
Abstract
Retroviral integration into germline DNA can result in the formation of a vertically inherited proviral sequence called an endogenous retrovirus (ERV). Over the course of their evolution, vertebrate genomes have accumulated many thousands of ERV loci. These sequences provide useful retrospective information about ancient retroviruses, and have also played an important role in shaping the evolution of vertebrate genomes. There is an immediate need for a unified system of nomenclature for ERV loci, not only to assist genome annotation, but also to facilitate research on ERVs and their impact on genome biology and evolution. In this review, we examine how ERV nomenclatures have developed, and consider the possibilities for the implementation of a systematic approach for naming ERV loci. We propose that such a nomenclature should not only provide unique identifiers for individual loci, but also denote orthologous relationships between ERVs in different species. In addition, we propose that—where possible—mnemonic links to previous, well-established names for ERV loci and groups should be retained. We show how this approach can be applied and integrated into existing taxonomic and nomenclature schemes for retroviruses, ERVs and transposable elements.
Similar content being viewed by others

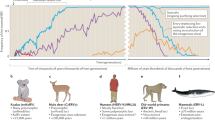

Background
Retroviruses (family Retroviridae) are characterized by a replication cycle in which the viral RNA genome is reverse-transcribed and integrated into the nuclear genome of the host cell. The principal determinants of the retroviral replication cycle are the enzymes reverse transcriptase (RT) and integrase (IN) [1]. These enzymes allow the conversion of single stranded viral RNA into double-stranded DNA, followed by integration of viral DNA into the nuclear genome of the infected cell to form the ‘provirus’. As a chromosomal insertion, the integrated provirus has a life-long association with the infected cell, and survives as long as that cell (or its progeny). When integration occurs in a germ cell (i.e. gametes or early embryo), the resultant provirus can be vertically inherited as a host allele (see Fig. 1). Such a provirus is called an endogenous retrovirus (ERV). Unless silenced or inactivated (e.g., by methylation [2] or mutation), ERV proviruses retain the potential to give rise to additional germline copies—either by infection of, or retrotransposition within further germ cells [3,4,5]. Selective forces operating at the level of the host population determine the fate of individual ERV loci. By far the most likely outcome for any newly generated ERV locus is that it will be purged from the gene pool. Despite this, however, vertebrate genomes typically contain thousands of ERV loci that have been genetically ‘fixed’—i.e. they occur in all members of the species [6].

Retroviral genome invasion and the fate of endogenous retrovirus (ERV) loci in the germline. The three panels show schematic diagrams illustrating how the distribution of ERVs is influenced by a host phylogeny; b activity of ERV lineages within the gene pool; c patterns of ERV locus inheritance within populations of host species. Panel a shows how ERV lineages originate when infection of an ancestral species by an ancient retrovirus causes a ‘germline colonisation’ event in which a retroviral provirus is integrated into the nuclear genome of a germline cell that then goes on to develop into a viable organism. This ‘founder’ ERV provirus can subsequently generate further copies within the germline (panel b). The fate of individual ERV loci is determined by selective forces at the level of the host population. Most ERV loci are quickly eliminated from the germline via selection or drift. However, some may increase in frequency from one host generation to the next, to the point where they become genetically ‘fixed’—i.e. they occur in all members of the species. The schematic in panel c illustrates this in a simplified way, showing an ERV locus (copy x) becoming fixed in over several host generations. As shown in panel a, fixed ERV loci persist in the host germline as ‘footprints’ of ERV activity, and the identification of orthologous ERV loci in multiple species indicates that those species diverged after the ERV was inserted. Thus, when host divergence dates have been estimated, they can be used to infer minimum ages for orthologous ERV loci. Importantly, extinction of host lineages eliminates swathes of ERV loci. In some rare cases, however, their sequences may still be recoverable (e.g. see [79]). Abbreviations: ERV endogenous retrovirus, NWM New World monkeys, OWM Old World monkeys
Studies over recent years have revealed the profound impact that ERVs have exerted on vertebrate evolution. For example, more of the human genome (~ 8%) is made up of the remnants of past retroviral infections than of sequences encoding the proteins necessary for life (~ 1–2%) [7]. Moreover, ERVs are not—as was once believed—mere ‘junk DNA’—some encode intact proteins that have been co-opted or exapted to perform physiological functions in host species, and even ERVs that are relatively degraded in terms of their coding capacity can perform important functions as components of gene regulatory networks [8,9,10,11,12,13].
ERV sequences also provide a unique source of retrospective information about retroviruses that circulated millions of years ago, and can therefore be used to explore the long-term history of evolutionary interaction between retroviruses and their hosts [14, 15]. Until quite recently, most investigations of this nature have of necessity been theoretical or comparative, but in recent years ‘investigators have utilized gene synthesis to ‘repair’ the mutated genes of ERVs and study their biological properties in vitro [16,17,18,19,20,21,22,23,24,25].
New vertebrate genome sequences are becoming available for study on an almost daily basis, providing a deluge of novel ERV data to drive further investigations of ERVs. There is therefore an urgent need for a unified system of nomenclature for ERV loci, not only to assist genome annotation, but also to facilitate research on ERVs and their impact on the genome biology and evolution of host species.
Insights into ERV biology in the genomic era
Modern genomics has allowed investigations of ERVs across a wide range of vertebrate whole genome sequences [26]. Together, these have provided a number of important insights into the general biology of ERV lineages that should be taken into consideration when constructing a nomenclature system.
Firstly, phylogenetic studies in humans and other species have shown that the multitudes of ERV sequences found in vertebrate genomes derive from a relatively small number of initial founder events [27, 28], and that distinct vertebrate lineages contain characteristic sets of ERVs that reflect their specific histories of; (1) retroviral germline invasion; (2) ERV copy number expansion; (3) and ERV locus fixation (see Fig. 1). However, establishing precisely the number of distinct retroviral germline invasion events that have occurred in the evolution of a host lineage is difficult. Significant germline invasions by retroviruses can presumably occur without any ERVs being fixed in descendant species, and even those ERV groups that do get fixed may be comprised entirely of partial and/or low copy number sequences that are problematic to detect. Moreover, even for the subset of ERVs that are detectable, phylogenetic approaches may not allow the number of separate invasion events to be determined with confidence—particularly when multiple invasions involving relatively similar viruses have occurred in the distant past. For example, estimates for the number of distinct germline invasion events that gave rise to the ERVs found in the human genome vary widely, from ~ 34 to ~ 80 [10, 73].
Secondly, it is clear from genomic studies that the vast majority of ERVs no longer encode functional proteins. Retroviral proviruses typically possess three principal coding domains (gag, pol and env), flanked at either side by long terminal-repeat sequences (the 5′ and 3′ LTRs) that are identical at the time of integration [29] (Fig. 2). A non-coding sequence containing a tRNA-specific primer-binding site (PBS) is usually present between the end of the 5′ LTR and the first codon of the gag gene. Without the purifying selection provided by replication, however, ERV sequences undergo mutational decay. Frequently, internal coding sequences are completely deleted through recombination between 5′ and 3′ LTRs, leaving behind a ‘solo LTR’ [30]. Indeed, solo LTR numbers are typically orders of magnitude more common than loci containing internal coding regions [31]. Other rearrangements of ERV genomes can also arise through processes such as LINE1-mediated retrotransposition, recombination, and deletion (Fig. 2b) [3]. Recombination can generate a diversity of ‘mosaic’ ERV forms [6], and can lead to genes and LTR sequences being ‘swapped’ between retroelement lineages [32].

Figure partly adapted from [80]
Genomic structure of ERV sequences. Panel a shows a schematic representation of a generalised retroviral provirus. The four coding domains found in all exogenous retroviruses are indicated. The precise organization of these domains varies among retrovirus lineages, and some viruses also encode additional genes. The long terminal repeat (LTR) sequences are comprised of three distinct subregions that are named according to their organization in the genomic RNA: unique 3′ region (U3), repeat region (R), and unique 5′ region (U5). Panel b shows a schematic representation of processes that modify ERV sequences. (1) Recombination between the two LTRs of a single provirus resulting in the formation of a solo LTR. (2) Recombination between the 3′ and 5′ LTRs of a given provirus leading to a tandem duplicated provirus. (3) Adaptation to intracellular retrotransposition, resulting in the loss of the envelope gene. (4) LINE1-mediated retrotransposition, resulting in loss of the 5′ U3 sequence, and the 3′ U5 sequence. Variants with larger 5′ truncations may also occur. Poly-A tails at the 3′ end and L1-typical target site duplications flanking the retrotransposed sequence are usually found for these forms.
Finally, comparative genomic studies have shown that in many cases, homologous ERV sequences are present at the same genomic locus in multiple species genomes. Since retroviral integration—while not random—is not site-specific [33, 34], such ‘orthologous’ ERV loci can be assumed to have been generated before the species they are found in diverged. Thus, if host divergence dates are known, they can be used to infer minimum ages to be inferred for individual ERV loci, and by extension the founding colonization events that generated ERV lineages [35]. In higher primates, for example, comparative studies show that most integration events are extremely ancient, having occurred after the separation between New World monkeys (Platyrrhini) and Old World monkeys (Catarrhini) but before the split between Old World monkeys and hominoids (Hominoidae) around 30–45 million years ago (Mya) [36]. It should be noted, however, that fixed ERV loci may significantly predate the divergence times of the host species they occur in. Furthermore, as shown in Fig. 1, fixed ERV loci can be much younger than the ERV lineage they belong to, and due to different patterns of inheritance in descendant hosts, ERVs can end up being fixed in one set of descendant species, and lost from another.
Existing ERV nomenclature schemes and history of their development
Existing nomenclature systems for ERVs have developed in a haphazard manner reflecting their history of discovery. ERVs were first discovered in the 1960s by virtue of the genetically controlled expression of viral antigens of replication-competent ERVs in chickens and mice [37]. These viruses were closely related to exogenous oncogenic viruses, prompting a decades long search for disease-associated ERVs in other species, especially man [38, 39]. Infectious human counterparts, however, have remained elusive.
Laboratory techniques employed to identify ERVs have included virus isolation by co-cultivation with cells from a variety of species [40], hybridization under low stringency conditions with retroviral probes followed by cloning [41, 42], and PCR with primers directed to conserved regions of RT [42,43,44,45,46]. These studies formed the initial context of ERV nomenclature schemes, but in more recent years, ERV nomenclature has been increasingly influenced by in silico mining of vertebrate genome sequences, based either on sequence similarity or predicted features of proviruses such as nearby LTRs.
Originally, endogenous proviruses were named after the most closely related exogenous retrovirus, such as murine leukemia virus (MLV), as well as subgroups, like xenotropic MLV (XMV) [47]. A common approach to naming ERVs in different species has been to add one or two letters before the designation ERV to indicate the species in which they were initially identified; thus, HERV indicates an ERV first seen in human DNA, and MERV or MuERV implies one originally found in the genomes of murine species [e.g. house mouse (Mus musculus)]. HERVs have been further classified on the basis of the tRNA that binds to the viral primer binding site (PBS) to prime reverse transcription (see Fig. 2a). Hence HERV-K implies a provirus or ERV lineage that use a lysine tRNA, no matter their relationship to one another. In some cases the PBS sequence was not available when novel elements were first discovered leading to the names based on neighboring genes (e.g. HERV-ADP [48]), clone number (e.g. HERV-S71 [49]), or amino acid motifs (e.g. HERV-FRD [42]). Additional designations based on the probe used for cloning, and sub-divisions based on sequence identity or phylogenetic reconstructions, have also been used [50].
The somewhat arbitrary manner in which these nomenclatures have evolved has created a number of anomalies. The first concerns the use of the initial letter(s) to designate species of origin. This presents difficulties with proviruses that were integrated prior to the divergence of their host species. Many of the ERVs present in humans and chimpanzees fall into this category—thus related proviruses in both species genomes can end up with quite different names (e.g. HERVxxx and CERVyyy) despite the fact that proviruses in the two species will be more closely related to one another (identical at the time of integration) than their paralogous siblings within the same phylogenetic grouping. This problem becomes even more acute when considering specific proviruses shared among multiple species (i.e., when the same integrated provirus has been inherited by two or more descendant species). A further difficulty arises when what would appear to be the generic name for ERVs from one species becomes the trivial name for a discrete lineage of proviruses within that species, as has occurred with the MLV-related PERVs (porcine endogenous retroviruses) of pigs [51].
The use of tRNA primer specificity as a basis for sub-classification is problematic because there are a number of instances where this sequence does not reflect the overall relationship between distinct ERV lineages. For example, the HERV-K(HML-5) group appears to use a tRNAMet as primer while the other HERV-K lineages use tRNALys [52]. Even very recently integrated proviruses, such as endogenous MLVs, can be found to use different tRNA primers. The frequent convergent evolution implied by these examples, and the limited number of tRNAs available, makes primer usage an unsuitable basis for retroviral taxonomy.
At the level of individual ERV lineages, it is necessary to distinguish among specific proviruses at discrete chromosomal locations (i.e. between different but related ERV loci), and several different systems have developed for this purpose. Most commonly, individual proviruses are simply numbered; e.g. as Xmv1, HERV-K 108, etc. In the case of HERVs, some investigators have chosen to use cytogenetic designations to distinguish among related proviruses [53, 54], as in HERV-K 11q22 (located on the q-arm, chromosomal band 22, of human chromosome 11). The need for this kind of locus-level ERV annotation is far more urgent now that large numbers vertebrate genomes have been sequenced. Indeed, in genomes that have been sequenced to a high degree of coverage, it is now feasible to identify and annotate the majority of ERVs using purely in silico approaches.
The most comprehensive source of repetitive element annotations is REPBASE [55]. REPBASE annotations, which include but are not limited to ERVs, are based on sequence similarity to a set of consensus elements. As such, the naming conventions used within REPBASE may not necessarily reflect phylogenetic relationships between ERVs. Also, REPBASE annotations distinguish LTRs and internal regions, but do not provide any further breakdown of the genomic features found within ERV proviruses. Software tools have also been developed specifically to assist in the identification and characterization of ERVs (for instance, see [56,57,58]), and these, more focused systems can be used to map ERVs to a fine scale of detail, demarcating genes, protein domains, and functional RNA sequences [6, 59]. Unfortunately, however, there is currently no straightforward way to link the ERV annotations generated by distinct systems with one another, or with the taxonomic groupings of ERVs that have been defined in broad-based phylogenetic studies [27, 28, 45, 60,61,62].
Integrating ERV classification with retrovirus taxonomy
A further problem is aligning ERV classification—which so far has been derived in large part from systems of repetitive element annotation—with retroviral taxonomy as agreed by the International Committee for Virus Taxonomy (ICTV). The Retroviridae family is grouped into the order Ortervirales (retro-transcribing viruses) [63], and comprises two sub-families, Orthoretrovirinae (orthoretroviruses) and Spumaretrovirinae (spumaviruses or ‘foamy viruses’). Spumaretrovirinae is currently a monogeric subfamily, whereas the Orthoretrovirinae comprises six exogenous genera. Endogenous representatives have now been identified for the majority of retroviral genera (Table 1). Some of these ERVs group robustly within the diversity of exogenous representatives in phylogenetic trees. Others group basal to contemporary isolates, but exhibit genomic or phylogenetic characteristics that argue for their inclusion within a particular genus (e.g. the presence of characteristic genomic features such as accessory genes and nucleotide composition biases) [64,65,66].
However, most ERV lineages are more problematic to place in current taxonomic systems, and as a consequence, many have become known by the relatively arbitrary names they have been assigned within repetitive element classification systems. In these systems, ERVs form part of a larger assemblage of LTR-retroelements [55, 67, 68] characterised by their “paired LTR” structure. TE classification systems conventionally group ERVs into three ‘classes’ (I, II and III), based on relatedness to the exogenous Gammaretrovirus, Betaretrovirus and Spumaretrovirus genera respectively. Individual ERV lineages (i.e. groups of ERVs that are assumed to derive from a single germline invasion event) have historically been referred to as ‘families’. This is problematic as the terms ‘class’ and ‘family’ have specific, taxonomic meanings and their use in this context is incompatible with existing retroviral taxonomy.
Taxonomy should ideally follow phylogeny [69]. Since the overwhelming evidence from genomic studies indicates that endogenous retroviruses derive from ancient exogenous retroviruses, integration of ERVs into retroviral classification schemes is both feasible and logical, following this principle. Any novel system of classification for ERVs should therefore take into account the phylogenetic relationships of ERVs to exogenous viruses. In addition, it seems likely that integration of ERV nomenclature with exogenous retroviral taxonomy will require the definition of new groups to represent lineages that existed as exogenous retroviruses in the past but now exist only as ERV “fossils” (i.e., extinct lineages).
ERV nomenclature proposal
It is clear that a standard system of nomenclature is required. Such a system would greatly facilitate communication and reproduction of results. For example, it could be used to provide unambiguous lists of loci in methods sections of manuscripts, or for the purposes of reproducing or comparing results of different studies. Ideally, a nomenclature system would provide a stable foundation for the development of increasingly accurate and finely detailed annotations. In addition, it could be used to nurture the establishment of a unified taxonomic system for retroviruses and ERVs.
We therefore propose that ERV loci be assigned standard, unique IDs composed of three elements, each separated by a hyphen, as shown in Fig. 3. The first element is a classifier that identifies the element as an ERV. The second element is itself comprised of two subcomponents—one denoting the lineage of retroviruses that the ERV belongs to, and the second being a numeric ID that uniquely identifies the specific ERV locus within that taxonomic group. The third element identifies the host lineage in which the ERV insertion occurs. The host lineage component may specify a species (i.e. we suggest using well-established abbreviations, such as HomSap for Homo sapiens). Alternatively, a higher taxonomic rank may be used to refer to the entire set of orthologous insertions that occurs in an order, family or genus. Examples of how these IDs would be applied to specific ERV loci are shown in Table 2.

Proposed ERV ID structure. The proposed ID consists of three components separated by hyphens. The second component consists of two subcomponents, separated by a period, that identify (1) the group the ERV belongs to, and (2) the unique numeric ID of the locus. The third component identifies the species or species group in which the element(s) being referred to occur
Applying the proposed ERV nomenclature in practice
There are a number of contingencies pertaining to way that each of the individual elements within the ID is defined. Firstly, only sequences that disclose robust phylogenetic evidence of having been directly derived from an exogenous retrovirus should receive the classifier ‘ERV’ in the first ID element. Thus, loci belonging to the ancient mammalian lineage ERV-L would be included (even though none of the canonical ERV-L sequences encode an env gene) because the ERV-L RT has been shown to group robustly within the diversity of the family Retroviridae [70]. By contrast, other LTR-retroelements that do not disclose an unambiguous link to retroviruses are excluded. These include, for example, the mammalian apparent retrotransposon (MaLR) elements, which are comprised of LTR-bounded internal sequences containing little or no similarity to retroviruses. Initially, the ‘ERV’ classifier should be reserved for clearly proviral elements that contain recognisable coding domains in their internal regions, and can be placed within a phylogeny of elements that can itself be placed within the Retroviridae family. Subsequently, solo LTR loci can be incorporated if: (1) they are allelic variants, and some proviral alleles also occur at the same locus; (2) they fall within a clade of LTR elements that is demonstrably associated with a particular lineage of ERV proviruses.
Since ERV sequences included in our classification scheme must by definition demonstrate phylogenetic links to exogenous retroviruses, it follows they can be integrated into a unified taxonomic scheme with a rational phylogenetic basis. This taxonomic scheme would provide the basis for assigning the ‘lineage’ component of the ID. Figure 4 illustrates a proposal for a unified scheme that integrates the classification of exogenous and endogenous retroviruses with minimal disruption to the existing schemas used for each. Within our proposed scheme, ERV loci should ideally be assigned IDs wherein the lineage component accurately reflects their position in such a unified schema. As discussed earlier, some ERVs exhibit phylogenetic and genomic characteristics that clearly identify them as endogenous representatives of contemporary virus groups (Table 1). However, the vast majority of ERVs fall outside the diversity defined by exogenous isolates. Thus, additional taxonomic groups would need to be created before the proposed nomenclature could be applied. These might be relatively broad to begin with—for example, the schema shown in Fig. 4 includes three ‘placeholder’ groups designed to act as temporary ‘bins’ for ERV loci that cannot be confidently placed within the existing taxonomic system approved by the ICTV. These groups correspond to three major divergences in orthoretroviral RT sequences [71], and are labelled as follows: Spumavirus-related (S), Gammaretrovirus/Epsilonretrovirus-related (GE), and Alpharetrovirus/Betaretrovirus-related (AB). Placeholder groups are reserved for ERVs that do not group within the diversity of established genera. Within these broad groups, additional subgroupings representing well-established ERV lineages can then be recognized. Wherever possible, ERVs should be assigned IDs that identify them at the level of individual lineages (i.e. monophyletic lineages of ERV sequences estimated to derive from a single germline colonisation event), or at the level of viral species for ERVs that show close relationships to exogenous viruses, such as some of those found in the mouse genome (see Table 2). Ultimately, some of the ERV lineages that lack exogenous counterparts might be recognised as fossil representatives of extinct lineages, and attributed genus status within the unified taxonomic scheme shown in Fig. 4.

Schematic phylogeny illustrating the basis for a unified ERV and retrovirus taxonomy. The top two brackets indicate taxonomic groupings. The ‘clade’ level reflects three major divergences in orthoretroviral reverse transcriptase genes [71]. The seven officially recognised genera are shown as coloured goblets at phylogeny tips. In addition, three placeholder groups are shown: Spumavirus-related (S), Gammaretrovirus/Epsilonretrovirus-related (GE), and Alpharetrovirus/Betaretrovirus-related (AB). Placeholder groups (indicated by coloured squares) are reserved for ERVs that do not group within the diversity of established genera. Within these broad groups, additional subgroupings representing well-established monophyletic ERV lineages may be recognized. Here, some examples are indicated, shown emerging from each of their parent groups. Ultimately, some of these lineages might be attributed genus status, and would be moved to the appropriate level within this classification scheme
With regard to the numeric ID component, each taxonomic level referenced by the nomenclature would require its own discrete numbering system, entirely independent of all other taxonomic levels, and within which numeric IDs are only assigned once. Inevitably, the taxonomic designations may be subject to a limited amount of change over time, since ERVs are often identified before their phylogenetic relationships are fully resolved. Similarly, the piecemeal task of identifying orthologs would be expected to cause ongoing adjustments to numeric IDs (e.g. as it becomes clear that an ERV in one species is orthologous to an ERV detected in another). Providing each adjustment generates a new key that is unique within the given taxonomic group, this can be accommodated.
Some ERV lineages have become known by particular names, and within these lineages, certain loci are also often known by particular numbers. We therefore propose that where ERV lineages or loci have established names or IDs that are well established and widely used, a mnemonic link to these should, where expedient, be retained. The examples shown in Table 2 illustrate how the proposed ID structure can support this.
The development of a consistent ERV nomenclature that uniquely identifies ERV loci would establish a basis for stably linking these loci to a wide range of relevant auxiliary information, such as cytogenetic location, or information about the genetic sub-structure of proviral insertions. This would compensate for the loss of such information from the ID itself, which would occur in some cases as a consequence of the standardization (see Table 2). Clearly, however, any auxiliary information attached to IDs would need to be collated and archived in a systematic way (i.e. using a database). Furthermore, ongoing maintenance of the nomenclature itself will be necessary, and a system of governance and oversight would need to be developed through which updates—e.g. addition, subtraction or merging of ERV loci, or reclassification of ERVs based on updated taxonomy—can be coordinated. An important aspect of nomenclature implementation will be the development of benchmarking procedures through which competing annotations can be assessed, as discussed more broadly for TEs in [72].
Conclusions
In this review, we have provided an account of how ERV nomenclature has developed, identifying the idiosyncrasies that have been generated in current nomenclature systems as a consequence of their historical development. We propose a novel, rational approach to naming ERV loci that is designed to unambiguously identify individual ERV loci, while accommodating as far as possible the contingencies and idiosyncrasies of ERV annotation. In addition, the proposed system allows for seamless integration into existing schemes for classification of transposable elements and viruses [55, 63, 67, 69, 73].
Abbreviations
- ERV:
-
endogenous retrovirus
- LTR:
-
long terminal repeat
- NWM:
-
New World monkey
- OWM:
-
Old World monkey
- PBS:
-
primer binding site
- tRNA:
-
transfer RNA
- HERV:
-
human endogenous retrovirus
- MLV:
-
murine leukemia virus
- ICTV:
-
International Committee for Virus Taxonomy
References
Vogt PK. Historical introduction to the general properties of retroviruses. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. New York: Cold Spring Harbour Laboratory Press; 1997.
Maksakova IA, Mager DL, Reiss D. Keeping active endogenous retroviral-like elements in check: the epigenetic perspective. Cell Mol Life Sci. 2008;65(21):3329–47.
de Parseval N, Heidmann T. Human endogenous retroviruses: from infectious elements to human genes. Cytogenet Genome Res. 2005;110(1–4):318–32.
Belshaw R, et al. High copy number in human endogenous retrovirus families is associated with copying mechanisms in addition to reinfection. Mol Biol Evol. 2005;22(4):814–7.
Ribet D, et al. An infectious progenitor for the murine IAP retrotransposon: emergence of an intracellular genetic parasite from an ancient retrovirus. Genome Res. 2008;18(4):597–609.
Vargiu L, et al. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology. 2016;13:7.
Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921.
Jern P, Coffin JM. Effects of retroviruses on host genome function. Annu Rev Genet. 2008;42:709–32.
Varela M, et al. Friendly viruses: the special relationship between endogenous retroviruses and their host. Ann N Y Acad Sci. 2009;1178:157–72.
Rowe HM, Trono D. Dynamic control of endogenous retroviruses during development. Virology. 2011;411(2):273–87.
Dupressoir A, Lavialle C, Heidmann T. From ancestral infectious retroviruses to bona fide cellular genes: role of the captured syncytins in placentation. Placenta. 2012;33(9):663–71.
Stoye JP. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol. 2012;10(6):395–406.
Wolf G, Greenberg D, Macfarlan TS. Spotting the enemy within: targeted silencing of foreign DNA in mammalian genomes by the Kruppel-associated box zinc finger protein family. Mob DNA. 2015;6:17.
Feschotte C, Gilbert C. Endogenous viruses: insights into viral evolution and impact on host biology. Nat Rev Genet. 2012;13(4):283–96.
Gifford RJ. Viral evolution in deep time: lentiviruses and mammals. Trends Genet. 2012;28(2):89–100.
Dewannieux M, et al. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 2006;16(12):1548–56.
Lee YN, Bieniasz PD. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 2007;3(1):e10.
Perez-Caballero D, et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139(3):499–511.
Soll SJ, Neil SJ, Bieniasz PD. Identification of a receptor for an extinct virus. Proc Natl Acad Sci USA. 2010;107(45):19496–501.
Brady T, et al. Integration target site selection by a resurrected human endogenous retrovirus. Genes Dev. 2009;23(5):633–42.
Goldstone DC, et al. Structural and functional analysis of prehistoric lentiviruses uncovers an ancient molecular interface. Cell Host Microbe. 2010;8(3):248–59.
Dewannieux M, et al. The mouse IAPE endogenous retrovirus can infect cells through any of the five GPI-anchored Ephrin A proteins. PLoS Pathog. 2011;7(10):e1002309.
Lemaitre C, et al. The HERV-K human endogenous retrovirus envelope protein antagonizes Tetherin antiviral activity. J Virol. 2014;88(23):13626–37.
Blanco-Melo D, Gifford RJ, Bieniasz PD. Reconstruction of a replication-competent ancestral murine endogenous retrovirus-L. Retrovirology. 2018;15(1):34.
Blanco-Melo D, Gifford RJ, Bieniasz PD. Co-option of an endogenous retrovirus envelope for host defense in hominid ancestors. Elife. 2017;6:e22519.
Johnson WE. Endogenous retroviruses in the genomics era. Annu Rev Virol. 2015;2(1):135–59.
Tristem M. Identification and characterisation of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database. J Virol. 2000;74:3715–30.
Bénit L, Dessen P, Heidmann T. Identification, phylogeny, and evolution of retroviral elements based on their envelope genes. J Virol. 2001;75(23):11709–19.
Coffin JM. Structure and classification of retroviruses. In: Levy JA, editor. The retroviridae. New York: Plenum Press; 1992. p. 19–49.
Sverdlov ED. Perpetually mobile footprints of ancient infections in human genome. FEBS Lett. 1998;428(1–2):1–6.
Belshaw R, et al. Rate of recombinational deletion among human endogenous retroviruses. J Virol. 2007;81(17):9437–42.
Lober U, et al. Degradation and remobilization of endogenous retroviruses by recombination during the earliest stages of a germ-line invasion. Proc Natl Acad Sci USA. 2018;115(34):8609–14. https://doi.org/10.1073/pnas.1807598115.
Bushman F, et al. Genome-wide analysis of retroviral DNA integration. Nat Rev Microbiol. 2005;3(11):848–58.
Kvaratskhelia M, et al. Molecular mechanisms of retroviral integration site selection. Nucleic Acids Res. 2014;42(16):10209–25.
Lee A, et al. Identification of an ancient endogenous retrovirus, predating the divergence of the placental mammals. Philos Trans R Soc Lond B Biol Sci. 2013;368(1626):20120503.
Bannert N, Kurth R. The evolutionary dynamics of human endogenous retroviral families. Annu Rev Genomics Hum Genet. 2006;7:149–73.
Weiss RA. The discovery of endogenous retroviruses. Retrovirology. 2006;3:67.
Löwer R. The pathogenic potential of endogenous retroviruses: facts and fantasies. Trends Microbiol. 1999;7(9):350–6.
Voisset C, Weiss RA, Griffiths DJ. Human RNA “rumor” viruses: the search for novel human retroviruses in chronic disease. Microbiol Mol Biol Rev. 2008;72(1):157–96.
Weiss RA, et al. Induction of avian tumor viruses in normal cells by physical and chemical carcinogens. Virology. 1971;46(3):920–38.
Dunwiddie CT, et al. Molecular cloning and characterization of gag-, pol-, and env-related gene sequences in the ev- chicken. J Virol. 1986;59(3):669–75.
Seifarth W, et al. Retrovirus-like particles released from the human breast cancer cell line T47-D display type B- and C-related endogenous retroviral sequences. J Virol. 1995;69(10):6408–16.
Medstrand P, Blomberg J. Characterization of novel reverse transcriptase encoding human endogenous retroviral sequences similar to type A and type B retroviruses: differential transcription in normal human tissues. J Virol. 1993;67(11):6778–87.
Cordonnier A, Casella JF, Heidmann T. Isolation of novel human endogenous retrovirus-like elements with foamy virus-related pol sequence. J Virol. 1995;69(9):5890–7.
Herniou E, et al. Retroviral diversity and distribution in vertebrates. J Virol. 1998;72(7):5955–66.
Gifford R, et al. Evolution and distribution of class II-related endogenous retroviruses. J Virol. 2005;79(10):6478–86.
Stoye JP, Coffin JM. The four classes of endogenous murine leukemia virus: structural relationships and potential for recombination. J Virol. 1987;61(9):2659–69.
Lyn D, et al. The polymorphic ADP-ribosyltransferase (NAD+) pseudogene 1 in humans interrupts an endogenous pol-like element on 13q34. Genomics. 1993;18(2):206–11.
Werner T, et al. S71 is a phylogenetically distinct human endogenous retroviral element with structural and sequence homology to simian sarcoma virus (SSV). Virology. 1990;174(1):225–38.
Subramanian RP, et al. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology. 2011;8:90.
Takeuchi Y, et al. Host range and interference studies of three classes of pig endogenous retrovirus. J Virol. 1998;72(12):9986–91.
Lavie L, et al. Human endogenous retrovirus family HERV-K(HML-5): status, evolution, and reconstruction of an ancient betaretrovirus in the human genome. J Virol. 2004;78(16):8788–98.
Hughes JF, Coffin JM. Evidence for genomic rearrangements mediated by human endogenous retroviruses during primate evolution. Nat Genet. 2001;29(4):487–9.
Macfarlane C, Simmonds P. Allelic variation of HERV-K(HML-2) endogenous retroviral elements in human populations. J Mol Evol. 2004;59(5):642–56.
Jurka J, et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–7.
Sperber GO, et al. Automated recognition of retroviral sequences in genomic data–RetroTector. Nucleic Acids Res. 2007;35(15):4964–76.
Sperber G, et al. RetroTector online, a rational tool for analysis of retroviral elements in small and medium size vertebrate genomic sequences. BMC Bioinform. 2009;10(Suppl 6):S4.
Lerat E. Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity (Edinb). 2010;104(6):520–33.
Grandi N, et al. Contribution of type W human endogenous retroviruses to the human genome: characterization of HERV-W proviral insertions and processed pseudogenes. Retrovirology. 2016;13(1):67.
Hayward A, Cornwallis CK, Jern P. Pan-vertebrate comparative genomics unmasks retrovirus macroevolution. Proc Natl Acad Sci USA. 2015;112(2):464–9.
Hayward A, Grabherr M, Jern P. Broad-scale phylogenomics provides insights into retrovirus-host evolution. Proc Natl Acad Sci USA. 2013;110(50):20146–51.
Xu X, et al. Endogenous retroviruses of non-avian/mammalian vertebrates illuminate diversity and deep history of retroviruses. PLoS Pathog. 2018;14(6):e1007072.
Krupovic M, et al. Ortervirales: new virus order unifying five families of reverse-transcribing viruses. J Virol. 2018;92(12):e00515–8.
Katzourakis A, et al. Discovery and analysis of the first endogenous lentivirus. Proc Natl Acad Sci USA. 2007;104(15):6261–5.
Katzourakis A, et al. Macroevolution of complex retroviruses. Science. 2009;325(5947):1512.
Farkasova H, et al. Discovery of an endogenous Deltaretrovirus in the genome of long-fingered bats (Chiroptera: Miniopteridae). Proc Natl Acad Sci USA. 2017;114(12):3145–50.
Wicker T, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8(12):973–82.
Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet. 2008;9(5):411–2.
Blomberg J, et al. Classification and nomenclature of endogenous retroviral sequences (ERVs): problems and recommendations. Gene. 2009;448(2):115–23.
Bénit L, et al. ERV-L elements: a family of endogenous retrovirus-like elements active throughout the evolution of mammals. J Virol. 1999;73(4):3301–8.
Llorens C, Fares MA, Moya A. Relationships of gag-pol diversity between Ty3/Gypsy and retroviridae LTR retroelements and the three kings hypothesis. BMC Evol Biol. 2008;8:276.
Hoen DR, et al. A call for benchmarking transposable element annotation methods. Mob DNA. 2015;6:13.
Seberg O, Petersen G. A unified classification system for eukaryotic transposable elements should reflect their phylogeny. Nat Rev Genet. 2009;10(4):276.
Green RG, Moosey MM, Bittner JJ. Serial transmission of the milk agent of mouse mammary carcinoma. Proc Soc Exp Biol Med. 1946;61:362.
Gross L. A filterable agent, recovered from Ak leukemic extracts, causing salivary gland carcinomas in C3H mice. Proc Soc Exp Biol Med. 1953;83(2):414–21.
Brown K, Emes RD, Tarlinton RE. Multiple groups of endogenous epsilon-like retroviruses conserved across primates. J Virol. 2014;88(21):12464–71.
Sverdlov ED, editor. Retroviruses and Primate Genome Evolution. Austin, TX: Landes Bioscience; 2005. p. 186–203.
Oja M, et al. Self-organizing map-based discovery and visualization of human endogenous retroviral sequence groups. Int J Neural Syst. 2005;15(3):163–79.
Greenwood AD, et al. Evolution of endogenous retrovirus-like elements of the woolly mammoth (Mammuthus primigenius) and its relatives. Mol Biol Evol. 2001;18(5):840–7.
Stoye JP. Endogenous retroviruses: still active after all these years? Curr Biol. 2001;11(22):R914–6.
Author’s contributions
RJG, JB, JM, HF, TH, JM, JS, MT, and WEJ wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Concepts for this nomenclature proposal were developed over several years in the retrovirus subcommittee of the International Committee on the Taxonomy of Viruses. We thank all members of the subcommittee for their contribution.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
In June 2014 a meeting was held in Missillac, France sponsored by a grant from the Borchard Foundation (awarded to HF) at which the final nomenclature system was developed. We thank the Foundation for its generous support of this effort. RJG was supported by a grant from the UK Medical Research Council (No. MC_UU_12014/10).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gifford, R.J., Blomberg, J., Coffin, J.M. et al. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology 15, 59 (2018). https://doi.org/10.1186/s12977-018-0442-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-018-0442-1