
Abstract
Background
The correlations between circulating tumour DNA (ctDNA)-derived genomic markers and treatment response and survival outcome in Chinese patients with advanced breast cancer (ABC) have not been extensively characterized.
Methods
Blood samples from 141 ABC patients who underwent first-line standard treatment in Peking University Cancer Hospital were collected. A next-generation sequencing based liquid biopsy assay (PredicineCARE) was used to detect somatic mutations and copy number variations (CNVs) in ctDNA. A subset of matched blood samples and tumour tissue biopsies were compared to evaluate the concordance.
Results
Overall, TP53 (44.0%) and PIK3CA (28.4%) were the top two altered genes. Frequent CNVs included amplifications of ERBB2 (24.8%) and FGFR1 (8.5%) and deletions of CDKN2A (3.5%). PIK3CA/TP53 and FGFR1/2/3 variants were associated with drug resistance in hormone receptor-positive (HR +) and human epidermal growth factor receptor 2-positive (HER2 +) patients. The comparison of genomic variants across matched tumour tissue and ctDNA samples revealed a moderate to high concordance that was gene dependent. Triple-negative breast cancer (TNBC) patients harbouring TP53 or PIK3CA alterations had a shorter overall survival than those without corresponding mutations (P = 0.03 and 0.008). A high ctDNA fraction was correlated with a shorter progression-free survival (PFS) (P = 0.005) in TNBC patients. High blood-based tumor mutation burden (bTMB) was associated with a shorter PFS for HER2 + and TNBC patients (P = 0.009 and 0.05). Moreover, disease monitoring revealed several acquired genomic variants such as ESR1 mutations, CDKN2A deletions, and FGFR1 amplifications.
Conclusions
This study revealed the molecular profiles of Chinese patients with ABC and the clinical validity of ctDNA-derived markers, including the ctDNA fraction and bTMB, for predicting treatment response, prognosis, and disease progression.
Trial registration: ClinicalTrials.gov ID: NCT03792529. Registered January 3rd 2019, https://clinicaltrials.gov/ct2/show/NCT03792529.
Similar content being viewed by others


Introduction
Female breast cancer has become the most commonly diagnosed malignancy worldwide according to global cancer statistics, with nearly 2.3 million new cases in 2020 [1]. Compared with other malignancies, such as lung cancer, patients with breast cancer have relatively favourable treatment response rates and therefore longer 5-year survival rates (89.7%, by SEER dataset) [2]. However, a significant percentage of breast cancer patients eventually develop incurable metastatic disease, which is characterized by an increasingly complex genomic landscape [3]. Traditional Sanger sequencing cannot fulfil the requirements of rapidly generating comprehensive correlations between genomic variants and treatment response in a large number of clinical samples [4]. Therefore, robust genomic profiling technologies should be applied to the characterization of advanced breast cancer (ABC) with the goal of defining genomic variation, evaluating prognosis and developing biomarkers to guide treatment selection.
Next generation sequencing (NGS) and molecular testing assays have evolved rapidly in recent years and are being successfully adopted in various clinical applications. When high coverage is achieved, targeted NGS enables the detection of low-frequency somatic mutations in heterogeneous tumour populations and circulating cell-free DNA (cfDNA) [5]. A number of gene mutations, such as those in ESR1, PIK3CA, and TP53, have been found to be associated with drug resistance and worse prognosis in breast cancer [6]. Given the strong detection capacity of NGS, it is now possible for clinicians to identify certain resistance-related mutations in a timely manner and make new therapeutic choices [7]. Despite remaining challenges and limitations, efficient high-throughput sequencing technologies are leading to real and unprecedented benefits for the medical care of cancer patients [8].
Circulating tumour DNA (ctDNA) is the small proportion of cfDNA that is released into the circulation from tumour cells and is detectable in a wide range of malignancies [9]. Several studies have demonstrated high concordance between alterations detected in ctDNA and tumour tissue biopsy in ABC [10,11,12,13,14]. Moreover, ctDNA can serve as a noninvasive biomarker for disease monitoring, predicting drug efficacy, and determining prognosis [15]. The ctDNA fraction is defined as the proportion of ctDNA in cfDNA. In other types of cancer, such as metastatic prostate cancer, the ctDNA fraction has been shown to be a prognostic factor [16]. Blood-based tumour mutation burden (bTMB) is a metric that represents the total number of mutations per coding area of a tumour genome, and is a metric that is frequently generated by NGS analysis [17]. The magnitude and prognostic impact of bTMB is significantly different across solid tumour types [18]. In triple-negative breast cancer (TNBC), bTMB is an emerging prognostic biomarker for immunotherapy [19, 20]. However, it remains unclear in ABC patients whether the ctDNA fraction and bTMB correlate with different subtypes, treatment response or prognosis.
Herein, we investigated the molecular profiles of samples from Chinese ABC patient with a targeted 152-gene NGS panel. A comparison of the mutation profiles across matched tumour tissue and plasma ctDNA samples was conducted. Then, several genomic aberrations related to the treatment response were identified. Moreover, we explored whether the ctDNA fraction and bTMB could be used to evaluate treatment response and prognosis, respectively. Finally, we evaluated the clinical validity of ctDNA sequencing for longitudinally monitoring disease progression.
Materials and methods
Patient cohort and clinical data collection
The study protocol strictly followed the Declaration of Helsinki and was approved by the Peking Universities Cancer Hospital Ethics Committee (CABC008, 2020KT75). All patients signed a written informed consent form prior to registration for ctDNA-analysis. Female patients with first-line metastatic or primary stage IV disease who were diagnosed at Peking University Cancer Hospital (PKUCH) were enrolled in this prospective study from December 2012 to June 2021.
The clinical data collected in this study included receptor status (oestrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2)), age, tumour grade, Ki67, primary TNM stage, adjuvant treatment, progression free-survival (PFS), metastasis site, disease status, and treatment response. The evaluation of the receptor status by immunohistochemistry (IHC) was based on the American Society of Clinical Oncology guidelines [21, 22]. A specimen with a minimum of 1% invasive tumour cells positive for ER/PR was considered hormone receptor-positive (HR +). HER2 positivity was defined as IHC staining 3 + or 2 + with gene amplification assessed by fluorescence in situ hybridization (FISH) with a HER2/CEP17 ratio ≥ 2.0 and average HER2 copy number ≥ 4.0 signals/cell. TNM staging was defined according to the American Joint Committee on Cancer staging manual for breast cancer [23]. All patients were treated with the current standard therapy defined by the NCCN clinical practice guidelines [24]. The treatment response was evaluated according to RECIST 1.1 every 2–3 months of treatment duration. When a patient’s first-line PFS was less than 3 months, the patient was designated as having drug resistance. Clinical information was collected by reviewing electronic medical records, and survival data were recorded using a follow-up database in PKUCH.
Sample collection
Baseline plasma samples were collected from all 141 patients to identify the mutation pattern of ABC. Metastatic tumour biopsies were obtained from 21 of the 141 patients to validate the concordance between plasma and tumour tissue. Moreover, extra plasma samples were collected from 31 of the 141 patients to dynamically monitor the disease. Blood samples (5–10 ml) were collected into EDTA-containing blood collection tubes and processed within 2 h of collection by centrifugation at 1600g for 20 min at room temperature. Plasma was separated from buffy coats and red blood cells, aliquoted, and stored at − 80 °C until ctDNA extraction.
ctDNA extraction, library construction, and sequencing
A highly sensitive NGS assay (PredicineCARE, a commercially available 152-gene cancer NGS panel, developed by Huidu Shanghai Medical Sciences Ltd.) was used to detect somatic mutations and copy number variations (CNVs) in ctDNA. The ctDNA was extracted from 1 to 2 ml of the isolated plasma samples using the QIAamp Circulating Nucleic Acid Kit (QIAamp, Venlo, NL). Then, the quantity and quality of the purified ctDNA were checked using the Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). A total of 5–30 ng of purified ctDNA was subjected to library construction, including end-repair dA-tailing and adapter ligation. Ligated library fragments with appropriate adapters were amplified via PCR. The amplified DNA libraries were then checked using the Bioanalyzer 2100. All the samples in this study had a yield > 700 ng and were processed for hybrid capture.
Library capture was conducted using biotin-labelled DNA probes (Thermo Fisher Scientific, Weltham, MA, USA). In brief, the library was hybridized using the PredicineCARE panel overnight and captured with Dynabeads M-270 Streptavidin (Thermo Fisher Scientific, Weltham, MA, USA). The unbound fragments were washed away, and the enriched fragments were amplified via PCR amplification. For library preparation, the purified product was assessed using the Bioanalyzer 2100 and loaded into the NextSeq 500 (Illumina, San Diego, CA, USA) for NGS with paired-end 2 × 150 bp sequencing kits. The average sequencing depth per sample was approximately 20,000 × before deduplication. NGS quality-assessment was performed by examining the percentage of targeted regions with sufficient unique coverage (greater than 1500x). Samples with > 80% regions having > 1500 × unique coverage were deemed to pass Quality Control and were included in this study.
Sequencing data analysis
Sequencing data were analysed using an in-house-developed NGS analysis pipeline developed by Huidu Shanghai Medical Sciences Ltd., as described in previous studies [16, 25]. This process begins with the analysis of the raw sequencing data (BCL files) and outputs the final mutation calls. First, the pipeline performed adaptor trimming, barcode checking, and correction. Cleaned paired FASTQ files were aligned to the human reference genome build hg19 using the Burrows–Wheeler Aligner alignment tool (version 0.7.15). Then, consensus bam files were derived by merging paired-end reads originating from the same molecules (based on mapping location and unique molecular identifiers) as single-strand fragments. Single-strand fragments from the same double stranded DNA molecules were merged to be double-stranded to reduce sequencing and PCR errors during this process. Detected variants were filtered based on the local variant background (defined by plasma samples from healthy Chinese donors and internal sample pools), log-odds threshold [26], base quality and mapping quality thresholds, and repeat regions. Variants with mutation allele frequency (MAF) ≥ 0.25% and hotspot variants with MAF ≥ 0.1% were reported. Benign and likely benign variants were excluded.
Statistical analysis
Categorical variables are described as frequencies (percentage), and continuous variables are presented as medians (range). Fisher’s exact test was used to evaluate differences in the rate of mutations across specific genes in different molecular subtypes. Survival analysis was used to evaluate the associations between ctDNA-based variables measured prior to the commencement of treatment and PFS and overall survival (OS). Associations between the variables and PFS and OS were estimated by plotting Kaplan–Meier curves and compared by log-rank test to determine significant differences among subgroups. A univariate Cox regression model was performed to compute medians and 95% confidence intervals (CIs) for prognostic variables. The Wilcoxon test or Kruskal–Wallis test was used to compare the ctDNA fraction and bTMB between different molecular subtypes. To dichotomize the variables for the survival analysis, the upper quartile cut-off was used for both the ctDNA fraction and bTMB. Cohen's kappa was used to evaluate the concordance of variants between plasma and tissue samples. All statistical analyses were performed using R language, version 3.5.3. A two-sided P value of < 0.05 was considered significant.
Results
Baseline characteristics of patients
In total, 141 ABC patients who underwent systematic treatment at PKUCH from December 2012 to June 2021 were included in this study. All patients were female. The median age was 48.0 years, and the age range was 21.0 to 77.0 years (Table 1). Among these patients, the proportions of patients with HR + , human epidermal growth factor receptor 2-positive (HER2 +), and triple-negative disease were 31.2% (44/141), 41.1% (58/141), and 27.7% (39/141), respectively. As shown in Table 2, all patients developed metastatic disease, and the metastatic sites included the chest wall, bone, lymph nodes, lung, liver, brain and others sites. More than half of these patients had visceral metastasis (52.5%, 74/141) and 71.0% (100/141) had ≥ 2 sites of metastasis. Current standard therapy was applied to all patients once metastatic/advanced disease was diagnosed. With respect to adjuvant therapy, 72.3% (102/141) of all patients had received chemotherapy. In the HR + patient subgroup, 62.1% (36/58) had received adjuvant endocrine therapy.
Molecular profiling of Chinese advanced breast cancer
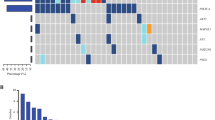
Among the 141 analysed baseline plasma samples, 111 (78.7%) contained at least 1 somatic mutation, and 112 (79.4%) contained single nucleotide variations (SNVs) or CNVs. Collectively, 267 somatic alterations were detected across 65 genes, among which TP53 (44.0%), PIK3CA (28.4%), ERBB2 (24.8%), and FGFR1 (8.5%) were most frequently altered (Fig. 1A). ESR1 hotspot mutations (D538G and Y537S/N/C) were also detected in a subset of HR + patients at a lower frequency (4.0%). CNVs were detected in 32 genes, including amplifications of ERBB2 (22.7%), FGFR1 (6.4%), and PIK3CA (4.3%) and deletions of CDKN2A (3.5%), CDH1 (3.5%), and BRCA1 (2.8%) (Fig. 1A). In the heatmap show in Fig. 1A, each column represents one patient and each row represents one gene. The mutation rates are displayed on the right side of the heatmap. Next, the detection rates of somatic mutations and CNVs were compared among the different clinical subtypes (HR + , HER2 + , and TNBC). The results indicated that HER2 + patients had the highest frequency of TP53 alterations, which was significantly higher than that observed in HR + patients (64% vs. 31%, P < 0.009; Fig. 1B). For ERBB2 CNVs, amplifications occurred in 31/32 (96.9%) HER2 + patients. One amplification was observed in a TNBC patient, and no ERBB2 CNVs were detected in HR + patients (P < 0.001; Fig. 1C).

CtDNA mutation profile of Chinese ABC patients and the distribution of variations among different IHC subtypes. A Somatic mutation landscape of 141 Chinese ABC patients. The bar chart above the heatmap shows the ctDNA fractions of all baseline samples in the cohort. In the heatmap, the top bars depict the number of mutations a patient carried and the bars below denote different IHC subtypes. Each column represents one patient, and each row represents one gene. The text on the left represents gene names. The values on the right represent the mutation rates of these genes. Distributions of (B) the top 10 somatic mutations and (C) the top 10 CNVs among the three IHC subtypes. ABC, advanced breast cancer; ctDNA, circulating tumour DNA; IHC, immunohistochemistry; CNVs, copy number variations
Comparison of plasma ctDNA and tumour tissue sequencing results
Targeted NGS sequencing was applied to analyse plasma samples from 21 patients and their matching tumour tissues from metastatic sites. PIK3CA and TP53 were the two most common genes with SNVs, and ERBB2 was the most common gene with CNVs (Fig. 2A, B). In the matrices, each column represents one patient and each row represents one gene. A square filled with two coloured triangles suggests an identical mutation in tissue and plasma. Kappa tests were performed to test the consistency between the plasma samples and tissue samples. Cohen's kappa is a robust statistic used to compare the ability of different raters to classify subjects into one of several groups and can range from − 1 to + 1 [27]. A kappa value ≤ 0 indicates no agreement, 0.01–0.20 indicates no to slight agreement, 0.21–0.40 indicates fair agreement, 0.41–0.60 indicates moderate agreement, 0.61–0.80 indicates substantial agreement, and 0.81–1.00 indicates almost perfect agreement. PIK3CA SNVs were found in ten tissue samples and six plasma samples, with a match number of six (kappa = 0.61; Fig. 2C). Nine tissue samples and seven plasma samples were shown to have TP53 SNVs, and five pairs of samples had the same variants (kappa = 0.41; Fig. 2D). In addition, ERBB2 CNVs were detected in six tissue samples and six plasma samples, with a match number of five (kappa = 0.77; Fig. 2E).

Concordance analysis of mutations between plasma and tissue samples. Concordance of (A) SNVs and (B) CNVs between tissue samples and plasma ctDNA. In the matrices, the top bar charts depict the number of mutations. The text below represents patient identities, and the text on the left represents gene names. Coloured triangles in the squares indicate mutations. If a square was filled with two coloured triangles, the same mutation was detected in the corresponding patient's plasma and tissue. Overlapping (C) PIK3CA SNVs, (D) TP53 SNVs, and (E) ERBB2 CNVs between tissue and plasma ctDNA. SNVs, single nucleotide variations; CNVs, copy number variations; ctDNA, circulating tumour DNA
Genomic aberrations related to treatment response and prognosis
All patients were treated with current standard first-line therapy, and more than 50% of patients responded to the therapy. Correlation analysis was conducted to find particular variants that were related to the drug response for different subtypes of patients. The results indicated that the presence of PIK3CA/TP53 and FGFR1/2/3 variants was related to drug resistance in HR + patients (P = 0.05; Fig. 3A) and HER2 + patients (P = 0.07; Fig. 3B), respectively. However, no potential response-related somatic mutations or CNVs were found for TNBC patients (Fig. 3C). The green bars and the orange bars below the heatmap indicate samples collected from patients who were sensitive and resistant to treatment, respectively. SNVs/Indels, amplifications, and deletions are represented by red, blue, and green squares, respectively. In terms of outcomes, TNBC patients harbouring TP53 mutations (29.8 vs. 52.8 months, P = 0.03; Fig. 3D) and PIK3CA mutations (21.5 vs. 50 months, P = 0.008; Fig. 3E) had worse OS than those without corresponding mutations, but no significant difference in PFS or OS was found in HER2 + and HR + patients (data not shown).

Oncoplots for drug-sensitive and drug-resistant (A) HR + samples, (B) HER2 + samples, and (C) TNBC samples. The green bars below the heatmap indicate samples collected from patients who are sensitive to treatment, while the orange bars indicate samples collected from patients who are resistant to treatment. Different colours of squares denote different types of mutations. Red represents SNVs/Indels, blue represents amplifications, and green represents deletions. Survival analyses of OS (D) between patients with or without TP53 mutations and (E) between patients with or without PIK3CA mutations. F Comparison of ctDNA fractions among the three IHC subtypes. The black lines represent the median of each group. HR + , hormone receptor-positive; HER2 + , human epidermal growth factor receptor 2-positive; TNBC, triple-negative breast cancer; SNVs, single nucleotide variations; Indels, insertions and deletions; OS, overall survival; ctDNA, circulating tumour DNA; IHC, immunohistochemistry
No significant difference in the ctDNA fraction between drug-sensitive and drug-resistant samples
As shown in Fig. 1A (the bar chart above the heatmap), the ctDNA fractions across all samples varied from 0.6% to 76%, with a median of 3.0%. HER2 + patients had a significantly higher ctDNA fraction (median, 13.0%) than HR + (median, 2.6%) and TNBC patients (median, 2.1%) (P < 0.02; Fig. 3F). To investigate whether the ctDNA fraction was associated with drug response, we compared the ctDNA fraction between drug-sensitive and drug-resistant patient samples among different subgroups. The results suggested that there was no significant difference in the ctDNA fraction between the two groups for all three subtypes (P > 0.05; Additional file 1: Fig. S1A).
A high ctDNA fraction was associated with worse prognosis in HR + and TNBC patients
Survival analyses of the three clinical subtypes of patients revealed a significantly shorter PFS (7.2 vs. 9 vs. 11 months, P = 0.007; Fig. 4A) and OS (41.6 vs. 65.7 vs. 87.2 months, P = 0.003; Fig. 4B) for TNBC patients compared to the other two subtypes. All patients were dichotomized into two groups with high and low ctDNA fractions by the 75th percentile (0.174), as previously described [28,29,30]. A high ctDNA fraction was shown to be associated with a shorter PFS and OS for all subtypes, but these trends were not significant for most patients (P > 0.05; Additional file 1: Fig. S1B and Additional file 2: Fig. S2). One exception was that for TNBC patients, as a high ctDNA fraction was significantly associated with a shorter PFS in these patients (2.9 vs. 7.3 months, P = 0.005; Fig. 5A).

Survival analyses of (A) PFS and (B) OS among the three IHC subtypes. C Comparison of bTMB among the three IHC subtypes. D Comparison of bTMB between HER2 + drug-sensitive and drug-resistant samples. The black lines represent the median of each group. Survival analyses of (E) PFS and (F) OS between patients with low and high bTMB. PFS, progression-free survival; OS, overall survival; IHC, immunohistochemistry; bTMB, blood-based tumour mutation burden; HER2 + , human epidermal growth factor receptor 2-positive

Survival analyses of PFS (A) between TNBC patients with low and high ctDNA fractions, (B) between HER2 + patients with low and high bTMB, and (C) between TNBC patients with low and high bTMB. PFS, progression-free survival; TNBC, triple-negative breast cancer; ctDNA, circulating tumour DNA; HER2 + , human epidermal growth factor receptor 2-positive; bTMB, blood-based tumour mutation burden
bTMB was elevated significantly in drug-resistant HER2 + samples
The values of bTMB across all samples ranged from 0 to 324.0, with a median of 24.8. Comparison of bTMB among the different IHC subtypes revealed no significant differences (P = 0.29, Fig. 4C). We also explored the association between bTMB and drug response. In the HER2 + subtype, bTMB in samples collected from drug-resistant patients was significantly higher than that of samples collected from drug-sensitive patients (P = 0.03; Fig. 4D), but this association was not found in HR + and TNBC subtypes (Additional file 1: Fig. S1C).
High bTMB was associated with worse prognosis in all patients
The patients were divided into high bTMB and low bTMB groups by the 75th percentile (43.3) as previously described [31,32,33,34]. Cox regression analysis revealed that patients with a high bTMB had a significantly shorter PFS (5 vs. 10 months, P = 0.05; Fig. 4E) and OS (40.6 vs. 70 months, P = 0.02; Fig. 4F) than those with a low bTMB. For different subtypes, HER2 + (5 vs. 20 months, P = 0.009; Fig. 5B) and TNBC patients (3 vs. 7.3 months, P = 0.05; Fig. 5C) with a high bTMB had a significantly shorter PFS than those with a low bTMB.
Disease monitoring with ctDNA sequencing
Plasma samples were collected at disease progression from 31 out of the 141 patients and were analysed to monitor genomic variants during clinical progression. Compared with the corresponding baseline samples, several acquired variants were detected in samples collected from HR + and TNBC patients at progression. These variants included mutations in TP53, PIK3CA, and ESR1, the deletion of CDKN2A, and the amplification of FGFR1. In contrast, some variants detected at baseline in HER2 + patients, such as TP53 and PIK3CA mutations, were not detected at disease progression (Additional file 3: Fig. S3).
Discussion
In recent years, efforts have been made to investigate the genomic complexity of breast cancer [2, 35,36,37,38]. However, these studies were either retrospective or lacked ctDNA-based dynamic disease monitoring and clinical management. Breast cancer is a heterogeneous disease that varies across different clinical subtypes, populations and geographical regions, all of which exhibit variable therapeutic responses [39, 40]. With the increasing incidence rate of breast cancer worldwide, it is critical to identify region-specific genomic variants and relevant therapeutic targets. In this prospective study, a novel 152-gene targeted NGS panel was used to evaluate the mutation landscape of ctDNA collected from 141 Chinese ABC patients. In addition, we profiled drug response-related genomic variants, evaluated the prognostic value of the ctDNA fraction and bTMB, and assessed the ability of ctDNA to monitor disease progression.
In this study, molecular profiling of ABC revealed genomic variants consistent with several previous reports, and TP53 (44.0%) and PIK3CA (28.4%) were the two most commonly altered genes [2, 35, 38, 41, 42]. TP53 is a well-characterized tumour suppressor gene that acts as a regulator of key cellular processes involved in controlling cell proliferation and maintaining genomic stability [43, 44]. Almost all hallmark features of cancer are impacted by the functions of the TP53 protein and correlate with genomic alterations in TP53 pathways [45]. In other words, the occurrence of many tumours requires TP53 mutations as a prerequisite. Indeed, TP53 was shown to be frequently altered in previous sequencing studies, with frequencies ranging from 38.24 to 74.11% [2, 35, 38, 41, 42, 46]. In particular, the mutation rates of TP53 were 38.24%, 41.67%, 64.1%, 43.27%, and 47.0% in Chinese populations [2, 35, 41, 42, 46]. Therefore, it is not surprising to find a high mutation rate of TP53 in this study. However, in regard to individual situations, more efforts are needed to reveal the specific role of TP53. HER2 + patients showed a significantly higher TP53 mutation rate than HR + patients. TNBC patients harbouring TP53 and PIK3CA mutations showed a significantly longer OS than those without these alterations. There is clear evidence that TP53 is associated with poor prognosis of HR + disease, while its clinical significance in HER2 + and TNBC patients remains controversial [47]. Considering the high degree of heterogeneity of TP53 mutations, further large-scale sequencing studies are needed to address its clinical impact and association with response to therapy in distinct subtypes. PIK3CA is the most frequently mutated oncogene in human cancers [48, 49]. The continuous activation of AKT that is induced by PIK3CA mutations can promote the growth and transformation of mammary epithelial cells and may explain the high mutation rate of PIK3CA [50]. The approval of alpelisib has rendered the detection of PIK3CA mutations clinically actionable, as everolimus, an mTOR inhibitor, does not directly target PIK3CA mutations. Thus, HR + /HER2- ABC patients harbouring PIK3CA mutations can be offered another targeted therapy choice after endocrine resistance occurs [51]. For TNBC, the prognostic impact of PIK3CA mutations remains debatable, and evidence for the clinical application of PIK3CA inhibitors is still lacking [52, 53]. The BELLE-4 study tested the efficacy of buparlisib (a pan-PIK3CA inhibitor) in combination with placebo or paclitaxel in HER2- ABC patients [54]. Approximately 25% of the patients were TNBC patients who showed a worse outcome with the addition of burpalisib than placebo (5.2 vs. 9.3 months; hazard ratio 1.86, 95% CI 0.91–3.79). Nonetheless, combination approaches targeting PIK3CA with other targeted drugs, such as androgen receptor and CDK4/6 inhibitors, may provide a potential therapeutic direction for specific subsets of TNBC patients [53]. Several ESR1 hotspot mutations within the ligand-binding domain, including D538G and Y537S/N/C, were also detected at a lower frequency (4%). Given established treatment-associated patterns of acquired mutations in ESR1, it is not surprising that the mutation rate was not high in the setting of first-line treatment [55].
ERBB2 was the gene with the most frequent occurrence of CNVs and had an amplification rate of 22.7% (32/141) in the entire cohort and 70.5% (31/44) in the HER2 + population. Compared with the study by Davis et al., the amplification rate in ERBB2 was higher in our cohort (70.5% vs. 44.0%) [38]. This difference may be explained by two factors. The first factor is the patient population. We enrolled Chinese patients in this study, while the study by Davis et al. was conducted in the United States. This explanation is supported by a previous study by Xiao et al. which also revealed a high amplification rate of ERBB2 (90.3%, 158/175) in HER2 + Chinese patients [46]. We (PredicineCARE, developed by Huidu Shanghai Medical Sciences Ltd.) and Davis et al. (Guardant360 assay, Guardant Health, Inc., Redwood City, CA) used different NGS panels to detect variants. The higher detection rate may indicate the improved ability of copy number detection from our NGS assay. Notably, one TNBC patient presented with an ERBB2 CNV, indicating tumour heterogeneity and the potential need to reassess HER2 status by ctDNA during the clinical management of TNBC [56]. These findings underscore the feasibility of tracing ERBB2 CNVs with liquid biopsy during disease progression and introduce the possibility for applying anti-HER2 therapy. In the analysis of drug response-related variants, the detection of individual SNVs or CNVs showed no correlation with drug response. However, the combination analysis of SNVs with CNVs revealed potential associations between the PIK3CA/TP53 and FGFR1/2/3 variants and drug resistance in HR + and HER2 + patients, respectively. These findings suggest that integrating multiple genetic alterations could improve the identification of treatment resistance-related mechanisms compared to measuring a single alteration [57].
CtDNA levels can dynamically reflect the tumour burden of a patient and predict disease progression prior to imaging [58]. Moreover, several studies have revealed the prognostic role of the ctDNA fraction in ABC patients [58,59,60]. Stover et al. found that a ctDNA fraction of ≥ 10% correlated with a worse metastatic OS (6.4 vs. 15.9 months) [59], while a cut-off of 0.5% ctDNA (MAF) was regarded as prognostic for both PFS and OS in another report [58]. In the present study, a cut-off of 0.174 was used to discriminate patients with high or low ctDNA fractions. Based on this grouping, we found a significant difference in PFS between TNBC patients with high vs. low ctDNA fractions (2.9 vs. 7.3 months, P = 0.005). However, no significant differences in PFS were observed in other subtypes. This could result from subject variations in disease onset, diagnosis, or intervention since this study contained all subtypes of patients and multiple treatment regimens. Moreover, this heterogeneity could result in opposite associations between the ctDNA fraction and prognosis among different subtypes. In addition to its prognostic role, the ctDNA fraction can reflect the panel sensitivity, as the panel sensitivity decreased if samples displayed low tumour fractions. Therefore, we evaluated the correlation between the ctDNA fraction and the number of mutations in all baseline plasma samples using the Pearson correlation coefficient, and the results suggested a significant positive correlation (R = 0.56, P < 0.05; Additional file 4: Fig. S4A). The number of mutations in samples with ctDNA fractions of 0–1% was significantly lower than that in samples with ctDNA fractions of 1–5% and > 5% (Additional file 4: Fig. S4B). These findings also illustrate the importance of improving sequencing sensitivity.
TMB is a measure of the somatic mutation frequency and mutation accumulation process of a tumour. In tumorigenesis, some nondriver mutations will lead to the activation of an antitumour response by generating neoantigens that are recognized by the immune system [61]. Consequently, TMB is regarded as a biomarker that is associated with the response to immune checkpoint inhibitors in several types of cancer [17]. Despite being a promising pancaner tool, the use of TMB is limited by several key remaining issues, including variable clinical impacts across cancer types and the lack of a standardized cut-off value [17]. In this study, a cut-off of 43.3 was adopted to define high vs. low bTMB, and patients with high bTMB showed a shorter PFS (5 vs. 10 months, P = 0.05) and OS (40.6 vs. 70 months, P = 0.02) than those with low bTMB. These findings are consistent with previous reports that suggested that TMB was a prognostic factor for poor outcome [19, 20]. Even so, the association between bTMB and prognosis should be interpreted with caution because of differences at the individual level, which may reduce its usability in practical situations. Indeed, in our further analyses based on subtypes, these associations were significant only in HER2 + (PFS 5 vs. 20 months, P = 0.009) and TNBC patients (PFS 3 vs. 7.3 months, P = 0.05). Hence, more research investigating the clinical utility of ctDNA-derived biomarkers in ABC is needed to address this issue. Interestingly, HER2 + samples displayed significantly elevated bTMB. Since recent evidence has indicated elevated programmed cell death-ligand 1 and tumour infiltrating lymphocyte expression in HER2 + disease [62], further studies exploring the possibility of implementing immunotherapy in such patients are warranted.
Previous sequencing studies have revealed the concordant detection of DNA variants across matched sets of ctDNA and tumour tissue [10,11,12,13,14]. Davis et al. described the genomic landscape of ctDNA in 255 ABC patients. They found an agreement of 79–91% between 105 pairs of ctDNA and tissue, with alterations in PIK3CA and TP53 showing moderate concordance (kappa = 0.5513 and 0.5809, respectively) [38]. In this study, we also found moderate concordance for the SNVs in PIK3CA and TP53 (kappa = 0.61 and 0.41, respectively). In addition, a high concordance was revealed for ERBB2 CNVs (kappa = 0.77), which was consistent with a report by Zhou et al. [35]. These findings suggest the equivalent utility of ctDNA with tissue sequencing in developing targeted therapeutic approaches for HER2 + patients.
We also performed longitudinal monitoring of disease progression to observe the dynamic changes in gene mutations and amplifications in ctDNA. The genomic variants detected in ctDNA at baseline and at disease progression were compared. Overall, the plasma ctDNA from samples collected at the time of clinical progression in HR + and TNBC patients had acquired genomic variants, indicating clonal and subclonal responses to treatment. In contrast, genomic variants detected at baseline in HER2 + patients were not detected at progression. In HR + patients, newly acquired ESR1 mutations at the time of disease progression were observed in four patients, which is consistent with previous findings that the emergence of ESR1 mutations is associated with the development of endocrine resistance [55]. Other emerging genomic variants included FGFR amplification and CDKN2A deletion events. Aberrant FGFR signalling has been identified as a mechanism that drives tumour growth and promotes angiogenesis [63]. The amplification of FGFR, which occurs in approximately 10–16% of HR + patients [60], has been shown to mediate endocrine resistance [64]. CDKN2A (cyclin-dependent kinase inhibitor 2A) is a tumour suppressor gene that was first reported in 1993, and it is negatively regulated by the CDK4/6/RB pathway [65]. Despite rare alterations of this gene in breast cancer (approximately 5.8%) [66], CDKN2A deletions were reported to be associated with poor outcomes in luminal B ER + patients [67]. Importantly, the deletion of this gene implicates CDK4/6 as a therapeutic target to some extent [68]. Taken together, the acquired FGFR amplification and CDKN2A deletion variants in progression samples may explain the disease progression in these HR + patients.
There are several limitations that should be mentioned. First, the sample size was relatively small, and only a subset of patients had matched blood and tissue samples. These factors may decrease the concordance of detected genomic variants. Second, the samples were collected in a single centre and sequenced by a single NGS assay. Hence, the results from this study need to be validated in other research centres using distinct sequencing assays. Third, since the NGS panel applied in this study only included 152 genes, its ability to detect tumour burden is theoretically inferior to genome-wide tests. Thus, the association between bTMB and prognosis needs more verification. Fourth, the sensitivity of this panel may decrease if samples display a lower ctDNA fraction, and the ctDNA fraction of all samples in this study varied greatly (Additional file 5). Finally, although treatment response-related variants were analysed, the effects of drug interventions guided by ctDNA profiling were not studied. Further studies will be required to evaluate the impact of ctDNA profiling-guided drug interventions on patient outcomes to establish the clinical utility of NGS liquid biopsy in the management of ABC patients.
In conclusion, this prospective study profiled the mutation landscape of Chinese ABC patients who underwent first-line standard treatment and demonstrated the clinical validity of ctDNA-based genomic analysis. Further studies are warranted to investigate the relationship between drug interventions and genomic changes.
Availability of data and materials
All data and materials supporting the conclusions of this article were available in the figures, tables, and supplementary materials, which are available to authorized users.
Abbreviations
- ABC:
-
Advanced breast cancer
- cfDNA:
-
Circulating cell-free DNA
- ctDNA:
-
Circulating tumor DNA
- NGS:
-
Next generation sequencing
- bTMB:
-
Blood-based tumor mutation burden
- HR + :
-
Hormone receptor-positive
- HER2 + :
-
Human epidermal growth factor receptor 2-positive
- TNBC:
-
Triple-negative breast cancer
- PKUCH:
-
Peking University Cancer Hospital
- ER:
-
Oestrogen receptor
- PR:
-
Progesterone receptor
- PFS:
-
Progression-free survival
- OS:
-
Overall survival
- IHC:
-
Immunohistochemistry
- FISH:
-
Fluorescence in situ hybridization
- SNVs:
-
Single nucleotide variations
- CNVs:
-
Copy number variations
- Indels:
-
Insertions and deletions
- MAF:
-
Mutation allele frequency
- CIs:
-
Confidence intervals
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–49.
Hu ZY, Xie N, Tian C, Yang X, Liu L, Li J, et al. Identifying circulating tumor DNA mutation profiles in metastatic breast cancer patients with multiline resistance. EBioMedicine. 2018;32:111–8.
Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic characterization of metastatic breast cancers. Nature. 2019;569(7757):560–4.
van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. The third revolution in sequencing technology. Trends Genet. 2018;34(9):666–81.
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–51.
Liao H, Li H. Advances in the detection technologies and clinical applications of circulating tumor DNA in metastatic breast cancer. Cancer Manag Res. 2020;12:3547–60.
Alimirzaie S, Bagherzadeh M, Akbari MR. Liquid biopsy in breast cancer: a comprehensive review. Clin Genet. 2019;95(6):643–60.
Gerratana L, Zhang Q, Shah AN, Davis AA, Zhang Y, Wehbe F, et al. Performance of a novel Next Generation Sequencing circulating tumor DNA (ctDNA) platform for the evaluation of samples from patients with metastatic breast cancer (MBC). Crit Rev Oncol Hematol. 2020;145: 102856.
Buono G, Gerratana L, Bulfoni M, Provinciali N, Basile D, Giuliano M, et al. Circulating tumor DNA analysis in breast cancer: is it ready for prime-time? Cancer Treat Rev. 2019;73:73–83.
Tzanikou E, Markou A, Politaki E, Koutsopoulos A, Psyrri A, Mavroudis D, et al. PIK3CA hotspot mutations in circulating tumor cells and paired circulating tumor DNA in breast cancer: a direct comparison study. Mol Oncol. 2019;13(12):2515–30.
Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(7):904–16.
Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8(1):1324.
Chung JH, Pavlick D, Hartmaier R, Schrock AB, Young L, Forcier B, et al. Hybrid capture-based genomic profiling of circulating tumor DNA from patients with estrogen receptor-positive metastatic breast cancer. Ann Oncol. 2017;28(11):2866–73.
Chae YK, Davis AA, Jain S, Santa-Maria C, Flaum L, Beaubier N, et al. Concordance of genomic alterations by next-generation sequencing in tumor tissue versus circulating tumor DNA in breast cancer. Mol Cancer Ther. 2017;16(7):1412–20.
Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20(2):71–88.
Kohli M, Tan W, Zheng T, Wang A, Montesinos C, Wong C, et al. Clinical and genomic insights into circulating tumor DNA-based alterations across the spectrum of metastatic hormone-sensitive and castrate-resistant prostate cancer. EBioMedicine. 2020;54: 102728.
Ritterhouse LL. Tumor mutational burden. Cancer Cytopathol. 2019;127(12):735–6.
McNamara MG, Jacobs T, Lamarca A, Hubner RA, Valle JW, Amir E. Impact of high tumor mutational burden in solid tumors and challenges for biomarker application. Cancer Treat Rev. 2020;89: 102084.
Zhang X, Li J, Yang Q, Wang Y, Li X, Liu Y, et al. Tumor mutation burden and JARID2 gene alteration are associated with short disease-free survival in locally advanced triple-negative breast cancer. Ann Transl Med. 2020;8(17):1052.
Gao C, Li H, Liu C, Xu X, Zhuang J, Zhou C, et al. Tumor mutation burden and immune invasion characteristics in triple negative breast cancer: genome high-throughput data analysis. Front Immunol. 2021;12: 650491.
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American society of clinical oncology/college of American pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28(16):2784–95.
Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol. 2018;36(20):2105–22.
Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17(6):1471–4.
Gradishar WJ, Anderson BO, Abraham J, Aft R, Agnese D, Allison KH, et al. Breast cancer, version 3.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2020;18(4):452–78.
Kwan EM, Dai C, Fettke H, Hauser C, Docanto MM, Bukczynska P, et al. Plasma cell-free DNA profiling of PTEN-PI3K-akt pathway aberrations in metastatic castration-resistant prostate cancer. Jco Precis Oncol. 2021;5:622–37.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9.
McHugh ML. Interrater reliability: the kappa statistic. Biochem Med (Zagreb). 2012;22(3):276–82.
Nygaard AD, Holdgaard PC, Spindler KLG, Pallisgaard N, Jakobsen A. The correlation between cell-free DNA and tumour burden was estimated by PET/CT in patients with advanced NSCLC. Brit J Cancer. 2014;110(2):363–8.
Spindler K-LG, Pallisgaard N, Vogelius I, Jakobsen A. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res. 2012;18(4):1177–85.
Schou JV, Larsen FO, Sørensen BS, Abrantes R, Boysen AK, Johansen JS, et al. Circulating cell-free DNA as predictor of treatment failure after neoadjuvant chemo-radiotherapy before surgery in patients with locally advanced rectal cancer. Ann Oncol. 2018;29(3):610–5.
Heeke S, Hofman P. Tumor mutational burden assessment as a predictive biomarker for immunotherapy in lung cancer patients: getting ready for prime-time or not? Transl Lung Cancer Res. 2018;7(6):631–8.
Stenzinger A, Allen JD, Maas J, Stewart MD, Merino DM, Wempe MM, et al. Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer. 2019;58(8):578–88.
Hendriks LE, Rouleau E, Besse B. Clinical utility of tumor mutational burden in patients with non-small cell lung cancer treated with immunotherapy. Transl Lung Cancer Res. 2018;7(6):647–60.
Zhang L, Chen Y, Wang H, Xu Z, Wang Y, Li S, et al. Massive PD-L1 and CD8 double positive TILs characterize an immunosuppressive microenvironment with high mutational burden in lung cancer. J Immunother Cancer. 2021;9(6):002356.
Zhou Y, Xu Y, Gong Y, Zhang Y, Lu Y, Wang C, et al. Clinical factors associated with circulating tumor DNA (ctDNA) in primary breast cancer. Mol Oncol. 2019;13(5):1033–46.
Angus L, Smid M, Wilting SM, van Riet J, Van Hoeck A, Nguyen L, et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet. 2019;51(10):1450–8.
Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575(7781):210–6.
Davis AA, Jacob S, Gerratana L, Shah AN, Wehbe F, Katam N, et al. Landscape of circulating tumour DNA in metastatic breast cancer. EBioMedicine. 2020;58: 102914.
Daly B, Olopade OI. A perfect storm: How tumor biology, genomics, and health care delivery patterns collide to create a racial survival disparity in breast cancer and proposed interventions for change. CA Cancer J Clin. 2015;65(3):221–38.
Warner ET, Tamimi RM, Hughes ME, Ottesen RA, Wong YN, Edge SB, et al. Racial and ethnic differences in breast cancer survival: mediating effect of tumor characteristics and sociodemographic and treatment factors. J Clin Oncol. 2015;33(20):2254–61.
Tao Z, Li T, Feng Z, Liu C, Shao Y, Zhu M, et al. Characterizations of cancer gene mutations in chinese metastatic breast cancer patients. Front Oncol. 2020;10:1023.
Wang Y, Lin L, Li L, Wen J, Chi Y, Hao R, et al. Genetic landscape of breast cancer and mutation tracking with circulating tumor DNA in Chinese women. Aging (Albany NY). 2021;13(8):11860–76.
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1): a001008.
Silwal-Pandit L, Vollan HKM, Chin S-F, Rueda OM, McKinney S, Osako T, et al. TP53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin Cancer Res. 2014;20(13):3569–80.
Aubrey BJ, Strasser A, Kelly GL. Tumor-suppressor functions of the TP53 pathway. Cold Spring Harb Perspect Med. 2016;6(5):a026062.
Xiao W, Zhang G, Chen B, Chen X, Wen L, Lai J, et al. Characterization of frequently mutated cancer genes and tumor mutation burden in chinese breast cancer. Front Oncol. 2021;11: 618767.
Meric-Bernstam F, Zheng X, Shariati M, Damodaran S, Wathoo C, Brusco L, et al. Survival outcomes by TP53 mutation status in metastatic breast cancer. Jco Precis Oncol. 2018;2018:1–5.
Kodahl AR, Ehmsen S, Pallisgaard N, Jylling AMB, Jensen JD, Laenkholm AV, et al. Correlation between circulating cell-free PIK3CA tumor DNA levels and treatment response in patients with PIK3CA-mutated metastatic breast cancer. Mol Oncol. 2018;12(6):925–35.
Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, et al. Double mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science. 2019;366(6466):714–23.
Dey N, De P, Leyland-Jones B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol Ther. 2017;175:91–105.
Narayan P, Prowell TM, Gao JJ, Fernandes LL, Li E, Jiang X, et al. FDA approval summary: alpelisib plus fulvestrant for patients with HR-positive, HER2-negative, PIK3CA-mutated, advanced or metastatic breast cancer. Clin Cancer Res. 2021;27(7):1842–9.
Elfgen C, Reeve K, Moskovszky L, Guth U, Bjelic-Radisic V, Fleisch M, et al. Prognostic impact of PIK3CA protein expression in triple negative breast cancer and its subtypes. J Cancer Res Clin Oncol. 2019;145(8):2051–9.
Pascual J, Turner NC. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann Oncol. 2019;30(7):1051–60.
Martin M, Chan A, Dirix L, O’Shaughnessy J, Hegg R, Manikhas A, et al. A randomized adaptive phase II/III study of buparlisib, a pan-class I PI3K inhibitor, combined with paclitaxel for the treatment of HER2- advanced breast cancer (BELLE-4). Ann Oncol. 2017;28(2):313–20.
Liao H, Huang W, Pei W, Li H. Detection of ESR1 mutations based on liquid biopsy in estrogen receptor-positive metastatic breast cancer: clinical impacts and prospects. Front Oncol. 2020;10: 587671.
Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–45.
Li Q, Guan X, Chen S, Yi Z, Lan B, Xing P, et al. Safety, efficacy, and biomarker analysis of pyrotinib in combination with capecitabine in HER2-positive metastatic breast cancer patients: a phase i clinical trial. Clin Cancer Res. 2019;25(17):5212–20.
Rossi G, Mu Z, Rademaker AW, Austin LK, Strickland KS, Costa RLB, et al. Cell-Free DNA and circulating tumor cells: comprehensive liquid biopsy analysis in advanced breast cancer. Clin Cancer Res. 2018;24(3):560–8.
Stover DG, Parsons HA, Ha G, Freeman SS, Barry WT, Guo H, et al. Association of cell-free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple-negative breast cancer. J Clin Oncol. 2018;36(6):543–53.
Bourrier C, Pierga JY, Xuereb L, Salaun H, Proudhon C, Speicher MR, et al. Shallow whole-genome sequencing from plasma identifies FGFR1 amplified breast cancers and predicts overall survival. Cancers. 2020;12(6):1481.
Klempner SJ, Fabrizio D, Bane S, Reinhart M, Peoples T, Ali SM, et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist. 2020;25(1):e147–59.
Krasniqi E, Barchiesi G, Pizzuti L, Mazzotta M, Venuti A, Maugeri-Sacca M, et al. Immunotherapy in HER2-positive breast cancer: state of the art and future perspectives. J Hematol Oncol. 2019;12(1):111.
Touat M, Ileana E, Postel-Vinay S, Andre F, Soria JC. Targeting FGFR signaling in cancer. Clin Cancer Res. 2015;21(12):2684–94.
Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019;10(1):1373.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70.
Aftab A, Shahzad S, Hussain HMJ, Khan R, Irum S, Tabassum S. CDKN2A/P16INK4A variants association with breast cancer and their in-silico analysis. Breast Cancer. 2019;26(1):11–28.
Araki K, Miyoshi Y. Mechanism of resistance to endocrine therapy in breast cancer: the important role of PI3K/Akt/mTOR in estrogen receptor-positive, HER2-negative breast cancer. Breast Cancer. 2018;25(4):392–401.
Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer. 2017;3(1):39–55.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
HaL, JiaZ, TZ, and XL collected the samples, conducted the ctDNA sequencing, and wrote the manuscript. JianZ and BS collected the clinical data. XD, AW, PD, BK, and SJ conducted the ctDNA sequencing and data analysis. JY and HuL revised the manuscript. All authors had full access to all the data in the study and accept responsibility to submit for publication. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in this study involving human participants were in accordance with the ethical standards of the Medical Ethics Committee of Peking University Cancer Hospital (No. 2020KT75). All patients signed a written informed consent prior to registration for ctDNA testing.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Figure S1. (A) Comparisons of ctDNA fractions between drug-sensitive and drug-resistant patients. (B) Survival analyses between all patients with low and high ctDNA fractions. (C) Comparisons of bTMB between drug-sensitive and drug-resistant samples. The black lines represent the median of each group. ctDNA, circulating tumour DNA; bTMB, blood-based tumour mutation burden.
Additional file 2.
Figure S2. Survival analyses between (A) HR + patients, (B) HER2 + patients, and (C) TNBC patients with low and high ctDNA fractions. HR + , hormone receptor-positive; HER2 + , human epidermal growth factor receptor 2-positive; TNBC, triple-negative breast cancer; ctDNA, circulating tumour DNA.
Additional file 3.
Figure S3. Survival analyses between (A) HR + patients, (B) HER2 + patients, and (C) TNBC patients with low and high bTMB. (D) Comparison of mutations between plasma samples at baseline and disease progression. In the heatmap, the top bars depict the total number of mutations a patient carried at baseline and at disease progression. The bars below denote different IHC subtypes. Each column represents one patient. Every two rows represent the status of one gene at baseline (upper) and at disease progression (lower). The text on the left represents gene names and sampling time points. The values on the right represent the mutation rates of these genes. Different colours of squares denote different types of mutations. Red represents SNVs/Indels, blue represents amplifications, and green represents deletions. HR + , hormone receptor-positive; HER2 + , human epidermal growth factor receptor 2-positive; TNBC, triple-negative breast cancer; bTMB, blood-based tumour mutation burden; IHC, immunohistochemistry; SNVs, single nucleotide variations; Indels, insertions and deletions.
Additional file 4.
Figure S4. Correlation between the ctDNA fraction and number of mutations. (A) Pearson correlation coefficient between the ctDNA fraction and number of mutations in 141 baseline plasma samples. The X-axis represents the ctDNA fraction, and the Y-axis represents the number of mutations. The blue slash indicates the correlation trend between the two sets of data. (B) Comparison of the number of mutations among samples with different ctDNA fractions. The X-axis represents the three groups divided according to ctDNA fraction and the Y-axis represents the number of mutations.
Additional file 5
. Original sequencing data including IHC type of all patients, SNVs, CNVs, and ctDNA fraction and bTMB of baseline plasma, SNVs and CNVs of plasma samples used for disease monitoring, and SNVs and CNVs of tissue samples. IHC, immunohistochemistry; SNVs, single nucleotide variations; CNVs, copy number variations; bTMB, blood-based tumor mutation burden.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liao, H., Zhang, J., Zheng, T. et al. Identification of mutation patterns and circulating tumour DNA-derived prognostic markers in advanced breast cancer patients. J Transl Med 20, 211 (2022). https://doi.org/10.1186/s12967-022-03421-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-022-03421-8