
Abstract
Background
Biomarkers for distinguishing psoriatic arthritis (PsA) from psoriasis without arthritis (PsO) are still lacking.
Methods
We applied isobaric tags for relative and absolute quantification (iTRAQ) and LC–MS/MS to analyze the proteome profile of peripheral blood mononuclear cells (PBMCs) collected from patients with PsO, patients with PsA, and healthy controls. Bioinformatics analysis and western blotting were performed to identify and validate differentially expressed proteins.
Results
We identified 389, 199, 291, and 60 significantly differentially expressed proteins (adj.p < 0.05) in the comparison of all psoriatic patients versus healthy controls, PsO group versus healthy controls, PsA group versus healthy controls, and PsA group versus PsO group, respectively. Among these proteins, 14 proteins may represent promising biomarkers for PsA: SIRT2, NAA50, ARF6, ADPRHL2, SF3B6, SH3KBP1, UBA3, SCP2, RPS5, NUDT5, NCBP1, SYNE1, NDUFB7, HTATSF1. Furthermore, western blotting confirmed that SIRT2 expression was significantly higher in PBMCs from PsA patients than PsO and healthy controls, and was negatively correlated with the phosphorylation of p38 mitogen-activated protein kinase (p-p38MAPK; p = 0.006, r = − 0.582).
Conclusions
This pilot study provided a broad characterization of the proteome of PBMCs in PsA as compared to PsO and healthy controls, which may help to provide prospective strategies for PsA diagnosis.
Similar content being viewed by others


Background
Psoriasis (PsO) is a common immune-mediated inflammatory disease mediated by dendritic and T cells, as key cell types, and type I interferons (TNF-α, IL-17, and IL-23), as key inflammatory cytokines [1]. Approximately 30% of patients with PsO finally develop psoriatic arthritis (PsA) [2], and they may experience a more significant reduction in quality of life [3]. As many as 29% of PsO patients attending dermatology practices might have undiagnosed PsA, and the diagnosis of early PsA remains a challenge for dermatologists [4]. Since a diagnostic delay of > 6 months may contribute to poor functional and radiographic outcomes, it is critical to detect biomarkers that can facilitate the diagnosis of PsA and predict therapeutic outcomes.
Almost 50% of PsA patients have a positive family history. In comparison to PsO, a cohort of PsA patients showed considerable difference in their predominant HLA alleles [5], indicating that genetics plays a major role in the predisposition to develop PsA. Despite a well-defined genetic cause, there is a growing consensus that because of splicing and protein modifications, systems-level approaches, including the analysis of proteomes, are now required for a more comprehensive understanding of the causes of PsA [6].
Proteomics, as the gene products, has been continuously developed for simultaneous identification and quantification of proteins associated with psoriasis and psoriatic arthritis, especially in the detection of various diagnostic markers for distinguishing PsA from PsO. In 2015, Cretu et al. [7]. first applied quantitative tandem mass-spectrometry to compare PsO and PsA; they identified 47 proteins that were elevated in the skin of 10 patients with PsA compared to that of 10 patients with PsO. Then, serum concentration of CXCL10, measured by Luminex assay, was reported to increase significantly after conversions from PsO to PsA [4] while Reindl et al. demonstrated that only the combination of cartilage oligomeric matrix protein (COMP) together with haptoglobin (HP) in plasma appeared to be appropriate for diagnosing PsA [8]. Furthermore, Matsuura et al. applied MALDI-TOF MS and Triple-TOF MS/MS to serum peptide profiles and found that the ion intensities of TMSB4X protein-derived peptides with 4964 m/z (p4964) and 4979 m/z (p4979) were 1.95- and 1.67-fold lower in the psoriatic arthritis group than their respective ion intensities in the psoriasis vulgaris group [9].
Peripheral blood mononuclear cell (PBMCs) are suitable samples for searching for biomarkers with proteomic techniques since they are easy to obtain and more stable than free-circulating plasma or serum. Nevertheless, the proteome profiling of PBMCs in patients with PsO and those with PsA remains to be elucidated. In the present study, as part of a relative and absolute quantitation (iTRAQ)-based quantitative proteomic approach, isobaric tags were employed to measure proteome changes of PBMCs in PsO and PsA groups, as well as healthy controls. The differentially expressed proteins were further analyzed by bioinformatics analysis and validated by Western blot.
Methods
Subjects
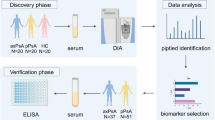
A total of 33 blood samples were collected, including 12 samples for proteomics analysis (PsO, n = 4; PsA, n = 4; healthy controls (HC), n = 4), and 21 samples for western blotting analysis (PsO, n = 8; PsA, n = 8; HC, n = 5) at the Huashan Hospital, Fudan University. The diagnosis of psoriasis was based on typical clinical and/or histopathological criteria. PsA patients had psoriasis and satisfied the CASPAR classification criteria [10]. Healthy controls were sex-and age-matched with patients. The study was conducted in accordance with the Declaration of Helsinki. The Medical Ethics Committee of Huashan Hospital, Fudan University, reviewed and approved the protocol, and all participants provided written informed consent. 15 ml of peripheral venous blood were collected by venipuncture into EDTA-coated tubes. The peripheral blood mononuclear cells (PBMCs) were isolated via density-gradient centrifugation method using Lymphoprep™ (LMP, Stemcell Technologies, Cambridge, UK) according to the manufacturer’s instructions. All PBMCs samples were frozen in liquid nitrogen and stored at − 80 °C. The workflow is shown in Fig. 1. More detailed procedures on the sample preparation, iTRAQ Labeling, High pH reverse phase fractionation (HPRP), and LC–MS/MS analysis could be found in Additional file 5.

Experimental workflow for iTRAQ labeling proteome analysis. Equal amounts of proteins from 12 samples were digested with trypsin. The resultant peptides were subjected to iTRAQ labeling, HPRP fractionation, and subsequent LC–MS/MS analysis
Western blotting
21 PBMCs samples for western blotting analysis were washed with cold PBS and lysed in RIPA lysis buffer (Beyotime, China) supplemented with protease and phosphatase inhibitors and PMSF (Beyotime, China). Protein concentrations of samples were determined by the BCA assay (Beyotime, China). The protein (40 μg/lane) were separated by 10% SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membrane (Millipore, USA). Membranes were blocking with 5% skim milk for 2 h and then incubated at 4 °C overnight with the different primary antibodies respectively. The antibodies were rabbit anti-SIRT2 (ab211033, 1:2000 dilutions; abcam), rabbit anti-phospho-P38 (# 4511, 1:1000 dilutions; CST), rabbit anti- GAPDH (# 5174, 1:1000 dilutions, CST, set as additional loading controls). Membranes were then incubated with the secondary fluorescent antibody (Beyotime, China) at 37 °C for 1 h. The blotting bands were analyzed using the Odyssey Infrared Imaging system (LI-COR, USA).
Statistical analysis
The limma method was performed to detect the difference of PBMCs proteome profiling among patients with PsO, patients with PsA, and healthy controls. Limma is a highly recommended tool for comparative proteomics analysis of data sets with low replicate numbers, which not only outperforms the standard t test in terms of number of differentially regulated features but also maintains the right estimate for false positives for sufficiently large feature numbers. Protein fold changes (on log scale) were computed and differential expression P values corrected for multiple testing using the Benjamini-Hochberg [false discovery rate (FDR)] method. Proteins with an adjusted P value of < 0.05 were considered as significantly differentially expressed between groups. All computations were performed in R statistical programming environment (version 3.6.3). Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway analysis was performed using DAVID Bioinformatics Resources version 6.8. Protein–protein interaction analysis was performed using STRING Version 11.0 (https://string-db.org/) and GeneMania (Version v3.5.2, https://genemania.org) [11]. The score of minimum required interaction in STRING was medium confidence (0.400).
Results
Patient characteristics
As shown in Additional file 1: Table S1, we recruited eight patients with psoriasis (four with PsO and four with PsA, five men, three women; mean age 55.5 years, range 36–86 years) and four age- sex-matched healthy controls for proteomics analysis. The average PASI score was 14.3 (range 9.3–22.5), and the average BSA score was 33.1 (range 13.0–53.0). Two patients had a history of hypertension; one patient had a history of diabetes mellitus; two patients had a history of hyperlipidemia; and four patients reported a history of smoking. As shown in Additional file 2: Table S2, 16 patients (8with PsO and 8 with PsA) and age-sex-matched healthy controls was recruited for western blotting validation of SIRT2 protein expression (Additional file 3: Figure S1).
Potential biomarkers for psoriatic arthritis
A total of 22,734 peptides and 3177 proteins were identified by iTRAQ and LC–MS/MS (false discovery rate, < 1%). After using limma for differential protein expression analysis, we identified 389 significantly changed proteins when comparing eight patients with psoriasis (PsO and PsA) and four healthy controls (HC). Similarly, we found 199, 291, and 60 significantly changed proteins when comparing PsO and HC, PsA and HC, and PsA and PsO, respectively. The list of all above significantly changed proteins including fold change and adjusted P values was uploaded as Additional file 4. By performing the Venn diagram of three significantly changed proteins group sets in Fig. 2, 14 proteins meeting the following criteria were marked in red: (1) in the overlapping section of “PsA vs. HC” or “PsA vs. PsO”; and (2) not in the section of “PsO vs. HC”. These were SIRT2, NAA50, ARF6, ADPRHL2, SF3B6, SH3KBP1, UBA3, SCP2, RPS5, NUDT5, NCBP1, SYNE1, NDUFB7, HTATSF1 (Table 1). Most of them were up-regulated in PsA (FC > 1), while RPS5 and HTATSF1 were down-regulated (FC > 1).

Venn diagram depicted the overlapping of significantly expressed proteins in the three datasets (PsO group versus HC group, PsA group versus HC group, PsA group versus PsO group). The red section included 14 proteins (protein SIRT2, NAA50, ARF6, ADPRHL2, SF3B6, SH3KBP1, UBA3, SCP2, RPS5, NUDT5, NCBP1, SYNE1, NDUFB7, HTATSF1) that met the criteria: (1) in the overlapping section of “PsA vs. HC” and “PsA vs. PsO”; (2) not in the section of “PsO vs. HC”
Volcano plots of four significantly changed protein group sets were generated in Fig. 3A–D. In addition, we investigated the relationship of differentially expressed proteins in the PsA group versus the PsO group using GeneMania (Version v3.5.2, https://genemania.org) and STRING Version 11.0 (https://string-db.org/). (Fig. 3E, F). According to GeneMania, we identified several relationships among these proteins at the gene expression level, including co-expression (46.00%), physical interaction (33.06%), genetic interactions (10.05%), and co-localization (1.09%). In addition, according to STRING, 12 of the 60 proteins did not connect to any type of network (interaction score = 0.4), and none of the 12 proteins represent in the 14 candidate biomarkers. Forty-eight of the differentially expressed proteins were connected to networks by complex relationships.

Volcano plot (fold change and significance) of protein expression changes in different group sets. A All patients with psoriasis versus HC group, B PsO group versus HC group, C PsA group versus HC group, and D PsA group versus PsO group. The fold change of all group sets were all log2 transformed and plotted against their -log10 transformed p-values. Each spot represents an identified protein. The threshold p = 0.05 is indicated as a horizontal line; the threshold of a ratio of fold change is indicated by vertical lines. The relationship of differentially expressed proteins in the PsA group versus PsO group was shown using E GeneMANIA and F STRING
Bioinformatics analysis of the differentially expressed proteins
To understand the biological characterization of differentially expressed proteins in four significantly changed proteins group sets, we performed KEGG pathway analysis using DAVID Bioinformatics Resources version 6.8 (Fig. 4).

KEGG pathway analysis of protein expression changes in different group sets. A Psoriasis group versus HC group, B PsO group versus HC group, C PsA group versus HC group, and D PsA group versus PsO group. The p-values of each pathway were all − log10 transformed
Comparing all eight patients with psoriasis (PsO and PsA) and four healthy controls, top 10 highly enriched terms included ribosome, antigen processing and presentation, spliceosome, Alzheimer’s disease, RNA transport, protein processing in endoplasmic reticulum, endocytosis, oxidative phosphorylation, glycolysis/gluconeogenesis, carbon metabolism. In addition, KEGG pathway analysis also revealed several essential pathways, including spliceosome, oxidative phosphorylation, endocytosis, phagosome, and protein export when comparing patients with PsA and PsO.
Western blotting validation of SIRT2 protein expression
To further investigate the expression of SIRT2 in PBMCs from patients with PsA and PsO, we compared the level of SIRT2 and the phosphorylation of p38 mitogen-activated protein kinase (p-p38MAPK) in 8 patients with PsO, 8 patients with PsA, and 5 healthy controls by western blot. Representative results were shown in Fig. 5A. As shown in Fig. 5B, SIRT2 expression was significantly higher in PsA compared with PsO (p = 0.024) and healthy controls (p = 0.001). Furthermore, the expression of SIRT2 was negatively correlated with the phosphorylation of p38MAPK (p = 0.006, r = − 0.582; Fig. 5C).

Western blotting validation of SIRT2 expression and its correlation with the phosphorylation of p38MAPK. A Representative western blot analysis of SIRT2, p-p38MAPK, and GAPDH as loading controls. B Relative SIRT2 protein expression was normalized to GAPDH. HC, healthy controls, n = 5; PsO, n = 8; PsA, n = 8. Data was expressed mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001. C The correlation between SIRT2 and p-p38MAPK was shown in all 21 samples
Discussion
In this study, we adopted iTRAQ technology in combination with bioinformatics analysis and western blotting to investigate the proteome in PBMCs of patients with PsA and patients with PsO as well as healthy controls. PBMCs samples could be easily collected from patients by a minimally-invasive intervention, and our study represents the first proteomic research to use PBMCs as samples for exploring the different expressed proteins between patients with PsA and PsO. Taken differential proteins analysis performed by limma and Venn diagram results, we finally narrowed down the list of candidate biomarkers that distinguish PsA from PsO.
In the present study, proteomics analysis showed SIRT2 expression in PBMCs from PsA patients was significantly higher than PsO patients and healthy controls, and this finding was further validated by western blotting. SIRT2 is a member of the sirtuin protein family, which is localized primarily in the cytoplasm and can also shuttle to the nucleus [12]. SIRT2 has been implicated in the progress of inflammation in arthritis. A previous study demonstrated that SIRT2 mRNA expression is increased in active RA compared to inactive RA [13]. In addition, SIRT2 expression was found to be negatively correlated with the phosphorylation of p38MAPK in this study, which is consistent with the finding of Kim et al. [14] that SIRT2 suppressed the expression of pro-inflammatory cytokines and inhibited phosphorylation of p38MAPK in murine macrophages cells. However, SIRT2 was reported to promote the phosphorylation of p38MAPK in activated NK cells [15], kidney and tubular epithelial cells [16], and rabbit articular chondrocytes [17]. The contradictory effect of SIRT2 on the phosphorylation of p38MAPK needs to be further elucidated.
In the present study, KEGG pathway analysis of protein expression changes in the Ps (namely PsO + PsA) group versus HC group, PsA group versus HC group, and PsO group versus HC group all revealed differences in the ribosome pathway. Ribosomal protein S6, which is both a key mediator of mTOR function and coordinate regulator of ribosome function and biogenesis [18], is hyperactivated and abnormally phosphorylated in epidermal lesions of patients with psoriasis [19]. A quantitative proteomic analysis of psoriasis vulgaris also revealed that the differentially expressed proteins of skin lesions biopsies were mainly enriched in ribosomal pathway [20], supporting our finding in PBMCs.
The present study has several limitations. The major limitation is the small sample size and lack of control for differences between the sexes. While data from our small cohort generated distinctive and actionable data streams, it is clear that further validation of these preliminary findings in larger cohorts is necessary. Additionally, different PsA subtypes were not taken into consideration. Moll and Wright described five clinical subtypes of PsA in 1973 [21]. In this study, all patients with PsA fulfilled the CASPAR criteria for the diagnosis, and we decided not to classify PsA according to phenotypes for two reasons: (i) the phenotypes of PsA have been shown to change over time [22]; (ii) the enrolled PsA patients, consisting of the distal subtype, the oligoarticular subtype, the polyarticular subtype, and the spondylitis subtype, all showed the similar high expression level of SIRT2 in PBMCs.
Conclusions
In conclusion, we developed iTRAQ-based quantitative proteomic approach to measure PBMCs protein expression profiling of PsA and PsO patients, and confirmed the higher expression of SIRT2 in PsA in an independent western blotting validation cohort. Although concerning a proof-of-concept study with limited sample size, these data provide a stepping-stone for follow-up research on the validation of the discovered biomarker candidates.
Availability of data and materials
Reasonable requests for data will be made available for review.
Abbreviations
- ACOT9:
-
Acyl-coenzyme A thioesterase 9
- ADD3:
-
Gamma-adducin
- ADPRHL2:
-
ADP-ribosylhydrolase like 2
- ARF6:
-
ADP-ribosylation factor 6
- BSA:
-
Body surface area
- CASPAR:
-
Classification criteria for psoriatic arthritis
- COMP:
-
Cartilage oligomeric matrix protein
- COMT:
-
Catechol O-methyltransferase
- COX7A2:
-
Cytochrome C oxidase subunit VIIa polypeptide 2
- CXCL10:
-
C–X–C motif chemokine 10
- FASP:
-
Filter assisted sample preparation
- FDR:
-
False discovery rate
- HC:
-
Healthy control
- HLA:
-
Human leukocyte antigen
- HP:
-
Haptoglobin
- HPRP:
-
High pH reverse phase fractionation
- HTATSF1:
-
HIV Tat-specific factor 1
- IAA:
-
Iodoacetamide
- IL-17:
-
Inter-leukin 17
- ITGA5:
-
Integrin alpha-5
- iTRAQ:
-
Isobaric tags for relative and absolute quantification
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LC–MS/MS:
-
Liquid chromatography tandem mass spectrometry
- MALDI-TOF MS:
-
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- MHC:
-
Major Histocompatibility Complex
- MMP:
-
Matrix metalloproteinases
- NAA50:
-
N-Alpha-acetyltransferase 50
- NAFLD:
-
Non-alcoholic fatty liver disease
- NDUFB7:
-
NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 7
- NUDT5:
-
ADP-sugar pyrophosphatase
- OGDH:
-
2-Oxoglutarate dehydrogenase
- PASI:
-
Psoriasis Area Severity Index
- PBMCs:
-
Peripheral blood mononuclear cells
- PsA:
-
Psoriasis with psoriasis arthritis
- PsO:
-
Psoriasis without psoriasis arthritis
- ROS:
-
Reactive oxygen species
- RP-HPLC:
-
Reverse-phase high-performance liquid chromatography
- RPLC:
-
Reversed-phase liquid chromatography
- RPS5:
-
Ribosomal protein S5
- SCP2:
-
Non-specific lipid-transfer protein
- SD:
-
Standard deviation
- SIRT2:
-
NAD-dependent protein deacetylase sirtuin-2
- TEAB:
-
Tetraethyl-ammonium bromide
- TLR:
-
Toll-like receptor
- TMSB4X:
-
Thymosin beta-4
- TNF-α:
-
Tumor necrosis factor-α
- Triple-TOF MS/MS:
-
Triple time-of-flight mass spectrometry
- TXNRD1:
-
Thioredoxin reductase 1
- UBA3:
-
NEDD8-activating enzyme E1 catalytic subunit
- UBE2V2:
-
Ubiquitin-conjugating enzyme E2 variant 2
References
Di Meglio P, Villanova F, Nestle FO. Psoriasis. Cold Spring Harb Perspect Med. 2014;4:a015354.
Barnas JL, Ritchlin CT. Etiology and pathogenesis of psoriatic arthritis. Rheum Dis Clin North Am. 2015;41:643–63.
Cretu D, Gao L, Liang K, Soosaipillai A, Diamandis EP, Chandran V. Differentiating psoriatic arthritis from psoriasis without psoriatic arthritis using novel serum biomarkers. Arthritis Care Res (Hoboken). 2018;70:454–61.
Abji F, Pollock RA, Liang K, Chandran V, Gladman DD. Brief report: CXCL10 is a possible biomarker for the development of psoriatic arthritis among patients with psoriasis. Arthritis Rheumatol. 2016;68:2911–6.
FitzGerald O, Haroon M, Giles JT, Winchester R. Concepts of pathogenesis in psoriatic arthritis: genotype determines clinical phenotype. Arthritis Res Ther. 2015;17:115.
Altelaar AF, Munoz J, Heck AJ. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet. 2013;14:35–48.
Cretu D, Liang K, Saraon P, Batruch I, Diamandis EP, Chandran V. Quantitative tandem mass-spectrometry of skin tissue reveals putative psoriatic arthritis biomarkers. Clin Proteomics. 2015;12:1.
Reindl J, Pesek J, Kruger T, Wendler S, Nemitz S, Muckova P, Buchler R, Opitz S, Krieg N, Norgauer J, Rhode H. Proteomic biomarkers for psoriasis and psoriasis arthritis. J Proteomics. 2016;140:55–61.
Matsuura T, Sato M, Nagai K, Sato T, Arito M, Omoteyama K, Suematsu N, Okamoto K, Kato T, Soma Y, Kurokawa MS. Serum peptides as putative modulators of inflammation in psoriasis. J Dermatol Sci. 2017;87:36–49.
Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H, Group CS. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum. 2006;54:2665–73.
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214-220.
North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–44.
Kara M, Yolbas S, Sahin C, Koca SS. Changes in sirtuin 2 and sirtuin 3 mRNA expressions in rheumatoid arthritis. Eur J Rheumatol. 2017;4:83–6.
Kim MJ, Kim DW, Park JH, Kim SJ, Lee CH, Yong JI, Ryu EJ, Cho SB, Yeo HJ, Hyeon J, et al. PEP-1-SIRT2 inhibits inflammatory response and oxidative stress-induced cell death via expression of antioxidant enzymes in murine macrophages. Free Radic Biol Med. 2013;63:432–45.
Chen M, Xu M, Zhu C, Wang H, Zhao Q, Zhou F. Sirtuin2 enhances the tumoricidal function of liver natural killer cells in a mouse hepatocellular carcinoma model. Cancer Immunol Immunother. 2019;68:961–71.
Jung YJ, Park W, Kang KP, Kim W. SIRT2 is involved in cisplatin-induced acute kidney injury through regulation of mitogen-activated protein kinase phosphatase-1. Nephrol Dial Transplant. 2020;35:1145–56.
Eo SH, Kim DW, Choi SY, Kim HA, Kim SJ. PEP-1-SIRT2 causes dedifferentiation and COX-2 expression via the MAPK pathways in rabbit articular chondrocytes. Exp Cell Res. 2015;339:351–9.
Jastrzebski K, Hannan KM, Tchoubrieva EB, Hannan RD, Pearson RB. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors. 2007;25:209–26.
Ruf MT, Andreoli A, Itin P, Pluschke G, Schmid P. Ribosomal protein S6 is hyperactivated and differentially phosphorylated in epidermal lesions of patients with psoriasis and atopic dermatitis. Br J Dermatol. 2014;171:1533–6.
Li Y, Lin P, Wang S, Li S, Wang R, Yang L, Wang H. Quantitative analysis of differentially expressed proteins in psoriasis vulgaris using tandem mass tags and parallel reaction monitoring. Clin Proteomics. 2020;17:30.
Moll JM, Wright V. Psoriatic arthritis. Semin Arthritis Rheum. 1973;3:55–78.
Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. 2017;376:957–70.
Acknowledgements
Not applicable.
Funding
This research was funded by the National Natural Science Foundation of China (Nos. 81773322, 81673054), the KeyProject in Basic Research Advocated by Shanghai Science and Technology Commission (13JC1402300), and Clinical Research Plan of SHDC (Nos. SHDC2020CR6022, SHDC2020CR1014B). The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manu-script; or in the decision to publish the results.
Author information
Authors and Affiliations
Contributions
Conceptualization-JZ, LH, and KXY; methodology-JZ and RLL; software-KY and GQH; validation-JZ and LH; formal analysis-JZ; investigation-JZ and RLL; resources-HD and KXY; data curation-JZ and QH; writing original draft preparation-JZ; writing review and editing-NY and KXY; visualization-JZ; supervision-ZHZ, ZZZ, XF, HD and KXY; project administration-LH, HD and KXY; funding acquisition-ZHZ, HD and KXY. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The Medical Ethics Committee of Huashan Hospital, Fudan University, reviewed and approved the protocol, and all participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Characteristics of patients for proteomics analysis.
Additional file 2: Table S2.
Characteristics of patients for western blotting analysis.
Additional file 3: Figure S1.
Raw Western blotting images for Fig.5 in manuscript. (A) Raw western blotting image for GAPDH (used as a loading control, 37 kDa); (B) Raw western blotting image for p-p38 (43 kDa); (C) Raw western blotting image for SIRT2 (43 kDa).
Additional file 4.
The list of significantly changed proteins in the comparison of all psoriatic patients versus healthy controls, PsO group versus healthy controls, PsA group versus healthy controls, and PsA group versus PsO group, respectively.
Additional file 5.
Sample processing of PBMCs for proteomics analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, J., Han, L., Liu, R. et al. Identification of proteins associated with development of psoriatic arthritis in peripheral blood mononuclear cells: a quantitative iTRAQ-based proteomics study. J Transl Med 19, 331 (2021). https://doi.org/10.1186/s12967-021-03006-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-021-03006-x