids were harvested from three eggs after incubation for 40 h and three other eggs were further incubated for 144 h to observe the lesions of embryos. Several blind passages were performed until the dwarfing and death of embryos were observed between 48 and 144 h after inoculation.
Primer design
Based on the cDNA sequences of IBVs from GenBank (M41, AY851295 and BJ, AY319651) and the sequenced A2 fragments (AY043312), 22 pairs of primers were designed and used to amplify the complete genome sequence of strain A2 except 5′ terminal. 5 primers were used for RACE PCR to amplify 5′ terminal sequence. Primer sequences were listed in Table 1.
Reverse transcription-polymerase chain reaction (RT-PCR)
Reverse transcription was carried out at 37°C for 1 h using 5 μl of total RNA, 1 μl random primer (Promega, USA) and 1 μl M-MLV reverse transcriptase (Promega, USA). PCR was performed in PCR Machine (Biometra, Germany) with 3 μl cDNA as template in a total of 25 μl reaction volume containing 0.5 μl of each primer and 12.5 μl PCR TaqMix (Meilaibo, China). Reactions were carried out at 94°C for 5 min followed by 35 cycles of 94°C for 45 s, 45–60°C (depending on the special PCR reaction) for 45 s, 72°C for 2 min, and finally, 72°C for 10 min.
5′ RACE of genome RNA
5′ terminal sequence of genome RNA was amplified using 5′-full race kit (TaKaRa, Japan). 5′ terminal cDNA was synthesized using 1 μl RT primer, 5 μl total RNA, and 1 μl AMV reverse transcriptase. After reverse transcription, the hybrided RNA was degenerated using RNase H and the cDNA were ligated with T4 RNA ligase at 16°C for 15–18 h. First PCR was carried out using 10-fold diluted ligated liquid as cDNA template. The first PCR product was 100-fold diluted and serve as template for the second PCR. Two PCR were carried out at 94°C for 5 min followed by 25 or 30 cycles of 94°C for 45 s, 50°C for 45 s, 72°C for 1 min, and finally, 72°C for 10 min.
Cloning and sequencing of the genome sequence
Polymerase chain reaction products were inserted into pGEM®-T Easy Vector (Promega, USA) and transformed into DH5α competent cell according to the manufacturer’s instructions. Positive clones were screened from three different PCRs through blue and white cloney. The nucleotide (nt) sequences of the positive clones were determined using T7 and SP6 sequence primers on a commercial service.
Sequence analysis of the genome sequence
The sequences of IBVs were downloaded from NCBI in order to analyze the relationship between A2 and other strains. Nucleotide and amino acid (aa) identity of complete and partial genome were computed using DNAstar computer software. Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 3.1 (Kumar, Tamura, Nei 2004).
Accession numbers of IBV sequences
The complete genome sequences used for comparisons are as follows: A2 (EU526388); Beaudette (NC_001451); BJ (AY319651); California 99 (AY514485); M41 (AY851295); S14 (AY646283); KQ6 (AY641576); LX4 (AY338732); SAIBK (DQ288927). Partial gene sequences used for comparisons have the following GenBank accession numbers: LKQ3 (AY702085); LDT3 (AY702975); Jilin (AY846833, AY839144); HK (AY761141); Vic (DQ490221); S (DQ490213); Tw1171/92 (DQ646406). The accession numbers of all sequences used for phylogenetic analysis were included in Table 2.
Results and discussion
After four passages in 10-day-old embryonated SPF eggs, pathological changes in A2-infected embryos including dwarfing and death of embryos between 48 and 144 h post-inoculation were clearly observed (Figure not shown). The allantoic fluids were collected and used for RNA extraction.
The A2 strain was tested in Reverse transcription-polymerase chain reaction (RT-PCR) and 5′RACE PCR using the gene specific primers. 23 overlapping fragments were produced ranging from 411 to 2024 bp in size (Figure not shown).
To obtain the complete genome sequence of A2 strain, 23 sequenced large fragments were assembled using DNAstar software. The complete genome of strain A2 consists of 27,715 nucleotides (nt), which encodes six different genes. Gene 1 has a length of 20,446 nt, consisting of two overlapping ORFs: 1a and 1b. Gene 2, encoding S protein, contains 3567 nt and has a single ORF with 3507 nt. S gene is cleaved into two subunit S1 and S2 with 1614 and 1890 nt long, respectively (potentially encoding 538 and 621 aa). There are 699 nt in gene 3, encoding non-structural protein 3a, 3b and structural protein E with 174nt, 192nt and 327 nt, respectively. Between gene 2 and gene 3, there are 22 nt overlap. Between 3a and 3b, 3b and 3c, there are 1 and 18 nt overlap. Gene 4 contains 757 nt, encoding M protein of 225 aa. Gene 3 has 115 nt of overlap with gene 4. Gene 5 has two ORFs: 5a (198 nt) and 5b (249 nt). Between gene 4 and ORF 5a, there is a 363 nt fragment which does not encode protein. Gene 6 contains 1332 nt, encoding N protein of 409 aa. After the N gene, the 3′UTR and polyA tail were observed.
The full genome sequence of strain A2 was compared with all the available complete genomic sequences from GenBank. The results revealed that the nucleotide identities between A2 and these strains ranged from 84.43 to 94.27%. A2 strain had the highest similarity to S14 at 94.27% identity and least like M41 with 84.43% identity. In our current study, point mutations, insertions, and deletions were distributed throughout the whole genome RNA. Nsp1, S1, gene 3, and ORF 5a were hypervariable, whereas nsp4 to nsp13, M and ORF 5b were relatively conserved.
Nucleotide identities between different fragments of gene 1 were compared using DNAstar software. All gene 1 fragments of A2 had highest identity with S14, except for nsp9 and nsp11-nsp13 (Table 3). For nsp9 and nsp11-nsp13, A2 was 97.1% and 93.2% identical to BJ, whereas was 97.0% and 92.2% identical to S14, respectively. Nsp2 is the putative 3C-like proteinase (3C-LP), also known as the main proteinase (Mpro), which plays a pivotal role in viral gene expression and replication as well as in proteolytic processing of gene 1-encoded polyproteins in coronaviruses. Between different IBVs 3C-LP is highly conserved [25–27]. But compared with other gene 1 fragments in this study, nucleotide identity between A2 and other strains for 3C-LP was relatively low.
To interpret relationship between A2 and other IBV strains in 3′-terminal 7.3 kb genome, 15 other IBV sequences were obtained from GenBank. Parewise comparisons were performed using DNAstar computer software, as shown in Table 4.
In the S1 gene, the A2 virus and other strains were 58.7–91.9% identical and the SAIBK strain was the most similar at the nt level (91.9% identical). There was an eight aa insertion between 75–82 in deduced aa sequences, in which a putative N-glycosylation site was found. The BJ S1 gene had the same eight aa insert identical of the A2 virus. The insertion was also found in SAIBK with one aa difference. The insertion position was located in or near HVR1, which had high relationship with neutralization antibody [21, 28]. So insertion of eight aa and change of glycosylation pattern may potentially affect the immunoreaction of the S1 protein. In addition, there were two aa deletions (aa 22 and 124) and 95 aa mutations in the A2 S1gene compared with M41. 70 out of 95 mutations were located in the N-terminal 300 aa which contained HVR1, HVR2 and HVR3 [18, 29, 30].
Compared to S1 sequence, S2 was less variable. Between A2 and the other IBVs, nucleotide and aa identity were 74.6–94.8% and 74.2–94.9%, respectively. The A2 S2 gene was 94.5% identical to LX4 (94.3% identical to SAIBK and LDT3 at aa level). At position of aa 15, 293, 377, 435, 493, 565, 621 of S2 protein, the aa were S, I, R, L, N, R, and A for A2, while all other strains were P, V, Q, V, D, W, and T.
The S glycoprotein of IBV is translated as a precursor protein (S0), then cleaved into two subunits S1 and S2. Previous studies reveal that the cleavage recognition site sequence, which consists of five basic aa residues, does not appear to correlate with increased cleavability, host cell range and increased virulence as it does with envelope glycoproteins in orthomyxoviruses and paramyxoviruses, but correlates with IBVs in different geographic regions [30, 31]. In the present study, strain A2 shared the same cleavage recognition site sequence with a majority of other strains (10 of 15 viruses): Arg-Arg- Phe -Arg-Arg, which was the most common cleavage recognition site observed [31, 32].
The 3a amino acid sequence of the A2 virus was 100% identical to those of the S14 and LDT3, except a substitution of Gln by an His at aa 54. For the 3b protein, strain A2 was 92.2% identical to those of S14, LDT3, and BJ at aa level. The third protein encoded by gene 3, E protein was also the most similar to S14 and LDT3 with 95.5% identity. In addition, all other strains were Leu and Trp at aa 12 and 52, except for A2 which were replaced by Gln and Arg, respectively.
Comparisons of the A2 M protein with other IBV strains showed that the aa identity ranged from 90.7 to 94.7%, with California 99 strain being the closest matches, and with BJ and Vic strains the least similar. The identity was obviously higher than any other structural proteins. The N-terminal 18 amino acids were highly variable in M protein. A2 together with most other strains had a putative N-glycosylation site in this 18 aa sequence. At aa 197, all other strains had a basic aa Lys, whereas A2 changed to an acidic aa Glu.
In gene 5, the 5a gene of the A2 strain was 90.5% identical to BJ strain at nt level. As for the 5b gene, A2 was 96.4% identity to those of LX4 and S14 strains.
The N protein is another conserved structural protein. The aa identity of the A2 N protein with other IBV strains ranged from 87.8 to 95.1% and a maximum 95.1% aa identity with S14 and LDT3 viruses. Point mutations were randomly distributed in the whole sequence. But no deletion or insertion occurred in the N protein of the A2 strain.
Transcription-regulating sequences (TRS) precede each gene and include a conserved core sequence (CS) surrounded by relatively variable sequences (5′TRS and 3′TRS). The consensus TRS of IBV, CTT/GAACAA, was highly conserved in IBV genome at the levels of nucleotide sequence and location in regarding to the initiation codon of individual genes [33, 34]. The canonical TRS of IBV was also found in strain A2 in the present study as shown in Fig. 1, except for gene 5. In gene 5, A2 had a six nt insertion (AAGAAA) in TRS compared to those corresponding regions of IBVs. The BJ virus had the same insert observed. The TRSs maybe serve as binding site of transcript complex and the distance between TRS and initiation codon may affect the subgenomic RNA synthesis and translation and in turn change the virus biologic activity [35–37]. The results in the present study also suggest that the TRS sequence is not essential for virus replication and multiplication.

Sequence comparisons of strain A2 with other strains in TRS of gene 5. Strain A2 and BJ have six nucleotides (AAGAAA) insertion and two nucleotides deletion compared to most strains
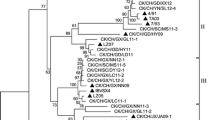
Phylogenetic trees were derived from the nucleotide sequences of specific gene of the A2 virus, as shown in Fig. 2. Phylogenetic tree constructed from the S1 gene sequences revealed six genetic groups. The analysis showed that A2 and most Chinese strains formed into the same groupI [21, 30]. Strains of groupIranged from 1996 to 2006, most of them were isolated between 2002 and 2006. The results revealed that A2 related IBVs were prevalent in China in recent years (Fig. 2a). A2 strain had a more close evolutionary distance to other group compared with other groupIstrains. We infer that A2 may play an important role in groupIIBVs evolution.


Phylogenetic trees of each fragment, generated by the neighbor-joining method with 1,000 bootstrap replicates. The horizontal bar indicates the nucleotide substitutions per site, while vertical distances are for clarity. Phylogenetic trees derived from all fragments revealed that most Chinese isolates were related to A2 strain and A2-like strains had a far distance to Mass and Conn serotype strains
In S2, E, M, and N genes, the same results were obtained and strain A2 grouped with most Chinese isolates into one subcluster (Fig. 2b–e).
For 3C-LP and the papain-like protease, the analysis was unclear because the limited strains in GenBank (Fig. 2f, g). In all phylogenetic trees, A2-like viruses and Massachusetts strains, such as H120 and H52, were distributed into different groups. Athough different genotypes of infectious bronchitis viruses co-circulated in China [21, 38], the results from our study revealed that A2-like viruses were the dominant IBV strains in China and had a far distance to Massachusetts strains, which were the most common vaccine strain in China.
Infectious bronchitis has occurred frequently in China in recent years. The results of this study indicate that the A2 virus, which is the first strain of 4/91 like serotype isolated in China, plays an important role in China IBV evolution. Due to vaccine of A2-like or 4/91 related serotype are not available, the A2-like IBV are still circulating in China. To prevent A2-like IBV outbreak frequently, attenuated or inactived vaccine are urgently needed, though the use of vaccine may contribute to the mutation and recombination of IBV.
References
D. Cavanagh, Vet. Res. 38, 281–297 (2007). doi:https://doi.org/10.1051/vetres:2006055
S.W. Liu, J.F. Chen, J.D. Chen, X.G. Kong, Y.H. Shao, Z.X. Han et al., J. Gen. Virol. 86, 719–725 (2005). doi:https://doi.org/10.1099/vir.0.80546-0
G.H. Zhang, J.D. Fu, T. Ren, W.S. Cao, K.J. Luo, C.G. Xu et al., Prog. Nat. Sci. 16, 1275–1280 (2006). doi:https://doi.org/10.1080/10020070612330141
D. Cavanagh, K. Mawditt, M. Sharma, S.E. Drury, H.L. Ainsworth, P. Britton et al., Avian Pathol. 30, 355–368 (2001). doi:https://doi.org/10.1080/03079450120066368
D. Cavanagh, K. Mawditt, D.B. Welchman, P. Britton, R.E. Gough, Avian Pathol. 31, 81–93 (2002). doi:https://doi.org/10.1080/03079450120106651
S. Dea, A.J. Verbeek, P. Tijssen, J. Virol. 64, 3112–3118 (1990)
E.J. Snijder, P.J. Bredenbeek, J.C. Dobbe, V. Thiel, J. Ziebuhr, L.L. Poon et al., J. Mol. Biol. 331, 991–1004 (2003). doi:https://doi.org/10.1016/S0022-2836(03)00865-9
M.M. Binns, J. Gen. Virol. 68, 57–77 (1987). doi:https://doi.org/10.1099/0022-1317-68-1-57
D.F. Stern, B.M. Sefton, J. Virol. 50, 22–29 (1984)
A.O. Pasternak, W.J.M. Spaan, E.J. Snijder, J. Gen. Virol. 87, 1403–1421 (2006). doi:https://doi.org/10.1099/vir.0.81611-0
W. Spaan, D. Cavanagh, M.C. Horzinek, J. Gen. Virol. 69, 2939–2952 (1988). doi:https://doi.org/10.1099/0022-1317-69-12-2939
S. Sutou, S. Sato, T. Okabe, M. Nakai, Virology 165, 589–595 (1988). doi:https://doi.org/10.1016/0042-6822(88)90603-4
S. Youn, J.L. Leibowitz, E.W. Collisson, Virology 332, 206–215 (2005). doi:https://doi.org/10.1016/j.virol.2004.10.045
T. Hodgson, P. Britton, D. Cavanagh, J. Virol. 80, 296–305 (2006). doi:https://doi.org/10.1128/JVI.80.1.296-305.2006
R. Casais, M. Davies, D. Cavanagh, P. Britton, J. Virol. 79, 8065–8078 (2005). doi:https://doi.org/10.1128/JVI.79.13.8065-8078.2005
S. Shen, Z.L. Wen, D.X. Liub, Virology 311, 16–27 (2003). doi:https://doi.org/10.1016/S0042-6822(03)00117-X
K.J. Worthington, R.C. Jones, Vet. Rec. 159, 291–292 (2006)
R. Smati, A. Silim, C. Guertin, M. Henrichon, M. Marandi, M. Arella et al., Virus Genes 25, 85–93 (2002). doi:https://doi.org/10.1023/A:1020178326531
S.W. Liu, X.G. Kong, Avian Pathol. 33, 321–327 (2004). doi:https://doi.org/10.1080/0307945042000220697
W.A. Nix, D.S. Troeber, B.F. Kingham, C.L. Keeler Jr., J. Gelb Jr., Avian Dis. 44, 568–581 (2000). doi:https://doi.org/10.2307/1593096
C.P. Xu, J.X. Zhao, X.D. Hu, G.Z. Zhang, Vet. Microbiol. 122, 61–71 (2007). doi:https://doi.org/10.1016/j.vetmic.2007.01.006
L. Yu, Y. Jiang, S. Low, Z. Wang, S.J. Nam, W. Liu et al., Avian Dis. 45, 416–424 (2001). doi:https://doi.org/10.2307/1592981
J. Gelb Jr., Y. Weisman, B.S. Ladman, R. Meir, Avian Pathol. 34, 194–203 (2005). doi:https://doi.org/10.1080/03079450500096539
D. Cavanagh, R. Casais, M. Armesto, T. Hodgson, S. Izadkhasti, M. Davies et al., Vaccine 25, 5558–5562 (2007). doi:https://doi.org/10.1016/j.vaccine.2007.02.046
S.P. Monda, C.J. Cardona, Virology 324, 238–248 (2004). doi:https://doi.org/10.1016/j.virol.2004.03.032
L.F. Ng, D.X. Liu, Virology 272, 27–39 (2000). doi:https://doi.org/10.1006/viro.2000.0330
H.Y. Xu, K.P. Lim, S. Shen, D.X. Liu, Virology 288, 212–222 (2001). doi:https://doi.org/10.1006/viro.2001.1098
D. Cavanagh, P.J. Davis, A.P. Mockett, Virus Res. 11, 141–150 (1988). doi:https://doi.org/10.1016/0168-1702(88)90039-1
C.H. Wang, Y.C. Huang, Arch. Virol. 145, 291–300 (2000). doi:https://doi.org/10.1007/s007050050024
S.W. Liu, Q.X. Zhang, J.D. Chen, Arch. Virol. 151, 1133–1148 (2006). doi:https://doi.org/10.1007/s00705-005-0695-6
M.W. Jackwood, D.A. Hilt, S.A. Callison, C.W. Lee, H. Plaza, E. Wade, Avian Dis. 45, 366–372 (2001). doi:https://doi.org/10.2307/1592976
D. Cavanagh, P.J. Davis, D.J. Pappin, M.M. Binns, M.E. Boursnell, T.D. Brown, Virus Res. 4, 133–143 (1986). doi:https://doi.org/10.1016/0168-1702(86)90037-7
T.L. Lin, C.C. Loa, C.C. Wu, Virus Res. 106, 61–70 (2004). doi:https://doi.org/10.1016/j.virusres.2004.06.003
I. Sola, J.L. Moreno, S. Zúñiga, S. Alonso, L. Enjuanes, J. Virol. 79, 2506–2516 (2005). doi:https://doi.org/10.1128/JVI.79.4.2506-2516.2005
A.O. Pasternak, E. van den Born, W.J. Spaan, E.J. Snijder, EMBO J. 20, 7220–7228 (2001). doi:https://doi.org/10.1093/emboj/20.24.7220
M.A. Ijms, D.D. Nedialkova, J.C. Zevenhoven-Dobbe, A.E. Gorbalenya, E.J. Snijder, J. Virol. 81, 10496–10505 (2007). doi:https://doi.org/10.1128/JVI.00683-07
P.S. Masters, Adv. Virus Res. 66, 193–292 (2006). doi:https://doi.org/10.1016/S0065-3527(06)66005-3
G.X. Bing, X. Liu, J. Pu, Q.F. Liu, Q.M. Wu, J.H. Liu, Virus Genes 35, 333–337 (2007). doi:https://doi.org/10.1007/s11262-007-0100-5
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Xl., Su, Jl., Zhao, Jx. et al. Complete genome sequence analysis of a predominant infectious bronchitis virus (IBV) strain in China. Virus Genes 38, 56–65 (2009). https://doi.org/10.1007/s11262-008-0282-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-008-0282-5