
Abstract
Inflammatory neuropathies are rare autoimmune-mediated disorders affecting the peripheral nervous system. Considerable progress has recently been made in understanding pathomechanisms of these disorders which will be essential for developing novel diagnostic and therapeutic strategies in the future. Here, we summarize our current understanding of antigenic targets and the relevance of new immunological concepts for inflammatory neuropathies. In addition, we provide an overview of available animal models of acute and chronic variants and how new diagnostic tools such as magnetic resonance imaging and novel therapeutic candidates will benefit patients with inflammatory neuropathies in the future. This review thus illustrates the gap between pre-clinical and clinical findings and aims to outline future directions of development.
Similar content being viewed by others

Background
Inflammatory neuropathies are a heterogeneous group of autoimmune disorders affecting the peripheral nervous system (PNS) that can exhibit an acute or chronic disease course. They are rare, but cause significant and often permanent disability in many affected patients [1, 2]. Although our understanding of disease mechanisms in inflammatory neuropathies has improved in recent years, several important aspects remain poorly understood and have not yet been experimentally addressed. The current review aims to summarize available data regarding pathomechanisms in inflammatory neuropathies and covers findings both in animal models and human patients.
The Guillain-Barré syndrome (GBS) is the prototypic acute inflammatory neuropathy usually featuring a rapidly progressive, symmetrical, often ascending weakness of all extremities and variable sensory deficits [3]. In typical cases, initial symptoms can include pain, numbness, and paresthesia, followed by a rapidly progressive flaccid paresis of all limbs [4, 5]. Very severe impairments and potentially lethal secondary complications can develop. GBS has been reported to occur with an annual incidence ranging between 0.8 and 1.9 per 100,000 worldwide. Men are approximately 1.5-fold more frequently affected and the incidence increases with age [6]. Although the majority of patients recover, the prognosis can remain poor with severe disability or death in 9–17% of all GBS patients [7]. In many cases, GBS develops subsequently to minor infections in otherwise healthy individuals but is not associated with other autoimmune or systemic disorders. An infection-triggered cross-reactive auto-immune response is thought to be aberrantly directed against the peripheral nervous system [5]. This concept of infection triggered cross-reactivity is well established in axonal GBS and suspected in demyelinating GBS (see below). Intravenous immunoglobulins and plasma exchange are effective in GBS and numerous other pharmacological candidates have been suggested in animal models, but their bench-to-bedside transfer is lacking [8].
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is the most common chronic inflammatory neuropathy and is usually characterized by slowly progressive, symmetric, proximal, and distal paresis and sensory dysfunction [9]. Symptoms develop within two or more months and the course can be either chronically progressive or relapsing with stepwise progression [10]. Different variants and atypical courses have also been described [11]. The prevalence of CIDP greatly depends on the diagnostic criteria and ranges between 1 and 9 cases per 100,000 individuals with annual incidence rates at 0.50–1.60 per 100,000 [12,13,14,15,16,17]. Cerebrospinal fluid protein levels are elevated up to sixfold while other parameters are typically normal. Nerve biopsy shows features of demyelination and remyelination, onion bulb formation, nerve oedema, occasional epineurial, or endoneurial T cells [18,19,20], and macrophages scattered either throughout the endoneurium or in small perivascular clusters in the endoneurium [21, 22]. Glucocorticoids, plasma exchange, and intravenous immunoglobulins (IVIg) are effective in CIDP. The randomized controlled “intravenous immune globulin for the treatment of CIDP (ICE) trial” provided the highest level of evidence that IVIg reduces the progression of impairments in CIDP patients [23].
Both acute and chronic inflammatory neuropathies exhibit considerable heterogeneity and can be divided into different subtypes. Subtypes of GBS have been classified as acute inflammatory demyelinating polyradiculoneuropathy (AIDP), acute motor axonal neuropathy (AMAN), acute motor and sensory axonal neuropathy (AMSAN), and Miller Fisher syndrome (MFS) [24]. Chronic idiopathic axonal polyneuropathy (CIAP), multifocal motor neuropathy (MMN), and paraproteinaemic demyelinating neuropathy (PDN) have overlap with CIDP or can be considered as its variants [25]. Such diversity impedes the diagnosis, treatment, and development of new therapeutic interventions.
The target antigen in GBS
Although the primary target antigen in GBS remains elusive, several recent studies have added interesting new candidates to the list of potential antigens [26]. Understanding the antigenic target could help to develop diagnostic or prognostic tests in the future and improve existing animal models. The hypothesis of microbial epitopes resembling endogenous peripheral nerve antigens, termed “molecular mimicry”, has been substantiated in the axonal subtypes of human GBS by several lines of evidence (reviewed in [27]). Briefly, there is a strong epidemiological association between GBS and preceding infections with Campylobacter jejuni (C. jejuni) and other infectious diseases [28]. Cell wall components of these infectious agents resemble endogenous lipids and may trigger a cross-reactive autoimmune response. Interestingly, a new infectious trigger for GBS was recently described.
Zika virus (ZIKV), a flavivirus such as yellow fever, dengue, Japanese encephalitis, and West Nile Virus, was recently identified as a novel trigger of GBS [29,30,31,32]. In one cohort, 98% of patients with GBS were tested positive for anti-ZIKV immunoglobulin M (IgM) or IgG. Most patients had a transient illness concordant with ZIKV infection several days before onset of GBS symptoms. The risk of GBS after ZIKV infection has therefore been estimated to be as high as 0.24/1000 infections, which is similar to the risk described for C. jejuni [33]. These data are supported by another study from Brazil during the recent major ZIKV outbreak. Between December 2015 and May 2016, evidence of recent ZIKV infection was found in serum and/or CSF of 77% of all GBS patients. Concurrently, GBS admission rates increased from an average of 1.0/month to 5.6/month [34]. First neurological symptoms were found to generally develop 6–10 days after symptoms of viral infection [33,34,35]. Only a minority of ZIKV-GBS patients had serological evidence of an autoimmune response against known GBS antigens [33]. Another study detected a considerable overlap between peptide sequences of ZIKV proteins and human structural myelin proteins [36]. These data support a molecular mimicry hypothesis in ZIKV-related GBS. Nerve biopsies show nerve fiber demyelination, axonal degeneration, and infiltration of mononuclear cells [37]. These findings raise concerns about a potential future health problem if endemic Zika virus infections could trigger clusters of GBS cases.
Autoantibodies binding components of the axonal membrane are found in a significant proportion of GBS cases [27]. Lipooligosaccharides from C. jejuni strains from a subset of axonal GBS patients carry ganglioside-like structures, resembling gangliosides enriched in axonal cell membranes (e.g., GM1, GD1a, GM1b, GalNAcGD1a) [28]. Therefore, the autoimmune reaction in axonal GBS is directed against axonal components. In support of this, some patients who received gangliosides as an experimental treatment for nonspecific pain syndromes subsequently developed an axonal GBS [38]. Recently, a novel glycoarray technique identified GM1, GA1, and GQ1b IgG antibodies in a high number of GBS patients and GQ1b antibodies were preferentially enriched in GBS patients with ophthalmoplegia [39]. In addition, some GBS sera were shown to strongly and specifically bind monoaminergic neurons in rat brain suggesting a potential interference of auto-antibodies with ion channels and neuronal receptors in GBS. This could explain neuropsychiatric and autonomic abnormalities in GBS [40].
Animal models for studying axonal GBS and anti-ganglioside antibodies in GBS have been described by repeatedly immunizing rabbits against axonal gangliosides GD1b (in ataxic sensory neuropathy) and GM1 (in acute motor axonal neuropathy) [41], causing axonal experimental allergic neuritis (EAN) with flaccid paresis [42], axonal damage, and ganglioside-directed antibody responses [43]. Some anti-ganglioside antibody-mediated neuropathies are characterized by a disruption of paranodal junctions and ion-channels at the nodes of Ranvier [44]. Ganglioside antibodies were speculated to cause a transient and initially reversible disruption of axonal impulse propagation. Only if secondary axonal degeneration ensues, disability will be permanent. A novel category of nodo-paranodopathies was therefore proposed for neuropathies associated with anti-ganglioside antibodies targeting nodal regions [45]. The applicability of this classification to standard clinical care remains to be determined.
Mice lacking complex gangliosides develop exaggerated humoral responses to gangliosides when immunized with C. jejuni, lipooligosaccharides [46], or gangliosides [47], indicating that tolerance-inducing mechanisms exist if endogenous gangliosides are present. In addition, passive transfer of ganglioside-directed antibodies induced axonal pathology in recipient animals [48], although sera from GBS patients did not exacerbate adoptive transfer EAN [49]. The available evidence thus supports that axonal GBS and corresponding ganglioside-induced animal models share a common antigenic target and disease mechanism.
The availability of animal models contrasts with the lack of novel therapeutic strategies in axonal GBS.
The target cellular structure or defined antigen in demyelinating human GBS remains unknown. Several studies have reported auto-antibodies or a cellular immune reaction against myelin proteins although this only holds true in a small proportion of demyelinating GBS patients [50,51,52,53]. Although mostly associated with axonal subtypes, lipids may also become target antigens in some demyelinating GBS cases [54]. Although myelin proteins can be used to elicit animal models of GBS (see below), no single antigenic target has been confirmed in a greater proportion of demyelinating GBS patients and no corresponding microbial structure with myelin-like properties has been identified [28]. Of note, a plethora of therapeutic strategies have been tested in demyelinating GBS models although the model fails to mirror the antigen specificity of the human disease [8]. Polymorphisms in the genes encoding interleukin 17 (IL-17) and intercellular adhesion molecule 1 (ICAM-1) lead to higher expression and may predispose to developing GBS [55]. Of note, both IL-17 and ICAM-1 have been associated with inflammatory neuropathies in animal models [56, 57].
Recent advances in understanding CIDP
Chronic inflammatory neuropathies such as CIDP lack strong evidence for a molecular mimicry-like mechanism described in GBS. The chronic and delayed onset of symptoms may potentially preclude the identification of triggering agents. CIDP can occur in autoimmune-susceptible patients with some patients also exhibiting inflammatory demyelination of the central nervous system with patchy regions of demyelination and oedema and inflammatory infiltrates seen in histology and magnetic resonance imaging [58]. When considering potential antigens, it is important to be aware that the majority of myelin proteins are specifically present in either central or peripheral nervous system but not both. Antibody responses against the myelin proteins P0 and P2 [59] and peripheral myelin protein 22 (PMP22) have been reported [50, 60], while other studies have not confirmed antibodies recognizing myelin I proteins in a relevant proportion of patients [53, 61]. Anti-ganglioside antibodies can be detected in some CIDP patients [62]. In addition, T cell reactivity against myelin proteins may occur in CIDP patients [51]. Thus, proteins of compact myelin can become target of chronic autoimmunity in some CIDP patients.
Several studies have interestingly identified the axonal node and paranode as novel potential targets of autoimmunity in chronic (and acute) inflammatory neuropathies [63]. Recent studies identified IgG4 antibodies against Neurofascin 155 (NF155) in a comparably homogenous group of CIDP patients. Patients with these antibodies were younger at onset, presented sensory ataxia, a disabling and characteristic tremor, and CNS (central nervous system) demyelination, and had a very poor response to IVIg treatment [64, 65]. A recent study also showed that immunoglobulins recognizing NF140/186 may cause reversible conduction block and demyelination which does respond to IVIg treatment. These findings suggest a complement-independent mechanism of NF140/186 IgGs [66]. Antibodies to Neurofascin have also been reported in EAN and as a potential target antigen in multiple sclerosis [67, 68]. Paranodal contactin-1 was identified as an additional potential antigenic target [69, 70]. Schwann cells exposed to sera from CIDP patients may be less effective at supporting growth of regenerating axons [71]. These observations indicate that different paranodal components can become targets of the immune response in CIDP and that the mechanisms leading to axonal degeneration may vary. Such novel antigenic targets may also reinvigorate the search for corresponding microbial antigens and molecular mimicry-like mechanisms.
Several studies have reported the endemic occurrence of a chronic inflammatory neuropathy in abattoir workers exposed to porcine brain tissue aerosols [72]. These cases provide interesting proof-of-principle, that active immunization against nervous system tissue by aerosol exposure can cause chronic inflammatory neuropathies in humans and argue for an antigenic target shared between central and peripheral nervous systems.
Primary structure and complementarity-determining region (CDR) 3 spectratyping of the T cell receptor (TCR) repertoire in CIDP patients has indicated that CD8+ T cells exhibit a much broader activation than CD4+ T cells and that IVIg preferentially ameliorates this. CD8+ T cells may thus have a crucial role in the immunopathogenesis of CIDP [73]. In accordance to these investigations, biopsies from CIDP patients showed a T cell receptor repertoire which has a strong monoclonal and oligoclonal restriction. Overlaps between CD8+ T cell clones in biopsies and blood have been observed, indicating an antigen-driven, major histocompatibility complex I restricted attack of peripheral nerve tissue components by CD8+ T cells [74]. Because of its CD8+ T cell-mediated autoimmune peripheral neuropathy, the L31 mouse strain (discussed in detail below) might be the appropriate model for this aspect [75]. Overall, several interesting new observations have been made in CIDP, but the antigenic specificity and the exact mechanisms of the autoimmune response in CIDP require future study.
Animal models of acute demyelinating inflammatory neuropathies
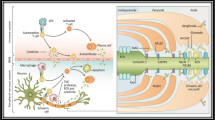
Animal models allow deciphering pathomechanisms of human disorders and enable preclinical testing of potential future treatments. The first available animal model of inflammatory peripheral neuropathies was generated by immunizing a susceptible rabbit strain with PNS myelin emulsified in complete Freund’s adjuvant (CFA) [76]. The disease resulting from such an induction protocol was named experimental autoimmune (initially “allergic”) neuritis and was later extended to various other animal species and strains including rats, mice, monkeys, and guinea pigs (Table 1). The Lewis rat strain enables most reliable EAN induction and has become a widely used GBS model. EAN induction in mice has proven to be problematic (see below).
Different components of PNS myelin can be used to trigger EAN. Lewis rats can be immunized with peripheral myelin homogenates, myelin proteins, or myelin protein-derived peptides to develop EAN (Fig. 1). The most abundant structural myelin protein is myelin protein zero (Mpz or P0) and can be used to induce EAN [77]. Less-severe forms of rat EAN are triggered by immunization against peripheral myelin protein of 22 kDa (PMP22) [78]. In addition to actively induced EAN, transfer of stimulated T lymphocytes that are reactive against various myelin antigens, including myelin proteins P2, P0, and derived peptides, evokes adoptive transfer EAN (AT-EAN) in receiving host animals (Table 1). These adoptive transfer studies have identified myelin-associated glycoprotein as another potential target in EAN [79]. Collectively, these studies have established the heterogeneity of potential antigenic targets in the PNS and allow understanding of general principles of PNS inflammation.
Actively induced EAN generates a PNS myelin-directed T cell response that triggers an acute monophasic cellular immune reaction within peripheral nerves [80]. Several aspects of human GBS are resembled in such actively induced EAN. Histologically, macrophages and T cells are the predominant infiltrating cells and cytokines mediate local tissue damage in the PNS and amplify the immune response by further attracting inflammatory cells [8, 80, 81]. Like EAN, GBS features an acute onset and monophasic course of disease that is triggered by exposure to proteins or microbial epitopes resembling self, respectively. Myelin protein autoreactivity, however, has been described in a small proportion of GBS patients (see above). Infiltration of mononuclear cells and immune cell-mediated myelin destruction in the PNS occurs at least in a proportion of GBS patients with a demyelinating disease phenotype [82]. Myelin component-induced EAN is thus a useful model of demyelinating GBS but fails to reproduce several aspects of the human disease.
Other evidence argues against the adequacy of myelin protein-induced EAN in reflecting GBS: A myelin directed autoimmune response is absent in most GBS patients [26, 52] and an evidence for a microbial epitope, resembling any human PNS myelin structure, is lacking. Furthermore, a long list of treatments was found effective in EAN but has not resulted in the establishment of novel therapeutic options in GBS (reviewed in [8]).
Of note, a recent publication described induction of a GBS-like disease in non-obese diabetic (NOD) mice after oral infection with C. jejuni strains from GBS patients. Mice developed autoantibodies against GM1, GQ1b, and GD1a gangliosides, inflammatory peripheral nerve lesions, axonal damage, and clinical impairments while antibiotic treatment exacerbated the symptoms. The authors also tested different NOD mutants and found that C. jejuni infection triggered T cell-dominated AIDP-like disease in the absence of B7-2 and axonal AMAN-like disease in the absence of IL-10. Therefore, NOD mice infected with C. jejuni may serve as a feasible novel GBS animal model enabling distinct disease phenotypes depending on host genotypes [83]. This tool appears especially suited to dissect intestinal triggers of PNS autoimmunity.
A recent publication using a rabbit model of axonal GBS demonstrated that the immunoglobulin G-degrading enzyme of Streptococcus pyogenes (IdeS) which cleaves IgG antibodies into fragments reduces complement-mediated axonal degeneration in anterior spinal roots. This is paralleled by a reduced disruption of voltage-dependent sodium (Nav) channels and less activated C3 at the nodes of Ranvier [84]. These data indicate that IdeS might be a promising therapeutic strategy for GBS.
Animal models of chronic inflammatory neuropathies
Animal models of CIDP or other chronic inflammatory neuropathies are less well established than acute models. Active immunization paradigms to induce chronic PNS inflammation have been reported: Rats of the Dark Agouti strain develop relapsing PNS inflammation and active myelin basic protein (MBP) immunization together with Cyclosporine A (CsA) treatment in Lewis rats, triggers a relapsing disease course with some CIDP-like features [85, 86]. Administration of low-dose CsA induces a relapsing or chronic persisting EAN course [86, 87]. The mechanisms for this chronification are still unknown but altered production of cytokines may be one potential explanation [88, 89]. IL-17A production was significantly enhanced after CsA cessation in relapsing murine EAE [90]. These data suggest a role of IL-17A in the relapses induced by CsA. One may also speculate that CsA suppresses Treg development by reducing IL-2 production [88, 89]. In fact, IL-2 is required for Treg proliferation, survival, and activity and inhibition of IL-2-dependent Treg proliferation together with increased IL-17A production due to CsA could trigger relapses [91,92,93].
Repeatedly performing adoptive transfer EAN may also reflect recurrent attacks in CIDP [94]. These induced models of CIDP, however, do not phenocopy the chronic progressive nature of CIDP. A number of studies have added several new animal models of chronic inflammatory neuropathies:
First, the so-called L31 mouse strain is characterized by constitutive expression of the costimulatory B7-2 (CD86) molecule on antigen-presenting cells. These mice develop spontaneous demyelination and immune cell infiltration of the peripheral nerves [95]. Depletion of CD4+ T cells in L31 mice expanded CD8+ T cells and accelerated and exacerbated the disease. CD8+ T cells are thus considered key drivers of the disease [96]. These mice exhibit deficits in motor and sensory functions with acute onset and continuous progression [75].
Second, mice on C57BL/6 background, transgenically expressing IL-10 under the control of the promotor of the vitelliform macular dystrophy 2 (VMD2) gene mutated in macular degeneration, spontaneously develop neuritis within 10 to 20 weeks of age. This neuritis is one of the very rare examples of a “pro-inflammatory function” of generally anti-inflammatory IL-10 and is caused by an IL-10-dependent ICAM-1 upregulation which triggers macrophage influx [97].
Third, several novel models of CIDP, based on the NOD mouse strain, have been established. The NOD mouse is prone to developing spontaneous autoimmunity such as a type 1 diabetes-like phenotype [98]. Initially, Salomon et al. reported in 2001 that NOD mice deficient in the costimulatory molecule B7-2 are protected from diabetes but instead spontaneously develop a CD4+ T cell-mediated, slowly progressive demyelinating peripheral neuritis predominantly in female (Fig. 1) [99]. Later studies found the spontaneous neuritis to be dependent on interferon γ (IFN-γ) and on a T-helper 1 skew in these mice [100]. The P0180–199 epitope of myelin protein zero was identified as a potential antigenic target, and a neuritogenic T cell receptor transgenic mouse line [101], recognizing the P01–25 epitope, was generated from this mouse strain [102]. In addition, an increased B cell reactivity to myelin protein zero (P0) and expansion of P0-specific plasmablasts was observed, while depletion of B cells with an anti-CD19 antibody led to decreased disease severity [103]. Another therapeutic option is FTY720, also termed Fingolimod, which was shown to intermit the progression of chronic neuritis in B7-2-deficient NOD mice by either depletion of T cells or indirect inhibition of pathogenic T cell expansion [104]. A recent study demonstrated lower frequencies and numbers of regulatory T cells (Tregs) and regulatory B cells (Bregs) in this mouse line and found these regulatory cell types to suppress spontaneous autoimmunity [105]. Summarizing, the B7-2-deficient NOD mouse mirrors the slowly and chronic progressive nature of CIDP, does not require immunization, and has been successfully used to study mechanisms and test several novel therapies of CIDP.

Representative animal models for GBS, CIDP, and underlying mechanisms. a Experimental autoimmune neuritis (EAN) is the animal model of acute peripheral neuropathies such as GBS. Induction is achieved by immunizing susceptible rodents (e.g., Lewis rats) with PNS myelin, peripheral myelin homogenates, myelin proteins (P0 or PMP22) or myelin-derived peptides. Immunization leads to a myelin-directed T cell response characterized by T cell and macrophage infiltration. Cytokine production mediates peripheral nerve damage and attraction of further inflammatory cells. b NOD B7-2−/− and NOD ICAM1−/− mice develop spontaneous CD4+ T cell-mediated neuritis with parallels to CIDP in humans. The neuritis is characterized by P0-specific CD4+ T cells that infiltrate into the PNS, IFN-γ dependency, an increased B cell reactivity to P0, expansion of P0-specific plasmablasts and lower frequencies of Tregs/Bregs. In the case of NOD ICAM1−/− mice, mechanisms are slightly different than depicted here. In this case, the neuritis is characterized by enhanced IL-17 production, macrophage and B cell infiltration without changes in Treg levels
Fourth, spontaneous autoimmune neuritis has also been reported in other mouse strains on the NOD genetic background. Antibody-mediated depletion of IL-2, required for the maintenance of Treg, caused spontaneous neuritis in NOD mice and antibody-mediated depletion of regulatory T cells exacerbated chronic neuritis [106, 107]. NOD mice carrying deficiency in programmed cell death 1 (PD-1)—an adhesion molecule with costimulatory function—develop diabetes with increased severity [108], while PD-1-deficient NOD mice, carrying the anti-diabetogenic major histocompatibility complex (MHC) haplotype H-2b, are protected from diabetes but develop spontaneous peripheral neuritis [109, 110]. NOD mice deficient in the inducible costimulator (ICOS) and ICOS ligand (ICOS-L) spontaneously develop multifocal autoimmunity in the nervous system and in muscle tissues including spontaneous neuritis [111]. NOD mice with a partial loss of Aire gene function (NOD.AireGW/+) spontaneously develop a peripheral nervous system autoimmunity comparable to CIDP. In these mice, a partial loss of Aire gene function leads to reduced P0 expression in the thymus resulting in an escape of P0 self-antigen-recognizing T cells from negative selection and thus central tolerance [112].
In addition, a recent study reported a spontaneous chronic neuritis in ICAM-1-deficient NOD mice. Interestingly, this neuritis is CD4+ T cell-dependent featuring enhanced production of pro-inflammatory IL-17 without any alternation in Treg numbers. T cells as well as macrophages and B cells infiltrate into peripheral nerves and show autoreactivity against P0 and an altered TCR repertoire. Thymus transplantation experiments identified an altered thymic selection of T cells rather than altered costimulation of T cells in the periphery to underlie the disease [56]. Altered costimulation may thus affect the thymic selection process and generally shift autoimmunity to peripheral nerves. It remains to be determined why the murine peripheral nervous system is preferentially prone to become an autoimmune target in specific pro-inflammatory settings and how this knowledge may be exploitable for human patients.
T cell subtypes in inflammatory neuropathies
The expansion of knowledge regarding CNS autoimmunity has not been paralleled by studies in the PNS. In particular, many novel subsets of CD4+ T cells have not been studied in inflammatory neuropathies at all.
For many years, attention focused on Th1 and Th2 helper T cells—two different differentiation programs of naïve CD4+ T cells. This long-accepted dichotomous model has been extended by the description of pro-inflammatory Th17 producing the cytokines IL-17A, IL-17F, and IL-22 [113]. Under physiological conditions, Th17 cells clear fungal pathogens. However, Th17 cells are also potent inducers of tissue inflammation and have been associated with many experimental and human autoimmune conditions [114]. The contribution of Th17 cells in the pathogenesis of inflammatory neuropathies in rodents and in humans has been poorly defined.
The presence of IL-17-producing cells has been described in murine EAN and recombinant IL-17 deteriorated the acute phase of rat EAN [115]. Interestingly, experimental treatments with Atorvastatin and Fingolimod reduced the accumulation of IL-17-producing cells in the PNS of experimental animals [116, 117].
In human CIDP patients, the percentage of peripheral Th17 cells and IL-17 plasma levels were increased specifically in patients with active disease [118]. The concentration of IL-17 and IL-6 (which induces IL-17) were increased in cerebrospinal fluid of CIDP patients [119], while another study reported an increased IL-17 production by blood mononuclear cells, although this was not specific for this cytokine [120]. In accordance, we recently found an increased number of IL-17-producing cells, preferentially in sural nerve biopsy sections of CIDP patients with high disease activity. The number of IL-17-producing T cells, before IVIg treatment, correlated with the beneficial response in ICAM-1−/−NOD mice. A high number of IL-17-producing cells thus predicted a good response to IVIg treatment while surprisingly IL-17 production in secondary lymphoid organs was unchanged [57]. These observations indicate IL-17 as a prognostic marker of disease activity and the efficacy of IVIg treatment.
In GBS patients, the frequency of circulating Th1, Th17, and Th22 cells was significantly increased and plasma and CSF (cerebrospinal fluid) levels of IL-17 and IL-22 were also elevated during the acute disease stage. The levels of both cytokines were directly correlated with the disability scale scores of the patients [121]. IVIg therapy downregulated these cells and respective cytokines [122], indicating a potential pathogenic role of Th17 cells in GBS. Different Th17 cell-inhibiting compounds have been described in experimental autoimmune encephalomyelitis (EAE) models [123, 124]. One may speculate that such findings could be transferable to inflammatory peripheral neuropathies and open new therapeutic avenues in the future.
Treg cells expressing the transcription factor forkhead box P3 (FoxP3) have been studied extensively and deficiency of Tregs causes a severe and lethal autoimmune syndrome in humans (termed IPEX, immunodysregulation polyendocrinopathy enteropathy X-linked syndrome) and mice [125, 126]. Treg dysfunction has been implicated in various human inflammatory disorders (reviewed in [127]). An impaired function or reduced numbers of Tregs have also been described in CIDP and GBS patients [128, 129]. Experimental studies have confirmed the presence of Tregs in EAN, and the beneficial effect of treatments in EAN has been attributed to increased Treg numbers [115, 130]. In EAN, Treg depletion with antibodies increased the clinical severity, the proliferation of myelin-specific T cells, and peripheral nerve inflammation in the initial priming phase of inflammation. Notably, systemic autoimmunity caused by lack of FoxpP3+ Tregs does not manifest with neuritis [131]. Lack of Tregs thus does not precipitate PNS-specific autoimmunity but exacerbates ongoing PNS inflammation triggered by other mechanisms.
IVIg treatment in GBS patients expanded Treg cells [132]. Protocols for the in vitro induction of Tregs from naïve CD4+ T cells of murine and human origin have been developed [133]. It is intriguing to speculate whether specific induction of stable Tregs would ameliorate EAN and pose a potential future treatment of GBS or CIDP. Another option is the suppression of leukocyte trafficking to the PNS. Preconditioning rats with forced exercise decreased EAN severity due to altered leukocyte composition and egress of autoreactive Th1 cells from draining lymph nodes [134]. These data argue for an immunosuppressive and beneficial effect of exercise in inflammatory neuropathies. In a recent study, treatment of EAN mice with PD-1 ligand (PD-L1) inhibited T cell proliferation and shifted T cell responses from a Th1/Th17 to a Th2/Treg phenotype [135]. PD-L1 may thus constitute a potential future treatment option.
Leukocyte trafficking across the blood-nerve barrier in GBS and CIDP
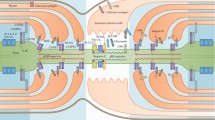
Trafficking of leukocytes across the blood-nerve barrier (BNB) is an important step in inflammatory neuropathies such as GBS and CIDP. Several different factors like selectins (E- and P-Selectin), chemokines and chemokine receptors, adhesion molecules, and integrins play a role in the process [136]. No animal model allows investigating trafficking of leukocytes at the BNB in real time in vivo. Therefore, different in vitro models using static and flow conditions have been established.
In vitro studies using primary endoneurial endothelial cells from human sciatic nerves showed that activation of endoneurial endothelial cells by the chemokines TNF-α and IFN-γ leads to induction of pro-inflammatory chemokines such as CCL2, CXCL9, CXCL11, and CCL20. The observed upregulation of CCL2 is thought to play a major role in mediating monocyte/macrophage trafficking through interaction with CCR2 in humans [137]. Studies on a mouse model of peripheral neuropathic pain support the importance of CCL2/CCR2 in monocyte/macrophage infiltration into peripheral nerves. Upregulation of CCL2 correlated with local monocyte/macrophage infiltration while sciatic intraneural microinjection of CCL2 lead to a recruitment of these immune cells [138]. Furthermore, in humans, CXCL2-3 and CXCL8 are thought to drive neutrophil trafficking through interaction with chemokine receptors CXCR1 and CXCR2. It was also demonstrated that the activation state of endothelial cells is more important determining cellular trafficking across the BNB than the activation state of leukocytes [137].
A multi-step paradigm for leukocyte trafficking at the BNB was proposed in parallel to leukocyte trafficking in other tissues. The initial step is the rolling of leukocytes on the endothelial monolayer surface, followed by arrest and firm adhesion at intercellular membranes and some transmigration via the paracellular route presumably at places with high chemokine presentation. Leukocytes extracted from AIDP patients showed an αM integrin-ICAM-1-dependent adherence to the BNB in vitro with CD14+ monocytes being the most prevalent subpopulation [137]. In fact, αM integrin (CD11b) was found in sural nerve biopsies of AIDP patients in large clusters of endoneurial CD11b+ leukocytes associated with demyelinating axons. These observations were paralleled in a murine EAN model with significant symptom amelioration using a monoclonal antibody against CD11b [139].
ICAM-1 blocking antibodies prevented or suppressed the development of EAN in the induction phase of disease rather than in the effector phase, indicating the importance of ICAM-1 in BNB trafficking [140, 141]. Other studies in murine EAN revealed that CCL2-CCR2 and CXCL10-CXCR3 have a direct effect on leukocyte transmigration to the peripheral nerve. Both receptors and ligands accumulate in the sciatic nerves of EAN mice [142]. Similar observations were made in sural nerve biopsies from GBS, AIDP and CIDP patients. Specific chemokine receptors are expressed by endoneurial macrophages (CCR1, CCR5) and by invading T cells (CCR2, CCR4, CXCR3) despite CCR5-deficient mice showing no difference in EAN severity [143,144,145]. Blocking of CXCR7 resulted in a significantly reduced EAN score, while blocking of CXCR4 lead to increased severity. This increase was accompanied by higher IFN-γ, IL-12, and TNF-α mRNA expression in regional lymph nodes and spleen as well as by increased serum levels of IFN-γ. The expression of ICAM-1 and VCAM-1 on vascular endothelial cells of the sciatic nerve was upregulated using CXCR4 blocking compounds which resulted in an increased infiltration of CD4+ T cells and macrophages to the sciatic nerve. Interestingly, blocking of both CXCR7 and CXCR4 lead to a significant disease suppression. These data suggest a functional hierarchy of these receptors [146].
Very Late Antigen-4 (VLA-4), a dimer of α4 and β1 integrin on leukocytes, plays an important role in pathogenic leukocyte trafficking in many chronic inflammatory diseases. A counterligand of α4 integrin is FNCS1 expressed by microvascular endothelial cells from CIDP patients in vitro and in situ. FNCS1-blocking antibodies inhibited leukocyte trafficking at the human BNB in vitro. Furthermore, these antibodies ameliorated the spontaneous neuritis in B7-2−/− NOD mice. FNCS1 is also expressed in microvessels in nerve biopsies of CIDP patients arguing for a potential role in leukocyte trafficking at the BNB in CIDP [147].
Targeting leukocyte trafficking across the BNB could be a promising therapeutic strategy in inflammatory neuropathies. Statins, for example, inhibit Rho-mediated transendothelial T cell migration and T cell activation and proliferation [148, 149]. In EAN, statins ameliorated clinical symptoms and nerve inflammation by decreasing the number of Th1 and Th17 cells in sciatic nerves and increasing the number of Tregs [116, 150]. In vitro experiments indicate that statins inhibit the TNF-α-mediated CCL2 release by endoneurial endothelial cells and reduce CD40 expression and thereby transendothelial migration [151,152,153,154].
Several studies using different statins showed that oral administration as well as multiple injections of statins are not optimal for efficacy. Therefore, a localized, controlled, and sustained release system based on statin-embedded nanoparticles was introduced [155]. Bilateral peri-neural administration of such a nanoparticle-based statin delivery system proofed to significantly attenuate EAN clinical severity and reduce peripheral nerve morphological and functional deficits [156]. Reducing migration of autoreactive leukocytes across the blood-nerve barrier using a peri-neural nanoparticle-based drug delivery platform might therefore be a future treatment option in localized nerve disorders.
To further decipher the mechanisms of leukocyte trafficking at the BNB, real-time in vivo models are needed. Using in vivo imaging techniques such as intravital microscopy or magnetic resonance neurography (MRN) (discussed in detail below) might help in observing the underlying mechanisms of peripheral nerve infiltration under physiological conditions.
MR imaging and neurography
Visualizing disease activity and immune cell infiltration and monitoring the effect of anti-inflammatory treatments require insight into the PNS. Electrophysiology is the mainstay of diagnosing and monitoring neuritis, but it has limited spatial resolution and often poorly correlates with clinical impairments especially in cases with a high level of axonal injury. Nerve biopsy is no option for monitoring treatment success as it is an invasive and one-time diagnostic procedure. MRI of peripheral nerves, also termed MRN, is therefore an interesting tool for repeatedly assessing PNS integrity and immune cell infiltration in inflammatory neuropathies.
MRN allows directly visualizing damage in the nervous system and pinpointing the exact localization of nerve lesions even in clinically unaffected nerves [157]. Therefore, this technique may help to improve therapy decisions of anti-inflammatory agents and targeting cellular trafficking (reviewed in [158, 159]). In a multiple sclerosis animal model, infiltration of cells labeled with iron particles has been monitored by iron oxide-enhanced MRI [160]. In an experimental setting, injection of labeled inflammatory cells thus allows for visualizing lesion formation and disease activity. Macrophage invasion in animals treated with Fingolimod has also been monitored with this technique as well as nerve repair [161, 162]. In adoptive transfer EAN, infiltration of macrophages into spinal nerves leading to myelin damage was detected with MRI and precedes clinical onset. Based on these findings, blocking invasion mechanisms of macrophages reduces clinical disease in an animal model of inflammatory neuropathies [163]. In the same model, using gadofluorine M-enhanced MRN, nerve injury and recovery was observed [164]. Beside injecting labeled cells, a specific uptake of intravenous injected iron oxide nanoparticles by innate immune cells as well as brain-resident cells (e.g., activated microglia, infiltrating macrophages, neutrophils) allows for studying disease activity in vivo without targeting adaptive immune cells [165].
Overall, MRN and imaging allow for directly observing lesion formation and if using certain contrast enhancing agents even the infiltration of cells. This technique can thus improve diagnosis and treatment monitoring and might help improving the effect of novel therapeutic strategies in inflammatory neuropathies.
Selected candidate therapeutic compounds
Plasma exchange and IVIg are effective in GBS while plasma exchange, IVIg, and glucocorticoids are the primary treatment options in CIDP and have been reviewed elsewhere [166, 167]. In addition to such established treatments, animal models of inflammatory neuropathies have enabled the preclinical testing of a multitude of potential novel therapeutic agents. Recently, several interesting new compounds have been introduced and may gain relevance in PNS autoimmunity.
Fingolimod
Fingolimod (FTY720) is a fungus-derived orally bioavailable immunomodulatory drug that therapeutically targets the egress of lymphoid cells from secondary lymphoid organs. Fingolimod binds to sphingosine-1-phosphate receptor (S1P) thereby sequestering lymphocytes in lymphoid organs [168]. Potential neuroprotective effects have additionally been suspected [169]. Fingolimod is an effective treatment in experimental autoimmune encephalomyelitis [170,171,172], the animal model of multiple sclerosis and has been approved for the treatment of relapsing-remitting multiple sclerosis based on successful randomized controlled trials [173, 174]. Fingolimod treatment also protects from EAN [175] and ameliorates the spontaneous chronic inflammatory neuropathy in B7-2-deficient NOD mice [104, 117]. Fingolimod constitutes a promising therapeutic candidate especially in chronic inflammatory neuropathies. Recent data indicate Fingolimod to induce repair by cultured adult Schwann cells. Therefore, Fingolimod might enhance regeneration of the PNS [176]. Furthermore, Fingolimod was shown to decrease the pro-inflammatory capabilities of antigen-presenting cells (APCs) and therefore reduce T cell activation, proliferation and differentiation [177]. A phase-III clinical trial, however, was aborted due to lack of a significant benefit of Fingolimod vs. placebo [178].
Rituximab/Ocrelizumab/Ofatumumab
Rituximab is a chimeric murine/human monoclonal antibody binding CD20, that is expressed on the surface of B cells of most developmental stages [179]. Rituximab is approved for the treatment of certain lymphomas and leukemia and is used off-label in some autoimmune disorders [180, 181]. Several reports describe efficacy of rituximab in CIDP [158], and B cell-directed treatment has been reported in the B7-2-deficient NOD animal model of CIDP [103]. Another anti-CD20 antibody is the humanized antibody Ocrelizumab (reviewed in [182]). A phase II, randomized, placebo-controlled multicentre trial indicated Ocrelizumab to be a promising therapeutic antibody for relapsing-remitting multiple sclerosis [183]. Three phase III clinical trials found Ocrelizumab to be efficacious in both relapsing-remitting multiple sclerosis (RRMS) and primary progressive multiple sclerosis (PPMS) [184, 185]. Clinical development of Ocrelizumab for the treatment of PPMS and RRMS is currently ongoing. Ofatumumab, a human monoclonal antibody binds to a distinct CD20 epitope. It is approved for chronic lymphocytic leukemia and tested for RRMS. A phase II trial indicated that Ofatumumab reduces the number of MRI lesions in multiple sclerosis [186]. A phase III trial in multiple sclerosis is being planned. Summarizing, the data suggest that treatments targeting CD20+ B cells are effective in multiple sclerosis and therefore also constitute interesting pharmaceutical candidates in inflammatory neuropathies.
Quinpramine
A novel group of chimeric compounds derived from the tricyclic antidepressant Imipramine and the malaria drug Quinacrine demonstrated antiprion effects [187], resulting from intracellular cholesterol redistribution [188]. Anti-inflammatory effects have been demonstrated in an animal model of human multiple sclerosis [189]. In EAN, Quinpramine ameliorated the clinical and histological severity with reduced immune cell infiltration and inflammatory myelin destruction in the PNS. In cell culture studies Quinpramine took effect on antigen-presenting cells by reducing their MHC class II surface expression and cell surface localization of cholesterol [190]. Thus, by redistributing cholesterol-rich membrane domains to intracellular compartments, Quinpramine may reduce cell surface availability of MHC class II and therefore inhibit autoimmune activation [187]. The anti-inflammatory compound Quinpramine may thus constitute a potential future therapeutic option in GBS.
Alemtuzumab
Alemtuzumab is a humanized monoclonal antibody against CD52 expressed on the surface of lymphocytes, monocytes, and macrophages. Application of this antibody leads to complement-dependent cell lysis, antibody-dependent cytotoxicity, and apoptosis which results in a 12-month-lasting lymphopenia. Alemtuzumab has been approved for treating multiple sclerosis and leukemia [191]. In IVIG-dependent CIDP, Alemtuzumab prolonged remission in young patients with short disease duration [192, 193]. Another study reported successful treatment of a 79-year-old patient with overlapping features of Miller Fisher syndrome and Bickerstaff brainstem encephalitis with Alemtuzumab [194]. Despite these promising results, potential autoimmune adverse effects such as autoimmune thyroiditis and other immunologic effects may limit the applicability of the drug [192, 195].
Etanercept
Etanercept is a dimeric, recombinant human tumor necrosis factor (TNF) receptor, fused to human IgG1 capable of binding TNF-α. It is used effectively as an antagonist of TNF-α in rheumatoid and psoriatic arthritis by reducing inflammation and pain [196]. In a study with 10 CIDP patients, three patients showed significant improvement, three other possible improvement, one complete resolution of weakness, one successfully discontinued prednisone, and one temporarily relapsed after treatment ended [197]. Etanercept as an anti-TNF-α drug may be a future treatment option for patients not responding to standard therapies. On the other hand, Etanercept treatment has been reported to trigger multiple sclerosis-like autoimmunity with demyelination of the central nervous system [198].
Complement inhibitors and modulators (e.g., Eculizumab)
Different lines of evidence support the role of complement in the pathogenesis of subtypes of human GBS. Complement deposition can be visualized on either Schwann cells or axonal membranes [199]. Anti-ganglioside antibodies can activate complement and cause complement-mediated disruption of ion channels and other nodal structures [200, 201]. Complement inhibitors and modulators ameliorate axonal injury in murine GBS models [48, 202]. Previous studies successfully demonstrated the principle of complement inhibition in GBS models through soluble complement receptors [203], complement depletion by cobra venom factor [204], and the complement inhibitor APT070 [205], although EAN severity was not reduced in complement component C6-deficient rats [206]. The monoclonal antibody Eculizumab for example is directed against human complement component C5 and is approved for treating paroxysmal nocturnal haemoglobinuria, in which attacks of complement-dependent haemolysis occur. This biological was effective in a murine axonal GBS model and appeared to be partially efficacious in a patient with severe GBS [48]. Therefore, Eculizumab is currently being investigated in patients with GBS in Japan (ClinicalTrials.gov Identifier: NCT02493725) and in a monocentric study in Scotland (ClinicalTrials.gov Identifier: NCT02029378). Therefore, complement-directed treatments constitute promising candidates for future GBS treatments and clinical trials are ongoing.
Conclusions
Our understanding of disease mechanisms in inflammatory neuropathies has improved in recent years. This is in part due to the introduction of novel animal models and development of new immunological concepts. In addition, new diagnostic techniques such as MRN and experimental new treatments are increasingly applied to patients who fail to respond to established therapies. Despite such progress, the following open questions remain to be addressed in the future.
Although several candidate genes conferring disease susceptibility to GBS have been reported, it remains a central question why some, but not other patients, develop GBS after exposure to a certain pathogen. Little is known about determinants of disease susceptibility in inflammatory neuropathies, and predicting or even influencing the risk of disease in GBS-prone patients would have high clinical relevance. Non-hypothesis-driven genetic susceptibility approaches—such as genome-wide association studies and TCR sequencing—could help to predict the risk of GBS and identify self-reactive T cell clones and antigens if sufficient patient numbers are collected despite the rareness of the disease [207].
Furthermore, molecular determinants of disease severity could add to established clinical predictors [1, 7]. It also remains to be addressed why PNS autoimmunity is monophasic in some but relapsing or chronic progressive in other patients. Animal models are genetically and immunologically far more homogenous than human patients and thus allow to address this aspect more specifically. If targeted inventions could “chronify” acute animal models (as described for CsA above), this could identify determinants of disease course in inflammatory neuropathies. As previously discussed in detail [8], converting experimental, animal-based science into clinical relevance appears as an insurmountable obstacle. Solving the bench-to-bedside transfer problem thus remains the most relevant challenge in inflammatory neuropathies.
Abbreviations
- AIDP:
-
Acute inflammatory demyelinating polyneuropathy
- AMAN:
-
Acute motor axonal neuropathy
- AMSAN:
-
Acute motor and sensory axonal neuropathy
- APC:
-
Antigen-presenting cell
- AT-EAN:
-
Adoptive transfer experimental allergic neuritis
- Breg:
-
Regulatory B cell
- CDR:
-
Primary structure and complementarity-determining region
- CFA:
-
Complete Freund’s adjuvant
- CIAP:
-
Chronic idiopathic axonal polyneuropathy
- CIDP:
-
Chronic inflammatory demyelinating polyradiculoneuropathy
- CNS:
-
Central nervous system
- CsA:
-
Cyclosporine A
- CSF:
-
Cerebrospinal fluid
- EAE:
-
Experimental autoimmune encephalomyelitis
- EAN:
-
Experimental allergic neuritis
- FNCS1:
-
Fibronectin connecting segment-1
- Fox:
-
Forkhead box
- GBS:
-
Guillain-Barré syndrome
- ICAM:
-
Intercellular adhesion molecule
- ICOS:
-
Inducible costimulator
- IdeS:
-
Immunoglobulin G-degrading enzyme of Streptococcus pyogenes
- IFN:
-
Interferon
- Ig:
-
Immunoglobulin
- IL:
-
Interleukin
- IPEX:
-
Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome
- IVIg:
-
Intravenous immunoglobulin
- MBP:
-
Myelin basic protein
- MFS:
-
Miller Fisher syndrome
- MHC:
-
Major histocompatibility complex
- MMN:
-
Multifocal motor neuropathy
- Mpz:
-
Myelin protein zero
- MRI:
-
Magnet resonance imaging
- MRN:
-
Magnet resonance neurography
- Nav:
-
Voltage-gated sodium channel
- NF:
-
Neurofascin
- NOD:
-
Non-obese diabetic
- PD:
-
Programmed death
- PDN:
-
Painful diabetic neuropathy
- PMP:
-
Peripheral myelin protein
- PNS:
-
Peripheral nervous system
- PPMS:
-
Primary progressive multiple sclerosis
- RRMS:
-
Relapsing-remitting multiple sclerosis
- S1P:
-
Sphingosine-1-phosphate
- TCR:
-
T cell receptor
- Teff:
-
Effector T cell
- Th:
-
T helper cell
- TNF:
-
Tumor necrosis factor
- Treg:
-
Regulatory T cell
- VLA-4:
-
Very Late Antigen
- VMD:
-
Vitelliform macular dystrophy
- ZIKV:
-
Zika virus
References
van Koningsveld R, Steyerberg EW, Hughes RA, Swan AV, van Doorn PA, Jacobs BC. A clinical prognostic scoring system for Guillain-Barre syndrome. Lancet Neurol. 2007;6:589–94.
Kuwabara S, Misawa S, Mori M, Tamura N, Kubota M, Hattori T. Long term prognosis of chronic inflammatory demyelinating polyneuropathy: a five year follow up of 38 cases. J Neurol Neurosurg Psychiatry. 2006;77:66–70.
van Doorn PA, Ruts L, Jacobs BC. Clinical features, pathogenesis, and treatment of Guillain-Barre syndrome. Lancet Neurol. 2008;7:939–50.
Griffin JW, Sheikh K. The Guillain-Barré Syndromes. In: Dyck PJ, Thomas PK, editors. Peripheral neuropathy, vol. 2. 4th ed. Philadelphia: Elsevier Saunders; 2005. p. 2197–220.
Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain. 2014;137:33–43.
Sejvar JJ, Baughman AL, Wise M, Morgan OW. Population incidence of Guillain-Barre syndrome: a systematic review and meta-analysis. Neuroepidemiology. 2011;36:123–33.
Walgaard C, Lingsma HF, Ruts L, van Doorn PA, Steyerberg EW, Jacobs BC. Early recognition of poor prognosis in Guillain-Barre syndrome. Neurology. 2011;76:968–75.
Meyer zu Horste G, Hartung HP, Kieseier BC. From bench to bedside--experimental rationale for immune-specific therapies in the inflamed peripheral nerve. Nat Clin Pract Neurol. 2007;3:198–211.
Dalakas MC. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat Rev Neurol. 2011;7:507–17.
Vallat JM, Sommer C, Magy L. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Lancet Neurol. 2010;9:402–12.
Koller H, Kieseier BC, Jander S, Hartung HP. Chronic inflammatory demyelinating polyneuropathy. N Engl J Med. 2005;352:1343–56.
Chio A, Cocito D, Bottacchi E, Buffa C, Leone M, Plano F, Mutani R, Calvo A, Parcidp. Idiopathic chronic inflammatory demyelinating polyneuropathy: an epidemiological study in Italy. J Neurol Neurosurg Psychiatry. 2007;78:1349–53.
Iijima M, Koike H, Hattori N, Tamakoshi A, Katsuno M, Tanaka F, Yamamoto M, Arimura K, Sobue G, Refractory Peripheral Neuropathy Study Group of J. Prevalence and incidence rates of chronic inflammatory demyelinating polyneuropathy in the Japanese population. J Neurol Neurosurg Psychiatry. 2008;79:1040–3.
Laughlin RS, Dyck PJ, Melton LJ 3rd, Leibson C, Ransom J, Dyck PJ. Incidence and prevalence of CIDP and the association of diabetes mellitus. Neurology. 2009;73:39–45.
Lunn MP, Manji H, Choudhary PP, Hughes RA, Thomas PK. Chronic inflammatory demyelinating polyradiculoneuropathy: a prevalence study in south east England. J Neurol Neurosurg Psychiatry. 1999;66:677–80.
Rajabally YA, Simpson BS, Beri S, Bankart J, Gosalakkal JA. Epidemiologic variability of chronic inflammatory demyelinating polyneuropathy with different diagnostic criteria: study of a UK population. Muscle Nerve. 2009;39:432–8.
Mahdi-Rogers M, Hughes RA. Epidemiology of chronic inflammatory neuropathies in southeast England. Eur J Neurol. 2014;21:28–33.
Dyck PJ, Lais AC, Ohta M, Bastron JA, Okazaki H, Groover RV. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc. 1975;50:621–37.
Prineas JW, McLeod JG. Chronic relapsing polyneuritis. J Neurol Sci. 1976;27:427–58.
Said G. Chronic inflammatory demyelinating polyneuropathy. Neuromuscul Disord. 2006;16:293–303.
Sommer C, Koch S, Lammens M, Gabreels-Festen A, Stoll G, Toyka KV. Macrophage clustering as a diagnostic marker in sural nerve biopsies of patients with CIDP. Neurology. 2005;65:1924–9.
Vital C, Vital A, Lagueny A, Ferrer X, Fontan D, Barat M, Gbikpi-Benissan G, Orgogozo JM, Henry P, Brechenmacher C, et al. Chronic inflammatory demyelinating polyneuropathy: immunopathological and ultrastructural study of peripheral nerve biopsy in 42 cases. Ultrastruct Pathol. 2000;24:363–9.
Hughes RA, Donofrio P, Bril V, Dalakas MC, Deng C, Hanna K, Hartung HP, Latov N, Merkies IS, van Doorn PA. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 2008;7:136–44.
van den Berg B, Walgaard C, Drenthen J, Fokke C, Jacobs BC, van Doorn PA. Guillain-Barre syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol. 2014;10:469–82.
Mathey EK, Park SB, Hughes RA, Pollard JD, Armati PJ, Barnett MH, Taylor BV, Dyck PJ, Kiernan MC, Lin CS. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry. 2015;86:973–85.
Hughes RA, Hadden RD, Gregson NA, Smith KJ. Pathogenesis of Guillain-Barre syndrome. J Neuroimmunol. 1999;100:74–97.
Yuki N. Ganglioside mimicry and peripheral nerve disease. Muscle Nerve. 2007;35:691–711.
Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005;366:1653–66.
Araujo LM, Ferreira ML, Nascimento OJ. Guillain-Barre syndrome associated with the Zika virus outbreak in Brazil. Arq Neuropsiquiatr. 2016;74:253–5.
de Oliveira WK, Carmo EH, Henriques CM, Coelho G, Vazquez E, Cortez-Escalante J, Molina J, Aldighieri S, Espinal MA, Dye C. Zika virus infection and associated neurologic disorders in Brazil. N Engl J Med. 2017;376:1591–3.
Song BH, Yun SI, Woolley M, Lee YM. Zika virus: history, epidemiology, transmission, and clinical presentation. J Neuroimmunol. 2017;308:50–64.
Krauer F, Riesen M, Reveiz L, Oladapo OT, Martinez-Vega R, Porgo TV, Haefliger A, Broutet NJ, Low N, Group WHOZCW. Zika virus infection as a cause of congenital brain abnormalities and Guillain-Barre syndrome: systematic review. PLoS Med. 2017;14:e1002203.
Cao-Lormeau VM, Blake A, Mons S, Lastere S, Roche C, Vanhomwegen J, Dub T, Baudouin L, Teissier A, Larre P, et al. Guillain-Barre syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet. 2016;387:1531–9.
da Silva IRF, Frontera JA, Bispo de Filippis AM, Nascimento O, Group R-G-ZR. Neurologic complications associated with the Zika virus in Brazilian adults. JAMA Neurol. 2017;74:1190–8.
Parra B, Lizarazo J, Jimenez-Arango JA, Zea-Vera AF, Gonzalez-Manrique G, Vargas J, Angarita JA, Zuniga G, Lopez-Gonzalez R, Beltran CL, et al. Guillain-Barre syndrome associated with Zika virus infection in Colombia. N Engl J Med. 2016;375:1513–23.
Lucchese G, Kanduc D. Zika virus and autoimmunity: from microcephaly to Guillain-Barre syndrome, and beyond. Autoimmun Rev. 2016;15:801–8.
Ritter JM, Martines RB, Zaki SR. Zika virus: pathology from the pandemic. Arch Pathol Lab Med. 2017;141:49–59.
Illa I, Ortiz N, Gallard E, Juarez C, Grau JM, Dalakas MC. Acute axonal Guillain-Barre syndrome with IgG antibodies against motor axons following parenteral gangliosides. Ann Neurol. 1995;38:218–24.
Morikawa M, Kuwahara M, Ueno R, Samukawa M, Hamada Y, Kusunoki S. Serological study using glycoarray for detecting antibodies to glycolipids and glycolipid complexes in immune-mediated neuropathies. J Neuroimmunol. 2016;301:35–40.
Rink C, Gortzen A, Veh RW, Pruss H. Serum antibodies targeting neurons of the monoaminergic systems in Guillain-Barre syndrome. J Neurol Sci. 2017;372:318–23.
Kusunoki S, Shimizu J, Chiba A, Ugawa Y, Hitoshi S, Kanazawa I. Experimental sensory neuropathy induced by sensitization with ganglioside GD1b. Ann Neurol. 1996;39:424–31.
Moyano AL, Comin R, Lardone RD, Alaniz ME, Theaux R, Irazoqui FJ, Nores GA. Validation of a rabbit model of neuropathy induced by immunization with gangliosides. J Neurol Sci. 2008;272:110–4.
Yuki N, Yamada M, Koga M, Odaka M, Susuki K, Tagawa Y, Ueda S, Kasama T, Ohnishi A, Hayashi S, et al. Animal model of axonal Guillain-Barre syndrome induced by sensitization with GM1 ganglioside. Ann Neurol. 2001;49:712–20.
Susuki K, Yuki N, Schafer DP, Hirata K, Zhang G, Funakoshi K, Rasband MN. Dysfunction of nodes of Ranvier: a mechanism for anti-ganglioside antibody-mediated neuropathies. Exp Neurol. 2012;233:534–42.
Uncini A, Susuki K, Yuki N. Nodo-paranodopathy: beyond the demyelinating and axonal classification in anti-ganglioside antibody-mediated neuropathies. Clin Neurophysiol. 2013;124:1928–34.
Bowes T, Wagner ER, Boffey J, Nicholl D, Cochrane L, Benboubetra M, Conner J, Furukawa K, Furukawa K, Willison HJ. Tolerance to self gangliosides is the major factor restricting the antibody response to lipopolysaccharide core oligosaccharides in campylobacter jejuni strains associated with Guillain-Barre syndrome. Infect Immun. 2002;70:5008–18.
Lunn MP, Johnson LA, Fromholt SE, Itonori S, Huang J, Vyas AA, Hildreth JE, Griffin JW, Schnaar RL, Sheikh KA. High-affinity anti-ganglioside IgG antibodies raised in complex ganglioside knockout mice: reexamination of GD1a immunolocalization. J Neurochem. 2000;75:404–12.
Halstead SK, Zitman FM, Humphreys PD, Greenshields K, Verschuuren JJ, Jacobs BC, Rother RP, Plomp JJ, Willison HJ. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain. 2008;131:1197–208.
Hadden RD, Gregson NA, Gold R, Willison HJ, Hughes RA. Guillain-Barre syndrome serum and anti-campylobacter antibody do not exacerbate experimental autoimmune neuritis. J Neuroimmunol. 2001;119:306–16.
Gabriel CM, Gregson NA, Hughes RA. Anti-PMP22 antibodies in patients with inflammatory neuropathy. J Neuroimmunol. 2000;104:139–46.
Csurhes PA, Sullivan AA, Green K, Pender MP, McCombe PA. T cell reactivity to P0, P2, PMP-22, and myelin basic protein in patients with Guillain-Barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry. 2005;76:1431–9.
Makowska A, Pritchard J, Sanvito L, Gregson N, Peakman M, Hayday A, Hughes R. Immune responses to myelin proteins in Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2008;79:664–71.
Inglis HR, Csurhes PA, McCombe PA. Antibody responses to peptides of peripheral nerve myelin proteins P0 and P2 in patients with inflammatory demyelinating neuropathy. J Neurol Neurosurg Psychiatry. 2007;78:419–22.
Samukawa M, Kuwahara M, Morikawa M, Ueno R, Hamada Y, Takada K, Hirano M, Mitsui Y, Sonoo M, Kusunoki S. Electrophysiological assessment of Guillain-Barre syndrome with both Gal-C and ganglioside antibodies; tendency for demyelinating type. J Neuroimmunol. 2016;301:61–4.
Kharwar NK, Prasad KN, Singh K, Paliwal VK, Modi DR. Polymorphisms of IL-17 and ICAM-1 and their expression in Guillain-Barre syndrome. Int J Neurosci. 2017;127:680–87.
Meyer zu Horste G, Mausberg AK, Cordes S, El-Haddad H, Partke HJ, Leussink VI, Roden M, Martin S, Steinman L, Hartung HP, Kieseier BC. Thymic epithelium determines a spontaneous chronic neuritis in Icam1(tm1Jcgr)NOD mice. J Immunol. 2014;193:2678–90.
Meyer zu Horste G, Cordes S, Pfaff J, Mathys C, Mausberg AK, Bendszus M, Pham M, Hartung HP, Kieseier BC. Predicting the response to intravenous Immunoglobulins in an animal model of chronic neuritis. PLoS One. 2016;11:e0164099.
Zephir H, Stojkovic T, Latour P, Lacour A, de Seze J, Outteryck O, Maurage CA, Monpeurt C, Chatelet P, Ovelacq E, Vermersch P. Relapsing demyelinating disease affecting both the central and peripheral nervous systems. J Neurol Neurosurg Psychiatry. 2008;79:1032–9.
Khalili-Shirazi A, Atkinson P, Gregson N, Hughes RA. Antibody responses to P0 and P2 myelin proteins in Guillain-Barre syndrome and chronic idiopathic demyelinating polyradiculoneuropathy. J Neuroimmunol. 1993;46:245–51.
Sanvito L, Makowska A, Mahdi-Rogers M, Hadden RD, Peakman M, Gregson N, Nemni R, Hughes RA. Humoral and cellular immune responses to myelin protein peptides in chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry. 2009;80:333–8.
Kwa MS, van Schaik IN, Brand A, Baas F, Vermeulen M. Investigation of serum response to PMP22, connexin 32 and P(0) in inflammatory neuropathies. J Neuroimmunol. 2001;116:220–5.
Kuwahara M, Suzuki S, Takada K, Kusunoki S. Antibodies to LM1 and LM1-containing ganglioside complexes in Guillain-Barre syndrome and chronic inflammatory demyelinating polyneuropathy. J Neuroimmunol. 2011;239:87–90.
Pollard JD, Armati PJ. CIDP - the relevance of recent advances in Schwann cell/axonal neurobiology. J Peripher Nerv Syst. 2011;16:15–23.
Querol L, Nogales-Gadea G, Rojas-Garcia R, Diaz-Manera J, Pardo J, Ortega-Moreno A, Sedano MJ, Gallardo E, Berciano J, Blesa R, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology. 2014;82:879–86.
Devaux JJ, Miura Y, Fukami Y, Inoue T, Manso C, Belghazi M, Sekiguchi K, Kokubun N, Ichikawa H, Wong AH, Yuki N. Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology. 2016;86:800–7.
Delmont E, Manso C, Querol L, Cortese A, Berardinelli A, Lozza A, Belghazi M, Malissart P, Labauge P, Taieb G, et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain. 2017;140:1851–58.
Yan W, Nguyen T, Yuki N, Ji Q, Yiannikas C, Pollard JD, Mathey EK. Antibodies to neurofascin exacerbate adoptive transfer experimental autoimmune neuritis. J Neuroimmunol. 2014;277:13–7.
Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR, Al-Hayani A, Davies SN, Rasband MN, Olsson T, et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med. 2007;204:2363–72.
Doppler K, Appeltshauser L, Wilhelmi K, Villmann C, Dib-Hajj SD, Waxman SG, Maurer M, Weishaupt A, Sommer C. Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J Neurol Neurosurg Psychiatry. 2015;86:720–8.
Manso C, Querol L, Mekaouche M, Illa I, Devaux JJ. Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain. 2016;139:1700–12.
Joshi AR, Holtmann L, Bobylev I, Schneider C, Ritter C, Weis J, Lehmann HC. Loss of Schwann cell plasticity in chronic inflammatory demyelinating polyneuropathy (CIDP). J Neuroinflammation. 2016;13:255.
Lachance DH, Lennon VA, Pittock SJ, Tracy JA, Krecke KN, Amrami KK, Poeschla EM, Orenstein R, Scheithauer BW, Sejvar JJ, et al. An outbreak of neurological autoimmunity with polyradiculoneuropathy in workers exposed to aerosolised porcine neural tissue: a descriptive study. Lancet Neurol. 2010;9:55–66.
St-Amour I, Pare I, Tremblay C, Coulombe K, Bazin R, Calon F. IVIg protects the 3xTg-AD mouse model of Alzheimer’s disease from memory deficit and Abeta pathology. J Neuroinflammation. 2014;11:54.
Schneider-Hohendorf T, Schwab N, Uceyler N, Gobel K, Sommer C, Wiendl H. CD8+ T-cell immunity in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2012;78:402–8.
Yang M, Rainone A, Shi XQ, Fournier S, Zhang J. A new animal model of spontaneous autoimmune peripheral polyneuropathy: implications for Guillain-Barre syndrome. Acta Neuropathol Commun. 2014;2:5.
Waksman BH, Adams RD. Allergic neuritis: an experimental disease of rabbits induced by the injection of peripheral nervous tissue and adjuvants. J Exp Med. 1955;102:213–36.
Patzig J, Jahn O, Tenzer S, Wichert SP, de Monasterio-Schrader P, Rosfa S, Kuharev J, Yan K, Bormuth I, Bremer J, et al. Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J Neurosci. 2011;31:16369–86.
Gabriel CM, Hughes RA, Moore SE, Smith KJ, Walsh FS. Induction of experimental autoimmune neuritis with peripheral myelin protein-22. Brain. 1998;121(Pt 10):1895–902.
Weerth S, Berger T, Lassmann H, Linington C. Encephalitogenic and neuritogenic T cell responses to the myelin-associated glycoprotein (MAG) in the Lewis rat. J Neuroimmunol. 1999;95:157–64.
Gold R, Stoll G, Kieseier BC, Hartung HP, Toyka KV. Experimental Autoimmune Neuritis. In: Dyck PJ, Thomas PK, editors. Peripheral neuropathy, vol. 2. 4th ed. Philadelphia: Elsevier Saunders; 2005.
Hartung HP, Schäfer B, Heininger K, Stoll G, Toyka KV. The role of macrophages and eicosanoids in the pathogenesis of experimental allergic neuritis. Serial clinical, electrophysiological, biochemical and morphological observations. Brain. 1988;111:1039–59.
Prineas JW. Acute idiopathic polyneuritis. An electron microscope study. Lab Investig. 1972;26:133–47.
St Charles JL, Bell JA, Gadsden BJ, Malik A, Cooke H, Van de Grift LK, Kim HY, Smith EJ, Mansfield LS: Guillain Barre syndrome is induced in non-obese diabetic (NOD) mice following campylobacter jejuni infection and is exacerbated by antibiotics. J Autoimmun. 2016;77:11–38.
Wang Y, Shi Q, Lv H, Hu M, Wang W, Wang Q, Qiao B, Zhang G, Lv Z, Kjellman C, et al. IgG-degrading enzyme of Streptococcus pyogenes (IdeS) prevents disease progression and facilitates improvement in a rabbit model of Guillain-Barre syndrome. Exp Neurol. 2017;291:134–40.
Jung S, Gaupp S, Korn T, Kollner G, Hartung HP, Toyka KV. Biphasic form of experimental autoimmune neuritis in dark agouti rats and its oral therapy by antigen-specific tolerization. J Neurosci Res. 2004;75:524–35.
McCombe PA, Harness J, Pender MP. Effects of cyclosporin A treatment on clinical course and inflammatory cell apoptosis in experimental autoimmune encephalomyelitis induced in Lewis rats by inoculation with myelin basic protein. J Neuroimmunol. 1999;97:60–9.
McCombe PA, van der Kreek SA, Pender MP. Neuropathological findings in chronic relapsing experimental allergic neuritis induced in the Lewis rat by inoculation with intradural root myelin and treatment with low dose cyclosporin A. Neuropathol Appl Neurobiol. 1992;18:171–87.
Harness J, Pender MP, McCombe PA. Cyclosporin a treatment modulates cytokine mRNA expression by inflammatory cells extracted from the spinal cord of rats with experimental autoimmune encephalomyelitis induced by inoculation with myelin basic protein. J Neurol Sci. 2001;187:7–16.
Granelli-Piperno A, Keane M, Steinman RM. Evidence that cyclosporine inhibits cell-mediated immunity primarily at the level of the T lymphocyte rather than the accessory cell. Transplantation. 1988;46:53S–60S.
Saitoh K, Kon S, Nakatsuru T, Inui K, Ihara T, Matsumoto N, Kitai Y, Muromoto R, Matsuda T. Anti-IL-17A blocking antibody reduces cyclosporin A-induced relapse in experimental autoimmune encephalomyelitis mice. Biochem Biophys Rep. 2016;8:139–45.
Kang HG, Zhang D, Degauque N, Mariat C, Alexopoulos S, Zheng XX. Effects of cyclosporine on transplant tolerance: the role of IL-2. Am J Transplant. 2007;7:1907–16.
Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169:4850–60.
Miroux C, Morales O, Ghazal K, Othman SB, de Launoit Y, Pancre V, Conti F, Delhem N. In vitro effects of cyclosporine A and tacrolimus on regulatory T-cell proliferation and function. Transplantation. 2012;94:123–31.
Lassmann H, Fierz W, Neuchrist C, Meyermann R. Chronic relapsing experimental allergic neuritis induced by repeated transfer of P2-protein reactive T cell lines. Brain. 1991;114(Pt 1B):429–42.
Zehntner SP, Brisebois M, Tran E, Owens T, Fournier S. Constitutive expression of a costimulatory ligand on antigen-presenting cells in the nervous system drives demyelinating disease. FASEB J. 2003;17:1910–2.
Brisebois M, Zehntner SP, Estrada J, Owens T, Fournier S. A pathogenic role for CD8+ T cells in a spontaneous model of demyelinating disease. J Immunol. 2006;177:2403–11.
Dace DS, Khan AA, Stark JL, Kelly J, Cross AH, Apte RS. Interleukin-10 overexpression promotes Fas-ligand-dependent chronic macrophage-mediated demyelinating polyneuropathy. PLoS One. 2009;4:e7121.
Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–85.
Salomon B, Rhee L, Bour-Jordan H, Hsin H, Montag A, Soliven B, Arcella J, Girvin AM, Padilla J, Miller SD, Bluestone JA. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-deficient NOD mice. J Exp Med. 2001;194:677–84.
Bour-Jordan H, Thompson HL, Bluestone JA. Distinct effector mechanisms in the development of autoimmune neuropathy versus diabetes in nonobese diabetic mice. J Immunol. 2005;175:5649–55.
Kim HJ, Jung CG, Jensen MA, Dukala D, Soliven B. Targeting of myelin protein zero in a spontaneous autoimmune polyneuropathy. J Immunol. 2008;181:8753–60.
Louvet C, Kabre BG, Davini DW, Martinier N, Su MA, DeVoss JJ, Rosenthal WL, Anderson MS, Bour-Jordan H, Bluestone JA. A novel myelin P0-specific T cell receptor transgenic mouse develops a fulminant autoimmune peripheral neuropathy. J Exp Med. 2009;206:507–14.
Abraham PM, Quan SH, Dukala D, Soliven B. CD19 as a therapeutic target in a spontaneous autoimmune polyneuropathy. Clin Exp Immunol. 2014;175:181–91.
Kim HJ, Jung CG, Dukala D, Bae H, Kakazu R, Wollmann R, Soliven B. Fingolimod and related compounds in a spontaneous autoimmune polyneuropathy. J Neuroimmunol. 2009;214:93–100.
Quan S, Sheng JR, Abraham PM, Soliven B. Regulatory T and B lymphocytes in a spontaneous autoimmune polyneuropathy. Clin Exp Immunol. 2016;184:50–61.
Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–35.
Meyer zu Horste G, Cordes S, Mausberg AK, Zozulya AL, Wessig C, Sparwasser T, Mathys C, Wiendl H, Hartung HP, Kieseier BC. FoxP3+ regulatory T cells determine disease severity in rodent models of inflammatory neuropathies. PLoS One. 2014;e108756:9.
Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A. 2005;102:11823–8.
Jiang F, Yoshida T, Nakaki F, Terawaki S, Chikuma S, Kato Y, Okazaki IM, Honjo T, Okazaki T. Identification of QTLs that modify peripheral neuropathy in NOD.H2b-Pdcd1−/− mice. Int Immunol. 2009;21:499–509.
Yoshida T, Jiang F, Honjo T, Okazaki T. PD-1 deficiency reveals various tissue-specific autoimmunity by H-2b and dose-dependent requirement of H-2g7 for diabetes in NOD mice. Proc Natl Acad Sci U S A. 2008;105:3533–8.
Prevot N, Briet C, Lassmann H, Tardivel I, Roy E, Morin J, Mak TW, Tafuri A, Boitard C. Abrogation of ICOS/ICOS ligand costimulation in NOD mice results in autoimmune deviation toward the neuromuscular system. Eur J Immunol. 2010;40:2267–76.
Zeng XL, Nagavalli A, Smith CJ, Howard JF, Su MA. Divergent effects of T cell costimulation and inflammatory cytokine production on autoimmune peripheral neuropathy provoked by Aire deficiency. J Immunol. 2013;190:3895–904.
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517.
Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol. 2016;34:335–68.
Wang X, Zheng XY, Ma C, Wang XK, Wu J, Adem A, Zhu J, Zhang HL. Mitigated Tregs and augmented Th17 cells and cytokines are associated with severity of experimental autoimmune neuritis. Scand J Immunol. 2014;80:180–90.
Li XL, Dou YC, Liu Y, Shi CW, Cao LL, Zhang XQ, Zhu J, Duan RS. Atorvastatin ameliorates experimental autoimmune neuritis by decreased Th1/Th17 cytokines and up-regulated T regulatory cells. Cell Immunol. 2011;271:455–61.
Zhang Z, Zhang ZY, Fauser U, Schluesener HJ. FTY720 ameliorates experimental autoimmune neuritis by inhibition of lymphocyte and monocyte infiltration into peripheral nerves. Exp Neurol. 2008;210:681–90.
Chi LJ, Xu WH, Zhang ZW, Huang HT, Zhang LM, Zhou J. Distribution of Th17 cells and Th1 cells in peripheral blood and cerebrospinal fluid in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2010;15:345–56.
Mei FJ, Ishizu T, Murai H, Osoegawa M, Minohara M, Zhang KN, Kira J. Th1 shift in CIDP versus Th2 shift in vasculitic neuropathy in CSF. J Neurol Sci. 2005;228:75–85.
Madia F, Frisullo G, Nociti V, Conte A, Luigetti M, Del Grande A, Patanella AK, Iorio R, Tonali PA, Batocchi AP, Sabatelli M. pSTAT1, pSTAT3, and T-bet as markers of disease activity in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2009;14:107–17.
Li S, Yu M, Li H, Zhang H, Jiang Y. IL-17 and IL-22 in cerebrospinal fluid and plasma are elevated in Guillain-Barre syndrome. Mediat Inflamm. 2012;2012:260473.
Li S, Jin T, Zhang HL, Yu H, Meng F, Concha Quezada H, Zhu J. Circulating Th17, Th22, and Th1 cells are elevated in the Guillain-Barre syndrome and downregulated by IVIg treatments. Mediat Inflamm. 2014;2014:740947.
Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, Chow J, Manel N, Ciofani M, Kim SV, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORgammat activity. Nature. 2011;472:486–90.
Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, Istrate MA, Kamenecka TM, Roush WR, Vidovic D, et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature. 2011;472:491–4.
Nie J, Li YY, Zheng SG, Tsun A, Li B. FOXP3(+) Treg cells and gender bias in autoimmune diseases. Front Immunol. 2015;6:493.
Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20.
Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849–59.
Chi LJ, Wang HB, Wang WZ. Impairment of circulating CD4+CD25+ regulatory T cells in patients with chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2008;13:54–63.
Harness J, McCombe PA. Increased levels of activated T-cells and reduced levels of CD4/CD25+ cells in peripheral blood of Guillain-Barre syndrome patients compared to controls. J Clin Neurosci. 2008;15:1031–5.
Zhang Z, Zhang ZY, Fauser U, Schluesener HJ. Distribution of Foxp3(+) T-regulatory cells in experimental autoimmune neuritis rats. Exp Neurol. 2009;216:75–82.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6.
Maddur MS, Trinath J, Rabin M, Bolgert F, Guy M, Vallat JM, Magy L, Balaji KN, Kaveri SV, Bayry J. Intravenous immunoglobulin-mediated expansion of regulatory T cells in autoimmune patients is associated with increased prostaglandin E2 levels in the circulation. Cell Mol Immunol. 2015;12:650–2.
Fantini MC, Dominitzki S, Rizzo A, Neurath MF, Becker C. In vitro generation of CD4+ CD25+ regulatory cells from murine naive T cells. Nat Protoc. 2007;2:1789–94.
Calik MW, Shankarappa SA, Langert KA, Stubbs EB Jr. Forced exercise preconditioning attenuates experimental autoimmune neuritis by altering Th1 lymphocyte composition and egress. ASN Neuro. 2015;7.
Ding Y, Han R, Jiang W, Xiao J, Liu H, Chen X, Li X, Hao J. Programmed death Ligand 1 plays a Neuroprotective role in experimental autoimmune neuritis by controlling peripheral nervous system inflammation of rats. J Immunol. 2016;197:3831–40.
Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–89.
Yosef N, Ubogu EE. alpha(M)beta(2)-integrin-intercellular adhesion molecule-1 interactions drive the flow-dependent trafficking of Guillain-Barre syndrome patient derived mononuclear leukocytes at the blood-nerve barrier in vitro. J Cell Physiol. 2012;227:3857–75.
Van Steenwinckel J, Auvynet C, Sapienza A, Reaux-Le Goazigo A, Combadiere C, Melik Parsadaniantz S. Stromal cell-derived CCL2 drives neuropathic pain states through myeloid cell infiltration in injured nerve. Brain Behav Immun. 2015;45:198–210.
Dong C, Palladino SP, Helton ES, Ubogu EE. The pathogenic relevance of alphaM-integrin in Guillain-Barre syndrome. Acta Neuropathol. 2016;132:739–52.
Archelos JJ, Maurer M, Jung S, Miyasaka M, Tamatani T, Toyka KV, Hartung HP. Inhibition of experimental autoimmune neuritis by an antibody to the lymphocyte function-associated antigen-1. Lab Investig. 1994;70:667–75.
Archelos JJ, Maurer M, Jung S, Toyka KV, Hartung HP. Suppression of experimental allergic neuritis by an antibody to the intracellular adhesion molecule ICAM-1. Brain. 1993;116(Pt 5):1043–58.
Xia RH, Yosef N, Ubogu EE. Selective expression and cellular localization of pro-inflammatory chemokine ligand/receptor pairs in the sciatic nerves of a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. Neuropathol Appl Neurobiol. 2010;36:388–98.
Kieseier BC, Tani M, Mahad D, Oka N, Ho T, Woodroofe N, Griffin JW, Toyka KV, Ransohoff RM, Hartung HP. Chemokines and chemokine receptors in inflammatory demyelinating neuropathies: a central role for IP-10. Brain. 2002;125:823–34.
Orlikowski D, Chazaud B, Plonquet A, Poron F, Sharshar T, Maison P, Raphael JC, Gherardi RK, Creange A. Monocyte chemoattractant protein 1 and chemokine receptor CCR2 productions in Guillain-Barre syndrome and experimental autoimmune neuritis. J Neuroimmunol. 2003;134:118–27.
Duan RS, Chen Z, Bao L, Quezada HC, Nennesmo I, Winblad B, Zhu J. CCR5 deficiency does not prevent P0 peptide 180-199 immunized mice from experimental autoimmune neuritis. Neurobiol Dis. 2004;16:630–7.
Brunn A, Utermohlen O, Mihelcic M, Sanchez-Ruiz M, Carstov M, Blau T, Ustinova I, Penfold M, Montesinos-Rongen M, Deckert M. Differential effects of CXCR4-CXCL12- and CXCR7-CXCL12-mediated immune reactions on murine P0106-125 -induced experimental autoimmune neuritis. Neuropathol Appl Neurobiol. 2013;39:772–87.
Dong C, Greathouse KM, Beacham RL, Palladino SP, Helton ES, Ubogu EE. Fibronectin connecting segment-1 peptide inhibits pathogenic leukocyte trafficking and inflammatory demyelination in experimental models of chronic inflammatory demyelinating polyradiculoneuropathy. Exp Neurol. 2017;292:35–45.
Greenwood J, Walters CE, Pryce G, Kanuga N, Beraud E, Baker D, Adamson P. Lovastatin inhibits brain endothelial cell rho-mediated lymphocyte migration and attenuates experimental autoimmune encephalomyelitis. FASEB J. 2003;17:905–7.
Danesh FR, Anel RL, Zeng L, Lomasney J, Sahai A, Kanwar YS. Immunomodulatory effects of HMG-CoA reductase inhibitors. Arch Immunol Ther Exp. 2003;51:139–48.
Sarkey JP, Richards MP, Stubbs EB Jr. Lovastatin attenuates nerve injury in an animal model of Guillain-Barre syndrome. J Neurochem. 2007;100:1265–77.
Langert KA, Pervan CL, Stubbs EB Jr. Novel role of Cdc42 and RalA GTPases in TNF-alpha mediated secretion of CCL2. Small GTPases. 2014;5.
Takeuchi S, Kawashima S, Rikitake Y, Ueyama T, Inoue N, Hirata K, Yokoyama M. Cerivastatin suppresses lipopolysaccharide-induced ICAM-1 expression through inhibition of rho GTPase in BAEC. Biochem Biophys Res Commun. 2000;269:97–102.
Lin R, Liu J, Peng N, Gan W, Wang W, Han C, Ding C. Lovastatin reduces apoptosis and downregulates the CD40 expression induced by TNF-alpha in cerebral vascular endothelial cells. Curr Neurovasc Res. 2006;3:41–7.
Stach K, Nguyen XD, Lang S, Elmas E, Weiss C, Borggrefe M, Fischer J, Kalsch T. Simvastatin and atorvastatin attenuate VCAM-1 and uPAR expression on human endothelial cells and platelet surface expression of CD40 ligand. Cardiol J. 2012;19:20–8.
Ho MH, Chiang CP, Liu YF, Kuo MY, Lin SK, Lai JY, Lee BS. Highly efficient release of lovastatin from poly(lactic-co-glycolic acid) nanoparticles enhances bone repair in rats. J Orthop Res. 2011;29:1504–10.
Langert KA, Goshu B, Stubbs EB Jr. Attenuation of experimental autoimmune neuritis with locally administered lovastatin-encapsulating poly(lactic-co-glycolic) acid nanoparticles. J Neurochem. 2017;140:334–46.
Baumer P, Weiler M, Ruetters M, Staub F, Dombert T, Heiland S, Bendszus M, Pham M. MR neurography in ulnar neuropathy as surrogate parameter for the presence of disseminated neuropathy. PLoS One. 2012;7:e49742.
Kollmer J, Bendszus M, Pham M. MR Neurography: diagnostic imaging in the PNS. Clin Neuroradiol. 2015;25(Suppl 2):283–9.
Stoll G, Bendszus M. Imaging of inflammation in the peripheral and central nervous system by magnetic resonance imaging. Neuroscience. 2009;158:1151–60.
Anderson SA, Shukaliak-Quandt J, Jordan EK, Arbab AS, Martin R, McFarland H, Frank JA. Magnetic resonance imaging of labeled T-cells in a mouse model of multiple sclerosis. Ann Neurol. 2004;55:654–9.
Rausch M, Hiestand P, Foster CA, Baumann DR, Cannet C, Rudin M. Predictability of FTY720 efficacy in experimental autoimmune encephalomyelitis by in vivo macrophage tracking: clinical implications for ultrasmall superparamagnetic iron oxide-enhanced magnetic resonance imaging. J Magn Reson Imaging. 2004;20:16–24.
Chen P, Piao X, Bonaldo P. Role of macrophages in Wallerian degeneration and axonal regeneration after peripheral nerve injury. Acta Neuropathol. 2015;130:605–18.
Stoll G, Wesemeier C, Gold R, Solymosi L, Toyka KV, Bendszus M. In vivo monitoring of macrophage infiltration in experimental autoimmune neuritis by magnetic resonance imaging. J Neuroimmunol. 2004;149:142–6.
Stoll G, Wessig C, Gold R, Bendszus M. Assessment of lesion evolution in experimental autoimmune neuritis by gadofluorine M-enhanced MR neurography. Exp Neurol. 2006;197:150–6.
Kirschbaum K, Sonner JK, Zeller MW, Deumelandt K, Bode J, Sharma R, Kruwel T, Fischer M, Hoffmann A, Costa da Silva M, et al. In vivo nanoparticle imaging of innate immune cells can serve as a marker of disease severity in a model of multiple sclerosis. Proc Natl Acad Sci U S A. 2016;113:13227–32.
Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barre syndrome. Lancet. 2016;388:717–27.
Kleyman I, Brannagan TH 3rd. Treatment of chronic inflammatory demyelinating polyneuropathy. Curr Neurol Neurosci Rep. 2015;15:47.
Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–9.
Miron VE, Jung CG, Kim HJ, Kennedy TE, Soliven B, Antel JP. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol. 2008;63:61–71.
Balatoni B, Storch MK, Swoboda EM, Schonborn V, Koziel A, Lambrou GN, Hiestand PC, Weissert R, Foster CA. FTY720 sustains and restores neuronal function in the DA rat model of MOG-induced experimental autoimmune encephalomyelitis. Brain Res Bull. 2007;74:307–16.
Fujino M, Funeshima N, Kitazawa Y, Kimura H, Amemiya H, Suzuki S, Li XK. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305:70–7.
Webb M, Tham CS, Lin FF, Lariosa-Willingham K, Yu N, Hale J, Mandala S, Chun J, Rao TS. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J Neuroimmunol. 2004;153:108–21.
Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P, Selmaj K, Agoropoulou C, Leyk M, Zhang-Auberson L, Burtin P. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401.
Khatri BO. Fingolimod in the treatment of relapsing-remitting multiple sclerosis: long-term experience and an update on the clinical evidence. Ther Adv Neurol Disord. 2016;9:130–47.
Ambrosius B, Pitarokoili K, Schrewe L, Pedreiturria X, Motte J, Gold R. Fingolimod attenuates experimental autoimmune neuritis and contributes to Schwann cell-mediated axonal protection. J Neuroinflammation. 2017;14:92.
Heinen A, Beyer F, Tzekova N, Hartung HP, Kury P. Fingolimod induces the transition to a nerve regeneration promoting Schwann cell phenotype. Exp Neurol. 2015;271:25–35.
Thomas K, Sehr T, Proschmann U, Rodriguez-Leal FA, Haase R, Ziemssen T. Fingolimod additionally acts as immunomodulator focused on the innate immune system beyond its prominent effects on lymphocyte recirculation. J Neuroinflammation. 2017;14:41.
Hartung H-P, Dalakas M, Merkies I, Latov N, Léger J-M, Nobile-Orazio E, Sobue G, Genge A, Merschhemke MB, Marie EC, et al. Oral Fingolimod in chronic inflammatory Demyelinating Polyradiculoneuropathy (FORCIDP): results from a phase III randomized placebo-controlled trial (S27.002). Neurology. 2017;88.
Binder M, Otto F, Mertelsmann R, Veelken H, Trepel M. The epitope recognized by rituximab. Blood. 2006;108:1975–8.
Barun B, Bar-Or A. Treatment of multiple sclerosis with anti-CD20 antibodies. Clin Immunol. 2011;142:31–37.
Vidal L, Gafter-Gvili A, Salles G, Dreyling MH, Ghielmini M, Hsu Schmitz SF, Pettengell R, Witzens-Harig M, Shpilberg O. Rituximab maintenance for the treatment of patients with follicular lymphoma: an updated systematic review and meta-analysis of randomized trials. J Natl Cancer Inst. 2011;103:1799–806.
Sorensen PS, Blinkenberg M. The potential role for ocrelizumab in the treatment of multiple sclerosis: current evidence and future prospects. Ther Adv Neurol Disord. 2016;9:44–52.
Kappos L, Li D, Calabresi PA, O’Connor P, Bar-Or A, Barkhof F, Yin M, Leppert D, Glanzman R, Tinbergen J, Hauser SL. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–87.
Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung HP, Hemmer B, Lublin F, Montalban X, Rammohan KW, Selmaj K, et al. Ocrelizumab versus interferon Beta-1a in relapsing multiple sclerosis. N Engl J Med. 2016;376:221–34.
Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, de Seze J, Giovannoni G, Hartung HP, Hemmer B, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376:209–20.
Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, Drulovic J, Filippi M. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology. 2014;82:573–81.
Meyer Zu Horste G, Mausberg AK, Korth C, Stuve O, Kieseier BC. Quinpramine-a promising compound for treating immune-mediated demyelination of the nervous system. Drug News Perspect. 2010;23:287–94.
Klingenstein R, Lober S, Kujala P, Godsave S, Leliveld SR, Gmeiner P, Peters PJ, Korth C. Tricyclic antidepressants, quinacrine and a novel, synthetic chimera thereof clear prions by destabilizing detergent-resistant membrane compartments. J Neurochem. 2006;98:748–59.
Singh MP, Meyer zu Horste G, Hu W, Mausberg AK, Cravens PD, Eagar T, Lober S, Klingenstein R, Gmeiner P, Korth C, et al. Quinpramine is a novel compound effective in ameliorating brain autoimmune disease. Exp Neurol. 2009;215:397–400.
Meyer Zu Horste G, Mausberg AK, Muller JI, Lehmann HC, Lober S, Gmeiner P, Hartung HP, Stuve O, Korth C, Kieseier BC. Quinpramine ameliorates rat experimental autoimmune neuritis and redistributes MHC class II molecules. PLoS One. 2011;6:e21223.
Milo R. Therapeutic strategies targeting B-cells in multiple sclerosis. Autoimmun Rev. 2016;15:714–8.
Marsh EA, Hirst CL, Llewelyn JG, Cossburn MD, Reilly MM, Krishnan A, Doran M, Ryan AM, Coles AJ, Jones JL, Robertson NP. Alemtuzumab in the treatment of IVIG-dependent chronic inflammatory demyelinating polyneuropathy. J Neurol. 2010;257:913–9.
Hirst C, Raasch S, Llewelyn G, Robertson N. Remission of chronic inflammatory demyelinating polyneuropathy after alemtuzumab (Campath 1H). J Neurol Neurosurg Psychiatry. 2006;77:800–2.
Tzachanis D, Hamdan A, Uhlmann EJ, Joyce RM. Successful treatment of refractory Guillain-Barre syndrome with alemtuzumab in a patient with chronic lymphocytic leukemia. Acta Haematol. 2014;132:240–3.
Thomas K, Eisele J, Rodriguez-Leal FA, Hainke U, Ziemssen T. Acute effects of alemtuzumab infusion in patients with active relapsing-remitting MS. Neurol Neuroimmunol Neuroinflamm. 2016;3:e228.
Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, Ettlinger RE, Cohen S, Koopman WJ, Mohler K, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. 1997;337:141–7.
Chin RL, Sherman WH, Sander HW, Hays AP, Latov N. Etanercept (Enbrel) therapy for chronic inflammatory demyelinating polyneuropathy. J Neurol Sci. 2003;210:19–21.
Andreadou E, Kemanetzoglou E, Brokalaki C, Evangelopoulos ME, Kilidireas C, Rombos A, Stamboulis E. Demyelinating disease following anti-TNFa treatment: a causal or coincidental association? Report of four cases and review of the literature. Case Rep Neurol Med. 2013;2013:671935.
Wanschitz J, Maier H, Lassmann H, Budka H, Berger T. Distinct time pattern of complement activation and cytotoxic T cell response in Guillain-Barre syndrome. Brain. 2003;126:2034–42.
Susuki K, Rasband MN, Tohyama K, Koibuchi K, Okamoto S, Funakoshi K, Hirata K, Baba H, Yuki N. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J Neurosci. 2007;27:3956–67.
McGonigal R, Rowan EG, Greenshields KN, Halstead SK, Humphreys PD, Rother RP, Furukawa K, Willison HJ. Anti-GD1a antibodies activate complement and calpain to injure distal motor nodes of Ranvier in mice. Brain. 2010;133:1944–60.
Willison HJ, Halstead SK, Beveridge E, Zitman FM, Greenshields KN, Morgan BP, Plomp JJ. The role of complement and complement regulators in mediating motor nerve terminal injury in murine models of Guillain-Barre syndrome. J Neuroimmunol. 2008;201-202:172–82.
Jung S, Toyka KV, Hartung H-P. Soluble complement receptor type 1 inhibits experimental autoimmune neuritis in Lewis rats. Neurosci Lett. 1995;200:167–70.
Vriesendorp FJ, Flynn RE, Malone MR, Pappolla MA. Systemic complement depletion reduces inflammation and demyelination in adoptive transfer experimental allergic neuritis. Acta Neuropathol (Berl). 1998;95:297–301.
Halstead SK, Humphreys PD, Goodfellow JA, Wagner ER, Smith RA, Willison HJ. Complement inhibition abrogates nerve terminal injury in Miller Fisher syndrome. Ann Neurol. 2005;58:203–10.
Tran GT, Hodgkinson SJ, Carter NM, Killingsworth M, Nomura M, Verma ND, Plain KM, Boyd R, Hall BM. Membrane attack complex of complement is not essential for immune mediated demyelination in experimental autoimmune neuritis. J Neuroimmunol. 2010;229:98–106.
Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–9.
Maurer M, Gold R. Animal models of immune-mediated neuropathies. Curr Opin Neurol. 2002;15:617–22.
Deretzi G, Zou LP, Pelidou SH, Nennesmo I, Levi M, Wahren B, Mix E, Zhu J. Nasal administration of recombinant rat IL-4 ameliorates ongoing experimental autoimmune neuritis and inhibits demyelination. J Autoimmun. 1999;12:81–9.
Miyamoto K, Oka N, Kawasaki T, Satoi H, Matsuo A, Akiguchi I. The action mechanism of cyclooxygenase-2 inhibitor for treatment of experimental allergic neuritis. Muscle Nerve. 1999;22:1704–9.
Abbas N, Zou LP, Pelidou SH, Winblad B, Zhu J. Protective effect of Rolipram in experimental autoimmune neuritis: protection is associated with down-regulation of IFN-gamma and inflammatory chemokines as well as up-regulation of IL-4 in peripheral nervous system. Autoimmunity. 2000;32:93–9.
Hoffman PM, Powers JM, Weise MJ, Brostoff SW. Experimental allergic neuritis. I. Rat strain differences in the response to bovine myelin antigens. Brain Res. 1980;195:355–62.
Miletic H, Utermohlen O, Wedekind C, Hermann M, Stenzel W, Lassmann H, Schluter D, Deckert M. P0(106-125) is a neuritogenic epitope of the peripheral myelin protein P0 and induces autoimmune neuritis in C57BL/6 mice. J Neuropathol Exp Neurol. 2005;64:66–73.
Zou LP, Ljunggren HG, Levi M, Nennesmo I, Wahren B, Mix E, Winblad B, Schalling M, Zhu J. P0 protein peptide 180-199 together with pertussis toxin induces experimental autoimmune neuritis in resistant C57BL/6 mice. J Neurosci Res. 2000;62:717–21.
Taylor WA, Hughes RA. Experimental allergic neuritis induced in SJL mice by bovine P2. J Neuroimmunol. 1985;8:153–7.
Calida DM, Kremlev SG, Fujioka T, Hilliard B, Ventura E, Constantinescu CS, Lavi E, Rostami A. Experimental allergic neuritis in the SJL/J mouse: induction of severe and reproducible disease with bovine peripheral nerve myelin and pertussis toxin with or without interleukin-12. J Neuroimmunol. 2000;107:1–7.
Abromson-Leeman S, Bronson R, Dorf ME. Experimental autoimmune peripheral neuritis induced in BALB/c mice by myelin basic protein-specific T cell clones. J Exp Med. 1995;182:587–92.
Snyder DH, Stone SH, Raine CS. Attempts to induce chronic experimental allergic neuritis in strain 13 and Hartley guinea pigs. J Neuropathol Exp Neurol. 1977;36:488–98.
Eylar EH, Toro-Goyco E, Kessler MJ, Szymanska I. Induction of allergic neuritis in rhesus monkeys. J Neuroimmunol. 1982;3:91–8.
Acknowledgements
Not applicable
Funding
This review was written without support or input from any funding source.
Availability of data and materials
References for this Review were identified by searches of PubMed and Google Scholar until November, 2017. Selected articles were also obtained from the reference lists of papers identified by previous searches. The search terms “Inflammatory Neuropathies”, “Guillain-Barré syndrome”, “Chronic inflammatory demyelinating polyradiculoneuropathy”, “experimental autoimmune encephalomyelitis”, “primary progressive multiple sclerosis”, “relapsing-remitting multiple sclerosis”, “spontaneous peripheral autoimmune polyneuropathy”, “Fingolimod”, “Rituximab”, “Ocrelizumab”, “Ofatumumab”, “Etanercept”, “Quinpramine”, “Alemtuzumab” and “Eculizumab” were used. Only reports published in English were included. The final reference list was generated on the basis of relevance to the topics covered in this Review.
Author information
Authors and Affiliations
Contributions
DS and GMzH performed literature search. DS, BCK, HW, and GMzH wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Schafflick, D., Kieseier, B.C., Wiendl, H. et al. Novel pathomechanisms in inflammatory neuropathies. J Neuroinflammation 14, 232 (2017). https://doi.org/10.1186/s12974-017-1001-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-017-1001-8