
Abstract
Background
Growing evidence has suggested that immune-related genes play crucial roles in the development and progression of hepatocellular carcinoma (HCC). Nevertheless, the utility of immune-related genes for evaluating the prognosis of HCC patients are still lacking. The study aimed to explore gene signatures and prognostic values of immune-related genes in HCC.
Methods
We comprehensively integrated gene expression data acquired from 374 HCC and 50 normal tissues in The Cancer Genome Atlas (TCGA). Differentially expressed genes (DEGs) analysis and univariate Cox regression analysis were performed to identify DEGs that related to overall survival. An immune prognostic model was constructed using the Lasso and multivariate Cox regression analyses. Furthermore, Cox regression analysis was applied to identify independent prognostic factors in HCC. The correlation analysis between immune-related signature and immune cells infiltration were also investigated. Finally, the signature was validated in an external independent dataset.
Results
A total of 329 differentially expressed immune‐related genes were detected. 64 immune‐related genes were identified to be markedly related to overall survival in HCC patients using univariate Cox regression analysis. Then we established a TF-mediated network for exploring the regulatory mechanisms of these genes. Lasso and multivariate Cox regression analyses were applied to construct the immune-based prognostic model, which consisted of nine immune‐related genes. Further analysis indicated that this immune-related prognostic model could be an independent prognostic indicator after adjusting to other clinical factors. The relationships between the risk score model and immune cell infiltration suggested that the nine-gene signature could reflect the status of tumor immune microenvironment. The prognostic value of this nine-gene prognostic model was further successfully validated in an independent database.
Conclusions
Together, our study screened potential prognostic immune-related genes and established a novel immune-based prognostic model of HCC, which not only provides new potential prognostic biomarkers and therapeutic targets, but also deepens our understanding of tumor immune microenvironment status and lays a theoretical foundation for immunotherapy.
Similar content being viewed by others
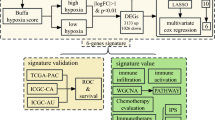


Background
Globally, liver cancer is known as the sixth leading cancer, and has the second-highest number of deaths [1]. All over the world, more than 600,000 people die of liver cancer each year, and nearly 850,000 new cases occur [1, 2]. Hepatocellular carcinoma (HCC) accounts for 85–90% of all liver cancers and has received public attention. Despite rapid advances in new tests and treatments, the 5-year survival rate for HCC is still less than one in five [3]. At present, surgery is still the main treatment for early liver cancer [4]. However, a significant proportion of patients will have postoperative recurrence or distant metastasis [5]. Recently, drugs such as sorafenib and regorafenib have been shown to be effective against advanced HCC [6, 7]. It is worth mentioning that patients with the same pathological type and clinical stage often have different outcomes after the same treatment, which is mainly due to the genetic heterogeneity of patients [8].
The immune system is thought to be a decisive factor in the development of cancer [9, 10], including HCC. Immune cells are major components of the tumor microenvironment and play a role in many key steps of HCC development from tumor growth to the development of metastasis [11, 12]. Besides, a large amount of inflammatory mediators were found to be associated with HCC development. IL-22, belongs to the cytokine family, was overexpressed in the HCC microenvironment and leading to tumor growth [13]. Chemokines (e.g. CXCL12, CCL20), as the immune molecules members of immune system, also play a crucial role in HCC growth, invasion and metastasis [14, 15]. It is proven that immunogenicity makes the immunotherapy of HCC a promising prospect [16]. Research progress discovery that programmed cell death-1 (PD-1) pathway is a new target for HCC immunotherapy [17]. As an anti-PD-1 monoclonal antibody, nivolumab can block PD-1 and restore the body’s anticancer immune response by interfering with the signaling pathway, thereby preventing T cell activation [18]. In HCC, nivolumab showed significant benefits in objective response rates and overall survival [19]. Therefore, nivolumab may provide a safe, effective and promising treatment for HCC [20]. Increasing studies have suggested that immune-related genes in HCC are closely related to the tumorigenesis and development of HCC [21]. However, there is currently no prognostic model based on immune-related genes to systematically evaluate tumor immune environment and predict the overall prognosis of HCC patients. Therefore, the construction of an immune-based prognosis model that can reliably predict HCC prognosis is of great clinical significance.
In the first step of this study, we screened differentially expressed immune-related genes closely related to HCC through bioinformatics analysis of large-scale sequencing database. Next, immune‐related genes significantly related to prognosis were further detected. Then we constructed an immune-related prognostic model by integrating immune-related genes for HCC. Moreover, the prognostic value of our immune-related prognostic model was further validated in an independent International Cancer Genome Consortium (ICGC) database. We here aimed to provide novel biomarkers that would be effective in predicting the prognosis and monitoring tumor immune microenvironment in HCC patients.
Methods
Data collection
Gene expression data and clinical information of HCC samples were acquired from TCGA data portal (https://portal.gdc.cancer.gov/cart; up to September 16, 2019). Processed RNA-Seq FPKM data of 374 HCC and 50 adjacent normal HCC tissues were downloaded for further analyses. After careful search and examination, 224 HCC patients were accompanied by hepatitis B virus. This included 81 HBsAg positive patients, 60 HBsAg and HBsAb both positive patients, and 83 patients whose history risk factors were hepatitis B. The International Cancer Genome Consortium (ICGC; https://dcc.icgc.org/search?filters=%7B%22donor%22:%7B%22projectId%22:%7B%22is%22:%5B%22LIRI-JP%22%5D%7D,%22availableDataTypes%22:%7B%22is%22:%5B%22exp_seq%22%5D%7D%7D%7D) was a web-based portal that provided comprehensive molecular genetic profiles of 50 different tumor types. ICGC represents a valuable database for analyzing cancer genome at the genomic and transcriptomic levels. For validation cohort, gene expression data and the corresponding survival information of 231 HCC patients were retrieved from the ICGC database. We download 1811 immune-related genes via the Immunology Database and Analysis Portal (ImmPort; https://www.immport.org/shared/genelists) database, which contains 17 immune categories based on various molecular function [22]. The cistrome Cancer (http://cistrome.org/CistromeCancer/CancerTarget/) represents a useful database for biomedical and genetic research and includes totally 318 transcription factors (TFs) [23]. In order to investigate the regulatory mechanism of immune-related genes, we extracted these TFs for subsequent research. Because our data were downloaded directly from public databases and we strictly abided by the publishing guidelines provided by TCGA and ICGC, there were no requirement for ethical approvals.
Differential expression analyses
The differentially expressed immune‐related genes and TFs in HCC and normal tissues were detected using the Wilcoxon test method in R. |log2 foldchange| > 1 and FDR < 0.05 were considered as significant. Heatmaps were generated using pheatmap package and volcano plots were also conducted in R software. To assess the potential biologic functions of differentially expressed immune‐related genes, Gene Ontology (GO) [24] and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis [25] were performed by the cluster Profiler package [26] in R. Functional categories with a adjusted P value < 0.05 were considered as significant pathways.
Survival analysis
Only patients with a follow-up time less than 2000 days were used for the survival analyses. To investigate the prognostic value of differentially expressed immune‐related genes in HCC patients, univariate Cox analysis was implemented by the survival package. Only these genes with a P value < 0.01 were considered as prognostic immune‐related genes. These prognostic immune‐related genes were further analyzed by GO and KEGG analysis. To evaluate how TFs regulating these immune‐related genes, we first screened prognosis-related TFs using univariate Cox analysis with a P value < 0.01 Then the correlation test between prognosis-related TFs and prognostic immune‐related genes was investigated. This step was performed using cor.test function in R, whose core method was Pearson test. The correlation coefficient and P value were calculated by cor.test. The cut-off criteria were set as correlation coefficient > 0.5 and P < 0.05. In order to make the picture clear, we only chose TFs that regulated more than nine immune‐related genes. Cytoscape was utilized for constructing and visualizing the regulatory network [27].
Construction of the immune-related signature for HCC
To develop a prognostic model, Lasso and multivariate Cox regression analyses were utilized to assess the relationship between prognostic immune‐related genes expressions and overall survival (OS). To avoid over-fitting and delete highly related genes, Lasso Cox regression was carried out using survival and glmnet package. Genes detected via Lasso algorithm were evaluated by step wise multivariate Cox regression analysis. Risk scores were acquired based on genes expression multiplied a linear combination of regression coefficient obtained from the multivariate Cox regression. Patients were assigned to high risk and low risk groups according to the median risk score. The Kaplan–Meier analysis was performed to compare overall survival between high risk and low risk groups via survival package in R. The receiver operating characteristic (ROC) curve was implemented by the R software package survival ROC. In addition, univariate and multivariate analyses were utilized to assess the effect of risk scores on overall survival and several clinical features.
Correlation analysis between immune-related signature and immune cells infiltration
To explore the associations between prognostic model and immune cells infiltration, we employed Tumor Immune Estimation Resource (TIMER) [28], a useful resource for comprehensive analysis of tumor-infiltrating immune cells. TIMER algorithm allows users to estimate the composition of six tumor-infiltrating immune cells subsets (B cells, CD4+ T cells, CD8+ T cells, macrophages, neutrophils, and dendritic cells). The immune infiltrate levels of HCC patients were derived from TIMER website and the correlation between the prognostic model and six tumor-infiltrating immune cells were conducted in R.
Genetical alteration of the immune-related signature
The cBio Cancer Genomics Portal (CBioPortal) represents an important online platform for visualization and analysis of various cancer genomics data [29, 30]. CBioPortal was conducted to analyse genetic alterations of prognostic genes in HCC patients (TCGA, Provisional). Anti-cancer drugs that target these genes were also identified.
External validation of the immune-related signature
To verify the prognostic value of immune-related signature risk score model, we used the ICGC database as the validation cohort. The same formula was used to calculate risk scores and patients were classified into high risk and low risk groups based on the optimal cut-off point. Kaplan–Meier and ROC curve analyses were carried out as described above.
Statistical analysis
All analyses were performed using Rversion 3.5.1. Unless otherwise noted, P < 0.05 was considered to be significant.
Results
Differentially expressed immune‐related genes and TFs in HCC
A total of 329 immune‐related genes (267 upregulated and 62 downregulated) and 117 TFs (108 upregulated and 9 downregulated) were identified as differentially expressed in HCC tissues compared with normal tissues. The heat maps revealed that HCC samples can be obviously distinguished from the normal samples according to differentially expressed immune‐related genes and TFs (Fig. 1a, b). Volcano plots shows the distribution of differentially expressed immune‐related genes and TFs between HCC and normal controls (Fig. 1c, d). The 329 differentially expressed immune‐related genes were further analyzed by GO and KEGG analysis. GO analysis revealed that primary functional categories in the biological processes (BP) were leukocyte migration, positive regulation of cytokine production and positive regulation of defense response. For cellular components (CC), the major enriched GO terms were receptor complex and external side of plasma membrane. The most enriched cellular components (CC) were receptor ligand activity, cytokine activity and cytokine receptor binding (Fig. 2a). KEGG pathway indicated that the differentially expressed immune‐related genes were mainly involved in Cytokine–cytokine receptor interaction, MAPK signaling pathway and PI3K-Akt signaling pathway (Fig. 2b).

Differentially expressed immune-related genes and transcription factors (TFs) in hepatocellular carcinoma (HCC). a Heatmap of significantly differentially expressed immune-related genes in HCC. The color from green to red represents the progression from low expression to high expression. b Volcano plot of differentially expressed immune-related genes. The red dots in the plot represents upregulated genes and green dots represents downregulated genes with statistical significance. Black dots represent no differentially expressed genes. c Heatmap of significantly differentially expressed TFs in HCC. Red represents higher expression while green represents lower expression. d Volcano plot of differentially expressed TFs in HCC. Colored dots represent differentially expressed TFs and black dots represent no differentially expressed TFs

Functional enrichment analysis of differentially expressed immune-related genes. a Gene ontology analysis; From top to bottom, the figure represents biological process, cellular component and molecular function, respectively. b The top 30 most significant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways
Screening of immune-related genes with prognostic value in HCC
To determine the differentially expressed immune‐related genes with prognostic characteristics, the 329 genes expression in the 337 HCC samples were evaluated by univariate Cox analysis. Totally, 64 immune‐related genes were found to be related to OS. The prognostic immune‐related genes were shown in Table 1. GO and KEGG analysis suggested that these prognostic immune‐related genes mainly participated in semaphorin–plexin signaling pathway, ErbB signaling pathway, Hepatitis C. These pathways were shown to be significantly correlated with the development of cancer (Fig. 3a, b).

Functional enrichment analysis of prognostic immune‐related genes. a Gene ontology analysis; The outer circle is a bar plot where the height of the bar indicates the significance of GO terms. The inner ring shows a scatter plot of the expression (logFC) of differentially expressed immune-related genes in each enriched gene ontology term. b Top 8 enriched KEGG pathways for the prognostic immune-related genes
TF regulatory network
To explore the regulatory mechanisms of prognostic immune‐related genes, we selected the prognosis-related TFs among HCC patients. Univariate Cox regression analysis detected that 54 TFs were correlated to patient overall survival (Table 2). We established a regulatory network according to 54 TFs and 64 immune‐related genes. We built the TF regulatory network in three steps: (1) 54 prognosis-related TFs and 64 prognostic immune‐related genes were selected. (2) The correlation test between each TF and each immune‐related gene was conducted using cor.test function, whose core method was Pearson test. Based on the cut-off criteria, 12 prognosis-related TFs and 32 prognostic immune‐related genes were identified to establish the network. (3) Here, we utilized Cytoscape to construct and visualize the main regulatory network. As shown in Fig. 4, HCFC1 regulated most of the immune‐related genes and occupied the dominant position. This transcriptional regulatory network revealed the regulatory relationships among these immune-related genes.

The main regulatory network constructed based on prognosis-related transcription factors and prognostic immune‐related genes. The red circular represent differentially expressed prognostic immune‐related genes and the green diamond represent prognosis-related transcription factors, respectively
Construction of immune-related prognostic model for HCC
64 prognostic immune‐related genes were subjected to Lasso Cox analysis and 21 genes were filtered out. Then multivariate Cox analysis were performed and nine genes were finally selected to establish a prognostic model. The formula was shown as: risk score = (0.2940 * expression level of ANGPT1) + (0.1753 * expression level of MAPT) + (0.1066 * expression level of DCK) + (0.0706 * expression level of SEMA3F) + (0.0703 * expression level of IL17D) + (0.0311* expression level of HSPA4) + (0.0204 * expression level of RBP2) + (0.0084 * expression level of NDRG1) + (0.0052 * expression level of OSGIN1). All the nine genes were risky prognostic genes with hazard ratio > 1. Risk scores were based on genes expression levels multiplied its corresponding regression coefficients. Regression coefficients were calculated by multivariate Cox regression. The risk scores were not only related to the expression levels of these genes, but also related to the correlation coefficients. Then 337 HCC samples were classified into a high risk group (n = 168) and low risk group (n = 169) based on the median risk score (Fig. 5a). The survival overview and gene expression heatmap were presented in Fig. 5b–c. Survival analysis indicated that patients in the high risk group showed markedly poorer overall survival than those in the low risk group (P < 0.0001; Fig. 5d). The area under the ROC curve for 1 year, 3 year, and 5 year OS were 0.811, 0.711, 0.734, suggesting that this prognostic model exhibited a good sensitivity and specificity (Fig. 5e). The relationships between the risk score model and immune cell infiltration were investigated. As shown in Fig. 6, dendritic cells, neutrophil and macrophage were positive correlated with risk score. However, no significant correlations were observed between B cells, CD4+ T cells, CD8+ T cells and risk score.

Construction of an immune-related prognostic signature for hepatocellular carcinoma. a The risk score distribution of HCC patients in the The Cancer Genome Atlas (TCGA) database. b Survival status and duration of patients. c Heatmap of the nine immune‐related genes expression in HCC patients. d Survival curves for the low risk and high risk groups. e Receiver operating characteristic curve (ROC) analysis predicted overall survival using the risk score

Relationships between the risk score model and infiltration abundances of six types of immune cells
Independence of immune-related prognostic signature from other clinical factors
207 HCC patients with clinical information containing gender, age, histological grade, pathologic stage and TNM stage were selected for further analysis. Univariate and multivariate Cox regression analyses were conducted to assess the independent predictive power of immune-related prognostic signature. Univariate analysis indicated that pathological stage (P < 0.0001), T classification (P < 0.0001) and immune-related prognostic model (P < 0.0001) were markedly correlated with overall survival. After the multivariate analysis, only immune-related prognostic model remained as an independent prognostic factor associated with OS (P < 0.0001; Table 3).
Genetic alterations of nine immune-related prognostic genes
The cBioPortal tool was employed to analyze genomic alternations and potential drugs of nine immune-related prognostic genes. As shown in Fig. 7a, ANGPT1 and NDRG1 were most commonly altered genes. Amplification was the main frequent genetic alterations and the nine immune‐related prognostic genes altered in 94 (25.2%) of 373 cases. Figure 7b illustrated the network built by nine immune-related prognostic genes and their 50 most frequently mutated neighbor genes. Anticancer drugs targeting these genes were exhibited. Among them, two genes (DCK and ANGPT1) were currently regarded as drugs targets. We considered that other genes might act as potential novel therapeutic targets.

Genetic alterations and biological function of nine prognostic immune‐related genes. a The genetic alteration of nine genes in HCC patients using the cBioPortal database. b The network contained 59 nodes, including nine query genes and the 50 most frequently altered neighbor genes (only five out of nine were correlated with the 50 genes). The relationship between key prognostic immune‐related and cancer drugs was illustrated
Validation of the immune-related prognostic signature by ICGC database
The ICGC database including 231 HCC samples were used for the validation of the immune-related signature. According to the median risk score, we divided patients into high risk (n = 115) and low risk groups (n = 116). In agreement with results of TCGA cohort, the Kaplan–Meier curve demonstrated that patients in the high risk group exhibited markedly poorer overall survival than those in the low risk group (P < 0.001; Fig. 8a). The AUCs for 1 year and 3 year OS were 0.781 and 0.783, demonstrating good performance of the immune-related signature in predicting OS (Fig. 8b). Because there was only one patient with a 5-year follow-up period, we did not plot a 5-year ROC curve.

ROC and Kaplan–Meier analysis of the nine-gene signature in International Cancer Genome Consortium (ICGC) datase. a The Kaplan–Meier curve of the overall survival between the high risk and low risk groups stratified by the median risk score in ICGC. b ROC analysis of the predictive efficiency of the nine-gene prognostic model on overall survival based on risk score
Discussion
Hepatocellular carcinoma still remains a lethal malignancy with extremely unfavorable prognosis globally. Precise prediction of HCC overall survival is of great significance for the choice of therapeutic methods and amelioration of prognosis. To date, there are no reliable and effective biomarkers to accurately predict the survival of HCC patients. There is a critical demand to identify robust biomarkers and prediction model to forecast HCC outcomes.
In the current study, based on the analysis of TCGA dataset, 329 differentially expressed immune‐related genes were screened out from 374 cases of HCC and 50 normal tissues. According to the results of the GO enrichment, the mentioned genes were primarily associated with immune response. The KEGG pathways were mostly concentrated on several cancer-related pathways (e.g., MAPK signaling pathway and PI3K–Akt signaling pathway). In univariate regression analysis on the differentially expressed immune‐related genes, 64 genes were detected to display significant association with OS. To delve into the regulatory mechanisms of the prognostic immune‐related genes, a TF-mediated network was built to reveal crucial TFs that are capable of regulating these immune‐related genes. The main network suggested that HCFC1 was the critical key regulator in the network. HCFC1 (host cell factor C1), belongs to the host cell factor family. It is noteworthy that a recent study reported HCFC1 as a clinically hub gene that was remarkably correlated with the survival time, grade and TNM stage of HCC patients [31]. To date, the contribution of HCFC1 to the development of HCC remains unclear. Further experimental evidence is needed to explore the molecular mechanisms of HCFC1 in HCC.
Recently, gene signatures according to aberrant mRNA have attracted wide attention and revealed the huge potential in prognosis prediction of HCC. For instance, Long et al. built a four-gene prognostic model that showed a good performance for HCC prognosis prediction [32]. Another study also constructed a six-gene prognostic signature for HCC overall survival prediction based on gene expression data from TCGA [33]. A recent study investigating the prognostic value of TP53-associated immune genes in HCC identified and validated a two-gene (TREM1 and EXO1) prognostic model [34]. However, these studies did not use a large number of samples to comprehensively explore the relationship between immune genes and prognosis of HCC. Compared with the previous researches, this study has several advantages: (1) we utilized the specialized immunology database, which allowed us to analyze as many immune genes as we can. To our knowledge, this is the first study to explore the relationships between a large number of immune-related genes and prognosis in HCC patients. (2) We obtained a number of prognostic immune-related genes and established a novel immune-related prognostic model. This prognostic model exhibited a prominent performance for OS prediction based on TCGA database. According to the in-depth analysis, the immune-related prognostic model was demonstrated to be an independent prognostic indicator after adjusting to other clinical factors. Subsequently, such model that consists of nine immune‐related genes was then successfully validated as a prognostic factor in an independent ICGC dataset. All the mentioned results revealed that immune-related prognostic model could act as an effective marker for HCC prognosis prediction.
To characterize the tumor immune microenvironment status, the relationships between immune-related prognostic model and immune cell infiltration were investigated. The data here indicated that higher infiltration levels of dendritic cells, neutrophil and macrophage may be observed in high risk patients. Dendritic cells, neutrophil and macrophage displayed positive correlation with immune-related prognostic model, revealing that the model may serve as predictor for increased immune cells infiltration. A recent study reported that intratumoral infiltration by dendritic cells had a close relation to the poor prognosis in HCC patients [35], which is consistent with our findings. An existing study reported that neutrophil infiltration within HCC might display an association with a poor clinical outcome [36]. Neutrophils contribute to the activation, regulation and effector function of immune cells [37]; they are also capable of HCC progression by secreting a wide range of cytokines [38], thereby demonstrating their crucial role in the pathogenesis of HCC. A number of studies have reported that increased macrophages were related to poor prognosis in HCC [39]. Macrophages infiltration within the tumor microenvironment could facilitate tumor growth, angiogenesis, invasion, as well as metastasis [40]. Targeting macrophages have been considered a promising adjuvant immunotherapy for HCC patients [41, 42]. The role of immune cells in HCC has not been fully elucidated. It may be a promising way to cure HCC by broadening the relationship between immune cells and tumor progression.
Nine immune-related genes that constituted the prognostic model were identified as potential biomarkers in HCC. Out of the nine genes, RBP2, NDRG1 and HSPA have been well studied in HCC compared to other immune-related genes. RBP2 (retinol binding protein 2), belongs to the Fatty-acid binding protein (FABP) family, was reported to be involved in the pathogenesis of diverse types of cancer [43]. It has been previously evidenced that RBP2 was overexpressed in HCC and was associated with unfavorable prognosis of HCC [44]. Overexpressed RBP2 was markedly correlated with AFP and TNM stage. RBP2 might be critical to the angiogenesis and progression of HCC. Elevated NDRG1 expression was observed in HCC and dramatically related to overall survival and tumor stage [45]. NDRG1 was suggested to play significant roles in the metastasis, recurrence and and prognosis of HCC [46]. Moreover, overexpressed NDRG1 displayed a significant association with hepatocarcinogenesis [47]. Accordingly, targeting NDRG1 might act as an attractive therapeutic strategy for HCC. HSPA4, also known as hsp70, was demonstrated to enhance the proliferation, invasion and metastasis of various cancers [48]. High Expression of HSPA4 was significantly correlated with worse overall survival of HCC; it was aslo an independent prognostic parameter for OS [49]. Given the findings here, HSPA4 demonstrated huge potential as a therapeutic target in HCC treatment. In the HCC tissues, up-regulated expression of ANGPT1 was detected as compared with normal liver tissues [50], whereas the prognostic implication in HCC was not studied. A previously study showed IL17D had a diagnostic value for HCC and the DNA methylation status of IL17D was related to OS [51]. Nevertheless, the specific role of IL17D in HCC has been rarely known. Only one study has reported OSGIN1 may be a tumor suppressor that was downregulated in HCC, which contradicted our findings [52]. Its exact role in HCC is as yet unclear. Thus far, no relevant research reported MAPT, DCK and SEMA3F in HCC. Further researches are required to elucidate the function of these potential immune-related genes in HCC.
Some shortcomings of this study should be addressed. First, this study was completely based on public databases and the results should be external validated by further experiments. Second, the efficiency of the immune-related prognostic model should be identified in a large number of HCC samples using experimental methods. Third, the biological functions of nine immune-related genes in HCC require further examined by a series of experiments.
Conclusion
In conclusion, for the first time, numerous immune-related genes were detected to be significantly related to HCC prognosis by comprehensive analyses. Moreover, we constructed a novel immune-related prognostic model as an independent prognostic predictor for HCC. Validation in an external ICGC database further confirmed the prognostic value of this model. This prognostic model may also serve as predictor for increased immune cells infiltration, proving its key role in tumor immune microenvironment. The current study deepens our understanding of immune-related genes in HCC and provides new potential prognostic and therapeutic biomarkers.
Availability of data and materials
Not applicable.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- TCGA:
-
The Cancer Genome Atlas
- DEGs:
-
Differentially expressed genes
- TFs:
-
Transcription factors
- PD-1:
-
Programmed cell death-1
- ICGC:
-
International Cancer Genome Consortium
- TIMER:
-
Tumor immune estimation resource
- GO:
-
Gene ontology database
- KEGG:
-
The Kyoto encyclopedia of genes and genomes
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the ROC curve
- OS:
-
Overall survival
References
Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, Gores G. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018.
Rao CV, Asch AS, Yamada HY. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis. 2017;38:2–11.
Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang XS, Bannon F, Ahn JV, Johnson CJ, Bonaventure A, et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. 2015;385:977–1010.
Joliat GR, Allemann P, Labgaa I, Demartines N, Halkic N. Treatment and outcomes of recurrent hepatocellular carcinomas. Langenbecks Arch Surg. 2017;402:737–44.
Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63:844–55.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90.
Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66.
Lin DC, Mayakonda A, Dinh HQ, Huang P, Lin L, Liu X, Ding LW, Wang J, Berman BP, Song EW, et al. Genomic and epigenomic heterogeneity of hepatocellular carcinoma. Cancer Res. 2017;77:2255–65.
Angell H, Galon J. From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Curr Opin Immunol. 2013;25:261–7.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99.
Schmidt N, Thimme R. Role of immunity in pathogenesis and treatment of hepatocellular carcinoma. Dig Dis. 2016;34:429–37.
Jiang R, Tan Z, Deng L, Chen Y, Xia Y, Gao Y, Wang X, Sun B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900–9.
Sutton A, Friand V, Brule-Donneger S, Chaigneau T, Ziol M, Sainte-Catherine O, Poire A, Saffar L, Kraemer M, Vassy J, et al. Stromal cell-derived factor-1/chemokine (C-X-C motif) ligand 12 stimulates human hepatoma cell growth, migration, and invasion. Mol Cancer Res. 2007;5:21–33.
Hou KZ, Fu ZQ, Gong H. Chemokine ligand 20 enhances progression of hepatocellular carcinoma via epithelial-mesenchymal transition. World J Gastroenterol. 2015;21:475–83.
Kabilova TO, Kovtonyuk LV, Zonov EV, Ryabchikova EI, Popova NA, Nikolin VP, Kaledin VI, Zenkova MA, Vlassov VV, Chernolovskaya EL. Immunotherapy of hepatocellular carcinoma with small double-stranded RNA. BMC Cancer. 2014;14:338.
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–14.
Sakurai K, Niitsuma S, Sato R, Takahashi K, Arihara Z. Painless thyroiditis and fulminant type 1 diabetes mellitus in a patient treated with an immune checkpoint inhibitor, Nivolumab. Tohoku J Exp Med. 2018;244:33–40.
El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling THR, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–502.
DeLeon TT, Ahn DH, Bogenberger JM, Anastasiadis PZ, Arora M, Ramanathan RK, Aqel BA, Vasmatzis G, Truty MJ, Oklu R, et al. Novel targeted therapy strategies for biliary tract cancers and hepatocellular carcinoma. Fut Oncol. 2018;14:553–66.
Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, Kammula US, Chen Y, Qin LX, Tang ZY, Wang XW. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10:99–111.
Bhattacharya S, Andorf S, Gomes L, Dunn P, Schaefer H, Pontius J, Berger P, Desborough V, Smith T, Campbell J, et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res. 2014;58:234–9.
Mei S, Meyer CA, Zheng R, Qin Q, Wu Q, Jiang P, Li B, Shi X, Wang B, Fan J, et al. Cistrome cancer: a web resource for integrative gene regulation modeling in cancer. Cancer Res. 2017;77:e19–22.
Ontology CG. The gene ontology (GO) project in 2006. Nucleic Acids Res. 2006;34:D322–6.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–d361.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, Liu XS. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77:e108–10.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Xiong DD, Feng ZB, Lai ZF, Qin Y, Liu LM, Fu HX, He RQ, Wu HY, Dang YW, Chen G, Luo DZ. High throughput circRNA sequencing analysis reveals novel insights into the mechanism of nitidine chloride against hepatocellular carcinoma. Cell Death Dis. 2019;10:658.
Long J, Zhang L, Wan X, Lin J, Bai Y, Xu W, Xiong J, Zhao H. A four-gene-based prognostic model predicts overall survival in patients with hepatocellular carcinoma. J Cell Mol Med. 2018;22:5928–38.
Wang Z, Teng D, Li Y, Hu Z, Liu L, Zheng H. A six-gene-based prognostic signature for hepatocellular carcinoma overall survival prediction. Life Sci. 2018;203:83–91.
Long J, Wang A, Bai Y, Lin J, Yang X, Wang D, Yang X, Jiang Y, Zhao H. Development and validation of a TP53-associated immune prognostic model for hepatocellular carcinoma. EBioMedicine. 2019;42:363–74.
Zhou ZJ, Xin HY, Li J, Hu ZQ, Luo CB, Zhou SL. Intratumoral plasmacytoid dendritic cells as a poor prognostic factor for hepatocellular carcinoma following curative resection. Cancer Immunol Immunother. 2019;68:1223–33.
Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, Yin XY, Zheng L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. 2011;54:948–55.
Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519–31.
Lu C, Rong D, Zhang B, Zheng W, Wang X, Chen Z, Tang W. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer. 2019;18:130.
Tian Z, Hou X, Liu W, Han Z, Wei L. Macrophages and hepatocellular carcinoma. Cell Biosci. 2019;9:79.
Wu SD, Ma YS, Fang Y, Liu LL, Fu D, Shen XZ. Role of the microenvironment in hepatocellular carcinoma development and progression. Cancer Treat Rev. 2012;38:218–25.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416.
Degroote H, Van Dierendonck A, Geerts A, Van Vlierberghe H, Devisscher L. Preclinical and clinical therapeutic strategies affecting tumor-associated macrophages in hepatocellular carcinoma. J Immunol Res. 2018;2018:7819520.
Liang X, Zeng J, Wang L, Fang M, Wang Q, Zhao M, Xu X, Liu Z, Li W, Liu S, et al. Histone demethylase retinoblastoma binding protein 2 is overexpressed in hepatocellular carcinoma and negatively regulated by hsa-miR-212. PLoS ONE. 2013;8:e69784.
Wang ZY, Yang J, Liu CK, Shen SQ. High expression of retinoblastoma-binding protein 2 (RBP2) in patients with hepatocellular carcinoma and its prognostic significance. Med Sci Monit. 2017;23:2736–44.
Chua MS, Sun H, Cheung ST, Mason V, Higgins J, Ross DT, Fan ST, So S. Overexpression of NDRG1 is an indicator of poor prognosis in hepatocellular carcinoma. Mod Pathol. 2007;20:76–83.
Cheng J, Xie HY, Xu X, Wu J, Wei X, Su R, Zhang W, Lv Z, Zheng S, Zhou L. NDRG1 as a biomarker for metastasis, recurrence and of poor prognosis in hepatocellular carcinoma. Cancer Lett. 2011;310:35–45.
Lu WJ, Chua MS, So SK. Suppressing N-Myc downstream regulated gene 1 reactivates senescence signaling and inhibits tumor growth in hepatocellular carcinoma. Carcinogenesis. 2014;35:915–22.
Xiong J, Jiang XM, Mao SS, Yu XN, Huang XX. Heat shock protein 70 downregulation inhibits proliferation, migration and tumorigenicity in hepatocellular carcinoma cells. Oncol Lett. 2017;14:2703–8.
Ma C, Xu T, Sun X, Zhang S, Liu S, Fan S, Lei C, Tang F, Zhai C, Li C, et al. Network pharmacology and bioinformatics approach reveals the therapeutic mechanism of action of baicalein in hepatocellular carcinoma. Evid Based Complement Alternat Med. 2019;2019:7518374.
Torimura T, Ueno T, Kin M, Harada R, Taniguchi E, Nakamura T, Sakata R, Hashimoto O, Sakamoto M, Kumashiro R, et al. Overexpression of angiopoietin-1 and angiopoietin-2 in hepatocellular carcinoma. J Hepatol. 2004;40:799–807.
Sun XJ, Wang MC, Zhang FH, Kong X. An integrated analysis of genome-wide DNA methylation and gene expression data in hepatocellular carcinoma. FEBS Open Bio. 2018;8:1093–103.
Liu M, Li Y, Chen L, Chan TH, Song Y, Fu L, Zeng TT, Dai YD, Zhu YH, Li Y, et al. Allele-specific imbalance of oxidative stress-induced growth inhibitor 1 associates with progression of hepatocellular carcinoma. Gastroenterology. 2014;146:1084–96.
Acknowledgements
The authors would like to express our sincere thanks for sharing the data from The Cancer Genome Atlas (TCGA) database and International Cancer Genome Consortium (ICGC) database.
Funding
Grant agency: Cultivating fund for Chinese National Natural Science Foundation of Shandong Provincial Qianfoshan Hospital.
Grant Number: QYPY2019NSFC0803.
Author information
Authors and Affiliations
Contributions
ZW conceived the study and performed the bioinformatics analyses. ZW and JZ downloaded and organized the clinical and gene expression data. ZW, YL and WW performed the statistical analyses. ZW, JZ and CL wrote the manuscript. FC and LM critically revised the article for essential intellectual content and administrative support. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Z., Zhu, J., Liu, Y. et al. Development and validation of a novel immune-related prognostic model in hepatocellular carcinoma. J Transl Med 18, 67 (2020). https://doi.org/10.1186/s12967-020-02255-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02255-6