
Abstract
Breast cancer has grown to be the second leading cause of cancer-related deaths in women. Only a few treatment options are available for breast cancer due to the widespread occurrence of chemoresistance, which emphasizes the need to discover and develop new methods to treat this disease. Signal transducer and activator of transcription 3 (STAT3) is an early tumor diagnostic marker and is known to promote breast cancer malignancy. Recent clinical and preclinical data indicate the involvement of overexpressed and constitutively activated STAT3 in the progression, proliferation, metastasis and chemoresistance of breast cancer. Moreover, new pathways comprised of upstream regulators and downstream targets of STAT3 have been discovered. In addition, small molecule inhibitors targeting STAT3 activation have been found to be efficient for therapeutic treatment of breast cancer. This systematic review discusses the advances in the discovery of the STAT3 pathways and drugs targeting STAT3 in breast cancer.
Video abstract
Similar content being viewed by others
Background
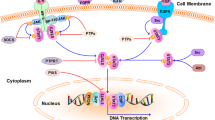
Transcription factors (TFs) are proteins possessing domains that bind to the DNA of promoter or enhancer regions of specific genes. Several TFs are directly involved in the development and progression of breast cancer. One of the most prominent TF families in breast cancer is the signal transducers and activators of transcription (STAT) family, which is comprised of seven structurally similar and highly conserved members, namely, STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6 [1, 2]. In general, these family members contain six common functional domains: an N-terminal domain (NH2) which is called STAT_int now, a coiled-coil domain (CCD), a DNA-binding domain (DBD), a linker domain, an SRC homology 2 domain (SH2) and a transactivation domain (TAD) [3]. Since the discovery of STAT3 in 1994, research has been primarily focused on its close association with cancer progression, proliferation, metastasis and multidrug resistance [4, 5]. Extensive reviews have described the classical STAT3 signaling pathways [6,7,8]. Here, we present a short overview of the STAT3 signaling pathways as depicted in Fig. 1. Briefly, STAT3 is activated through several cytokines, including interleukin 6 (IL-6) and interleukin 10 (IL-10), and growth factors, including epidermal growth factor (EGF), fibroblast growth factor (FGF) and insulin-like growth factor (IGF) [9, 10]. Once these factors bind to their corresponding receptors, Janus kinases (JAKs) are activated [11]. JAKs phosphorylate the cytoplasmic tail of the cognate receptor and STAT3 via its SH2 domain binds to phosphorylated tyrosine residues. The phosphorylated STAT3 forms homodimers and translocate into nucleus and, thus, can exchange signals between the cytoplasm and nucleus. Upon translocation into the nucleus, pSTAT3 forms a complex with some coactivators, including p68, and binds to the promotor region of target genes to activate their transcription [12]. This review aims to explore the mechanism of STAT3 in breast cancer development and summarize the latest advancements made.

The classical IL-6/STAT3 signaling pathway in cancer cells. IL-6 binds to the membrane-bound IL-6 receptor α (IL-6R) and IL-6 receptor β (also known as gp130). The IL-6/IL-6R/gp130 complex activate the phosphorylation of JAKs, followed by STAT3 phosphorylation and activation. Growth factors, such as FGF, IGF and EGF, can also phosphorylate STAT3 by binding to their cognate membrane receptors. Then, phosphorylated STAT3 forms a homodimer and translocates into the nucleus to bind to the promotor region of target genes and activates target gene transcription
Advances in the study of STAT3 signaling pathways in breast cancer
The role of STAT3 in breast cancer progression
An illustration of the advances in our understanding of the STAT3 signaling pathways in breast cancer progression is shown in Fig. 2. A member of the IL-6 family of cytokines, Oncostatin M (OSM) can induce IL-6 upregulation and STAT3 phosphorylation to promote breast cancer progression [13] and to activate STAT3 and hypoxia inducible factor 1 alpha (HIF-1α) in estrogen receptor (ER)- breast cancer cells or in ER+ breast cancer cells in cooperation with IL-6 [14]. Additionally, other interleukins, such as IL-35 and IL-8, are also found to promote breast cancer progression by activating STAT3. IL-35 is found to inhibit conventional T (T-conv) cells and promote breast cancer progression via activation of STAT1 and STAT3 [15], whereas IL-8 and growth-regulated oncogene (GRO) chemokines are found to activate STAT3 and promote the progression of inflammatory breast cancer [16]. In contrast, low expression of IL-17 is found to inhibit STAT3 activation [17].

Advances of the STAT3 signaling pathways involved in breast cancer progression. Interleukins, including IL-6, IL-8 and IL-35, can bind to their receptors and activate the phosphorylation of JAKs and STAT3, OSM can increase IL-6-mediated activation, and IL-17 binding to its receptor leads to inhibition of STAT3 phosphorylation. STAT3 phosphorylated by EGF can be inhibited by PTPN2. COX2 and prostaglandin E2 upregulated by HDAC6 can activate STAT3 phosphorylation, and SMYD2 has a similar effect. Additionally, STAT3 and NEAT1 can form a loop to activate the phosphorylation of STAT3, which is inhibited by miR-124. The activated and phosphorylated STAT3 dimers translocate into the nucleus and activate the transcription of target genes involved in breast cancer progression
Other mediators of STAT3 expression and activation include activators, such as prostaglandin E2, cyclooxygenase-2 (COX2) and SET and MYND (myeloid-Nervy-DEAF-1) domain-containing protein (SMYD2), as well as its inhibitors, such as microRNA and protein tyrosine phosphatase 2 (PTPN2). Epigenetic regulators have been widely investigated and discovered to regulate STAT3 activation in breast cancers in recent years. Li et al. have found that histone deacetylase 6 (HDAC6), a class II histone deacetylase, and prostaglandin E2 and COX2, can upregulate STAT3 activation in breast cancer [18]. In addition, lysine methyltransferase SMYD2 can activate the methylation and phosphorylation of STAT3 to promote breast cancer progression [19]. MicroRNA (miR) has become a hot topic in the fields of cancer biology and development in recent years. Pang et al. have demonstrated that nuclear enriched abundant transcript 1 (NEAT1) forms a feedback loop with STAT3 to promote breast cancer progression. However, NEAT1 is suppressed by miR-124 [20]. Interestingly, glucosamine is found to suppress the activation of STAT3 and decrease breast cancer stemness and progression [21]. Additionally, knockdown of PTPN2 leads to EGF-mediated STAT3 activation [22]. The association of chronic inflammation with breast cancer progression is widely recognized, but it can be inhibited by blocking STAT3 [23]. Other mediators of STAT3 signaling pathways are also extensively studied. Kim et al. have found that the IL-6/STAT3/ROS pathway can not only promote breast cancer progression and inflammation but also increase the formation of breast cancer stem cells [24]. Moreover, TGFβ-regulated FAM3C/Interleukin-like EMT Inducer (ILEI), an oncogenic member of the FAM3 cytokine family, can mediate STAT3 signaling pathway to drive breast cancer stem cell formation and promote breast cancer progression [25]. In addition, TNFRSF1A, a gene encoding a transmembrane receptor for TNF-α, can be modulated by STAT3 and promote NF-κB signaling in breast cancer [26].
There were also some STAT3 co-factors influenced the proliferation and progress of breast cancer. Progranulin (PGRN), was seen to associate with chemoresistance and worse prognosis in breast cancer [27, 28], and the use of a specific progranulin antisense oligonucleotide was recently seen to hamper STAT3 oncogenic functions in CRC cells [29], suggesting a similar effect also in breast cancer cells. The cyclin dependent kinase 5 (CDK5) regulatory subunit-associated protein 3 (CDK5RAP3, also called C53/LZAP) was originally regarded as a p53 co-activator [30]. A recent research reported that CDK5RAP3 was associated with primary breast cancer progression and proliferation, and also enhanced the expression of STAT3-dependent genes [31]. Thus, targeting the co-factor of STAT3 maybe a potential therapeutic approach in breast cancer management.
The role of STAT3 in breast cancer proliferation and apoptosis
The illustration with advances of STAT3 signaling pathways in breast cancer proliferation and apoptosis is shown in Fig. 3. A recent research has reported that downregulation of zinc-finger gene DPF3 (also known as CERD4) promotes proliferation and motility of breast cancer via activating JAK2/STAT3 pathway [32]. It has been reported earlier that STAT3 can upregulate cyclin D-1, c-myc, and bcl-2 to suppress the apoptosis of breast cancer cells, indicating a potential involvement of STAT3 in cell cycle and survival [33]. Moreover, STAT3 activated by IL-6/JAK2 pathway can inhibit Bax/Bcl-2-related caspase-dependent apoptosis [34]. However, overexpression of WW domain-containing oxidoreductase (Wwox) blocks the combination of STAT3 and IL-6R, resulting in inhibition of proliferation [35]. Another research shows that IL-32θ targets chemokine ligand (CCL)18/STAT3 pathway to suppress macrophage-promoted breast cancer progression [36]. In addition, miRNAs are also widely investigated in breast cancer proliferation and invasion. Park et al. have found that miR-125a and let-7e could inhibit IL-6/STAT3 pathway to mediate the breast cancer proliferation and vasculogenic mimicry formation [37], and Shi et al. have found that miR-124 could suppress the mRNA and protein levels of STAT3 and inhibit the proliferation and invasion of breast cancer [38]. Similarly, miR-9 is reported to inhibit STAT3 activation and breast cancer proliferation [39]. In contrast, miR-93-5p and miR-25-3p are found to mediate STAT3 and promote breast cancer proliferation [40, 41]. Since the discovery of Warburg effects, metabolism is strongly linked with proliferation of cancer cells. It has been suggested that let-7a-5p, Stat3, and hnRNP-A1 form a feedback loop to regulate PKM2 expression and modulate glucose metabolism in breast cancer cells, suggesting that inhibiting STAT3-related metabolism may inhibit breast cancer proliferation [42].

Advances of the STAT3 signaling pathways involving breast cancer proliferation and apoptosis. Classical IL-6 /JAK/STAT3 pathways can activate the transcription of cyclin D-1, c-myc, bcl-2 and Bax to promote the proliferation and inhibit the apoptosis of breast cancer. miR-125a, miR-25-3p and p16 can promote the binding of IL-6 to its receptors, whereas Wwox has the opposite effect. CCL-18 binding to its receptor can activate the phosphorylation of STAT3, which can be inhibited by IL-32θ. The circuit loop of phosphorylated STAT3, TMEM16A and EGF leads to continuous activation of STAT3. miR-93-5p, SMYD2, TRIM14 and PKT-M2 induce the activation of STAT3, whereas miR-124 and miR-9 inhibit the activation of STAT3 and breast cancer proliferation. Let-7a-5p, hnRN-A and phosphorylated STAT3 dimers form a circuit loop to upregulate PKM2 and promote the proliferation and inhibit the apoptosis of breast cancer cells. DPF3 suppressed by phosphorylated STAT3 can promote breast cancer proliferation. Additionally, transcription factor EB (TFEB) can combine with phosphorylated STAT3 dimers to promote the transcription of target genes involved in breast cancer proliferation
There are several new pathways associated with STAT3 and breast cancer that have been minimally studied to date. It has been revealed that the Ca2+ activated chloride channel TMEM16A forms an activation loop with EGFR/STAT3 to promote breast cancer proliferation [43]. Moreover, tripartite motif-containing 14 (TRIM14) is found to increase the expression of p-STAT3 to promote breast cancer proliferation [44]. In addition, it is reported that pyruvate kinase type M2 (PKT-M2) regulates phosphorylation of STAT3 in breast cancer [45], whereas cystathionine-lyase (CSE) suppresses the expression of STAT3/matrix metallopeptidases-2 (MMP2), MMP9, p-protein kinase B and B-cell lymphoma 2 [46].
The role of STAT3 in breast cancer metastasis
An illustration of the advances of the STAT3 signaling pathways in breast cancer metastasis is shown in Fig. 4. Matrix metallopeptidases (MMPs) are known to play important roles in breast cancer metastasis. A well-studied mechanism of STAT3-mediated cell metastasis is through upregulating MMP2, MMP9, Twist, Snail, Slug and vimentin [47,48,49]. Ma et al. have reported that inhibition of STAT3 phosphorylation could reduce the expression of vasodilator-stimulated phosphoprotein (VASP), MMP2 and MMP9 in breast cancer [50]. As mentioned previously, STAT3 signaling is usually activated upon binding of cytokines and growth factors to their cognate receptors on the plasma membrane. The previously mentioned Wwox can inhibit breast cancer metastasis by preventing receptor binding [35]. Furthermore, Kim et al. have demonstrated that Mesoderm-specific transcript (MEST) induces Twist expression by activating the JAK/STAT3 signaling pathway [51], whereas Khanna et al. have shown the inhibition of GRAM domain-containing protein 1B (GRAMD1B) in breast cancer migration via the suppression of the JAK/STAT3 and protein kinase B (Akt) pathway [52]. Instead of classical ligand/receptor binding in the plasma membrane for STAT3 activation, a new pathway is found in which OSM/SMAD3 could also activate STAT3 and mediate Snail expression and promote epithelial-mesenchymal transition (EMT) in breast cancer, indicating a distinct route of STAT3 activation through cytoplasmic molecules and endogenous signaling [53]. Other signaling molecules, including miRNA, proto-oncogene serine/threonine-protein kinase (PIM1), Mucin-1-C (MUC1-C), natriuretic peptide receptor A (NPRA) and RhoU, were also discovered to participate in STAT3-mediated breast cancer metastasis. miR-30d is found to mediate migration and invasion in breast cancer cells by regulating Krüppel-like factor 11 (KLF-11), a new exogenous signaling pathway that can activate STAT3 by binding to its transmembrane receptor KLF-11R [54]. In addition, IL-11 is also found to regulate the JAK/STAT3 pathway in breast cancer-bone metastasis [55]. PIM1, a proto-oncogene responsible for promoting cell invasion and upregulating EMT expression in breast cancer, is found to be regulated by the IL-6/STAT3 signaling pathway [56]. MUC1-C, an oncogenic protein, can activate STAT3 and induce Twist transactivation to promote EMT [57]. Moreover, NPRA, one of the natriuretic peptide receptors, is found to increase the expression of STAT3 and MMP9 to promote the migration and invasion of breast cancer cells [58]. STAT3, by cooperating with Specificity Protein 1 (SP1), is found to induce high Ras Homolog Family Member U (RhoU) expression and breast cancer cell migration [59]. Additionally, some enzymes are also found to participate in breast cancer metastasis by the posttranscriptional modification of STAT3. ARHGAP24, a Rac-specific Rho GTPase-activating protein (Rho GAP), is found to promote phosphorylation of STAT3 and to increase the expression of MMP2 and MMP9 in breast cancer cells [60]. GCN5, a histone acetyltransferase, is found to upregulate the expression of p-STAT3, p-AKT, MMP9 and E2F1 and promote breast cancer migration and invasion [61].

Advances of the STAT3 signaling pathways involving breast cancer metastasis. Classical IL-6/JAK/STAT3 pathways activate the transcription of MMP2, MMP9, Twist, Snail, Slug and vimentin to promote breast cancer metastasis, which can be suppressed by MEST and activated by GRAMD1B. Wwox can inhibit the binding of IL-6 and IL-6R/gp130. IL-11 and KLF-11 can also activate STAT3 to promote breast cancer metastasis by binding to their receptors. ARHGAP24, MUC1-C, NPRA and OSM-mediated SMAD3 function to upregulate the phosphorylation of STAT3. Estrogen related receptor alpha (ERR-α) can be transcriptionally activated by STAT3 and promote breast cancer metastasis. Phosphorylated STAT3 induces the activation of VASP to inhibit the metastasis of breast cancer, whereas PIM1 induced by phosphorylated STAT3 may have the opposite effect. The combination of phosphorylated STAT3 and RhoU inhibits breast cancer metastasis. Additionally, TFEB can activate the phosphorylation of STAT3 and AKT to promote breast cancer metastasis
Hypoxia is a stressed state that is extensively studied in cancers. Abyaneh et al. have found that hypoxia can significantly induce the activation of STAT3 to promote breast cancer stemness and metastasis [62]. This phenomenon provides us with a new direction for STAT3 research and targeted STAT3 therapy in breast cancer. Moreover, our recent research has found that estrogen related receptor alpha could promote the metastasis of triple negative breast cancer as a target gene of STAT3 [63].
The role of STAT3 in breast cancer chemoresistance
An illustration of the advances of the STAT3 signaling pathways in breast cancer chemoresistance is shown in Fig. 5. Tzeng et al. have indicated that the Src/STAT3 signaling pathway is involved in multidrug resistance in triple negative breast cancer cells [64]. It is also found that crosstalk between breast cancer cells and macrophages can induce tamoxifen and ICI 182,780 resistance through the NF-κB/STAT3/ERK pathways [65].

Advances of the STAT3 signaling pathways involving breast cancer chemoresistance. Classical IL-6/JAK/STAT3 pathways can induce chemoresistance in breast cancer, while miR-4532 may attenuate this effect by inhibiting HIC-1 and IL-6/STAT3 pathways. Leukemia inhibitory factor (LIF) binding to its receptor LIFR can increase the activation of STAT3. STAT3 and pSTAT3 levels are regulated by GRP78. Then, phosphorylated STAT3 activates cellular molecules including FAO, CPT1B and MAPK/AKT to induce the chemoresistance of breast cancer. Oct-4 and c-Myc form a signaling loop to promote STAT3/NF-κB activation and chemoresistance in breast cancer. Additionally, miR-124 can inhibit HIF-1 and promote breast cancer chemoresistance
The newly discovered downstream targets of STAT3-mediated chemoresistance include fatty acid beta-oxidation (FAO), carnitine palmitoyltransferase 1B (CPT1B), mitogen-activated protein kinase (MAPK)/AKT, HIF-1 and octamer-binding transcription factor-4 (Oct-4). It has been found that the JAK2/STAT3 signaling pathway increases CPT1B and FAO to increase chemoresistance in breast cancer [66]. Wang et al. found that IL-22 can promote JAK-STAT3/MAPKs/AKT pathway activation to induce breast cancer migration and paclitaxel resistance [67]. Moreover, miR-124 has been identified to reverse doxorubicin (DOX) resistance of breast cancer cells through targeting the STAT3/hypoxia-inducible factor 1 (HIF-1) pathway [68]. A recent study shows that Oct-4 and c-myc can form a signal circuit to increase Adriamycin resistance in breast cancer [69]. Meanwhile, Kim et al. have discovered that Oct-4 confers radiation resistance via STAT3 and NF-B-mediated IL-24 production in breast cancer cells [70]. In addition, paclitaxel is widely used as a clinical drug of breast cancer treatment, and phosphorylated STAT3 could mediate Survivin to promote paclitaxel resistance [71].
There are several upstream regulators of STAT3-mediated chemoresistance that have been identified in recent years. The COOH-terminal proline-rich region of 78-kDa glucose-regulated protein (GRP78), by regulating STAT3, is found to play a crucial role in the development of tamoxifen-resistant breast cancer cells [72]. Wang et al. have found that leukemia inhibitory factor receptor (LIFR) could promote STAT3 activation and contribute to breast cancer resistance to Trastuzumab-emtansine (T-DM1) [73]. Furthermore, miR-4532 is found to suppress hypermethylated in cancer-1 (HIC-1) and IL-6/STAT3 to promote Adriamycin resistance in breast cancer [74].
Some small molecules have also been found to contribute to chemoresistance mediated by STAT3. Piperlongumine combined with DOX is also found to induce apoptosis and inhibit DOX resistance of breast cancer cells via the JAK/STAT3 pathway [75]. In addition, targeting IL6/STAT3 activity using STAT3 inhibitor combined with a poly ADP-ribose polymerase (PARP) inhibitor could effectively treat palbociclib resistance in breast cancer cells [76].
Advances in the study of compounds targeting STAT3 in breast cancer
Compounds inhibiting the upstream of STAT3 in breast cancer
Several compounds are found to inhibit the upstream mediators of STAT3 in breast cancer since 2018 (Table 1). Many of these compounds target the IL-6/STAT3 signaling pathway. Ilamycin C is found to induce apoptosis and inhibit migration and invasion by suppressing the IL-6/STAT3 pathway [34]. A small molecule, bazedoxifene, is a novel IL-6/GP130 inhibitor that reduces breast cancer proliferation and migration [77]. Moreover, Esparza-Lopez et al. have discovered the inhibitory effect of metformin in IL-6-induced proliferation and EMT through the STAT3/NF-κB pathway in breast cancer [89]. DT-13, the saponin monomer 13 of the Dwarf lilyturf tuber, has been identified as a suppressor of breast cancer metastasis that acts by inhibiting both JAK/STAT3 and PI3K/AKT signaling pathways [81]. Furthermore, a natural compound called esculentoside A, a triterpene saponin derived from the root of Phytolacca esculenta, can also inhibit the IL-6/STAT3 pathway [78]. Meanwhile, another nature compound called catechol, which is derived from Aronia juice, shows similar effects in breast cancer cells [79]. In addition, scorpion venom can decrease IL-6, RhoC, ERK (1/2), and STAT3 and inhibit breast cancer proliferation [80]. As discussed previously, dihydrotanshinone inhibits breast cancer cells progression and stem cell formation through the IL-6/STAT3 pathway [24].
Other compounds target different signaling pathways, including the JAK2/STAT3 and Akt pathways. Both ganoderic acid A, which is isolated from ganoderma, and methylseleninic acid are found to suppress breast cancer proliferation via the JAK2/STAT3 pathway [82, 83]. A compound called caffeic acid p-nitro-phenethyl ester (CAPE-pNO2) is found to inhibit the EGFR/STAT3/Akt pathway and suppress breast cancer proliferation and metastasis [88]. Moreover, I157172, a novel inhibitor of cystathionine-lyase, is found to inhibit the proliferation and migration of breast cancer cells via upregulation of SIRT1 and inhibition of STAT3 signaling pathway [46].
Other compounds target the regulation of STAT3 expression. Alantolactone, a sesquiterpene lactone, can significantly decrease the expression of STAT3 and NF-κB in breast cancer [90]. Similarly, cyanidin-3-glucoside (C3G) can increase miR-124 expression and attenuate breast cancer proliferation by downregulating STAT3 expression [87].
Compounds inhibiting the activation of STAT3 in breast cancer
In recent years, various novel compounds have been found to inhibit the phosphorylation and activation of STAT3. A sesquiterpenoid from Farfarae Flos (ECN) is found to inhibit the phosphorylation and dimerization of STAT3 in the JAK/STAT3 pathway [84]. Moreover, (−)-galiellalactone and its novel analogues, SG-1709 and SG-1721, are found to inhibit STAT3 phosphorylation and suppress the dimerization and DNA-binding of STAT3 in breast cancer [92]. Similarly, schisandrin A is found to reverse doxorubicin resistance via inhibition of STAT3 phosphorylation in breast cancer [93]. Chun et al. have found that the hexane fraction from I. helenium (HFIH) can inhibit STAT3 phosphorylation at tyrosine 705 [33]. Niclosamide, that was reported to be a potent STAT3 inhibitor in TNBC cells, was found to overcome the radioresistance in TNBC cells via inhibition of STAT3 and Bcl-2 activation and induction of reactive oxygen species (ROS) [97]. In addition, flubendazole (FLU), a widely used anthelmintic agent, eupalinolide J, a Michael-reaction acceptor extracted from Eupatorium lindleyanum, and betulinic acid are found to inhibit STAT3 activation in breast cancer cells [98,99,100]. As an upstream activator of STAT3, inhibition of JAK2 can undoubtedly suppress STAT3 activation. The classical JAK2 inhibitor is known as AG490. Recently, ruxolitinib is found to have a potential to be a new selective JAK2 inhibitor and to block STAT3 activation [94]. Furthermore, tagalide A and tagalol A are also found to inhibit the phosphorylation of STAT3 and JAK2 in breast cancer [86]. Additionally, sabutoclax, a pan-active BCL-2 protein family antagonist, is found to inhibit the IL-6/STAT3 pathway and thereby overcome multidrug resistance in breast cancer [85], whereas isoharringtonine (IHT) is found to suppress the STAT3/Nanong pathway to inhibit breast cancer proliferation [91].
Notably, some STAT3 inhibitors are found to function in many biological processes. Sravanthi et al. have screened 29,388 ligands docking with STAT3 and found that Risedronate Sodium (RES) and Zoledronic acid (ZOL) could tightly combine with STAT3 and show significant cytotoxicity in breast cancer cells [105]. Moreover, a new synthetic derivative of cryptotanshinone KYZ3 is found to directly bind to the SH2 domain of STAT3 and act as a new STAT3 inhibitor [101]. Napabucasin and its angularly anellated isomer could also combine with SH2 domain of STAT3 [102]. One of coumarin-benzo [b] thiophene 1, 1-dioxide conjugates, compound 7a, could also combine with SH2 domain of STAT3 [103]. 15-Keto prostaglandin E-2 could bind to the Cys 251 and Cys 259 sites of STAT3 protein to inhibit the migration and proliferation of breast cancer [104]. Furthermore, pyrimethamine, a classic anti-microbial drug, is found to be a new STAT3 inhibitor and shows strong anti-cancer effects [95]. In addition, osthole, via binding to STAT3 protein, is found to suppress STAT3 activity and inhibit breast cancer cells apoptosis [106], whereas another STAT3 inhibitor, stattic, is found to promote the Bax/Bcl-2-mediated apoptosis in breast cancer and to increase the therapeutic effects of doxorubicin [96].
Conclusions
In summary, evidence discussed in this review highlights the potential value of discovering new biological and physiological mechanisms in breast cancer. STAT3 acts as a transcriptional activator in breast cancer, which regulates several target oncogenes and affects breast cancer progression, proliferation, apoptosis, metastasis and chemoresistance. It is intriguing that various upstream regulators and downstream target genes have been newly discovered, suggesting potential targets that can be used for breast cancer therapy. Among these pathways, circuit loops and network crosstalk are notable. Together with the development of neural networks, these phenomena remind us that signaling pathways may not be regulated only in sequential order, suggesting that findings regarding the feedback-loops and networks still need our continuous attention. Using Bayesian inference, a mathematic framework, researchers have found that combination therapy targeting mTOR and STAT3 may be the best therapeutic target in breast cancer [107]. There were also several efficient and available clinical trials targeting STAT3, which was recently reported by Qin et al. [108]. Notably, several new specific STAT3 inhibitors have been found in recent years. Structure optimization of these inhibitors for reduced cytotoxicity to normal tissues and higher stability may be an interesting direction for researchers. Treatment with STAT3 inhibitors alone or combined with other clinical therapeutic drugs may provide more promising effects on suppressing or reversing chemoresistance in breast cancer. Especially for breast cancer patients suffering from doxorubicin or capecitabine resistance, STAT3 inhibitors instead of expensive monoclonal antibodies may be more beneficial. Therefore, STAT3 remains to be a strong clinical target for breast cancer prevention and therapy, which is worth continuous research.
Availability of data and materials
Not applicable
Abbreviations
- AKT:
-
Protein kinase B
- CCL:
-
Chemokine ligand
- COX:
-
Cyclooxygenase
- CPT:
-
Carnitine palmitoyltransferase
- EGF:
-
Epidermal growth factor
- FAO:
-
Fatty acid beta-oxidation
- FGF:
-
Fibroblast growth factor
- GRAMD:
-
GRAM domain-containing protein
- GRP:
-
Glucose-regulated protein
- HIF:
-
Hypoxia inducible factor
- IGF:
-
Insulin-like growth factor
- IL:
-
Interleukin
- JAK:
-
Janus kinase
- KLF:
-
Krüppel-like factor
- LIF:
-
Leukemia inhibitory factor
- MAPK:
-
Mitogen-activated protein kinase
- MEST:
-
Mesoderm-specific transcript
- MMP:
-
Matrix metallopeptidases
- MUC:
-
Mucin-1-C
- NEAT:
-
Nuclear enriched abundant transcript
- NPRA:
-
Natriuretic peptide receptor A
- OSM:
-
Oncostatin M
- PTPN:
-
Protein tyrosine phosphatase
- ROS:
-
Reactive oxygen species
- SMYD:
-
SET and MYND ( myeloid-Nervy-DEAF-1) domain-containing protein
- STAT:
-
Signal transducer and activator of transcription
- TFEB:
-
Transcription factor EB
- VASP:
-
Vasodilator-stimulated phosphoprotein
References
Levy DE, Lee CK. What does Stat3 do? J Clin Investig. 2002;109:1143–8.
Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol. 2016;11:308–18.
Mitchell TJ, John S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology. 2005;114:301–12.
Darnell JE, Kerr IM, Stark GR. JAK-stat pathways and transcriptional activation in response to IFNS and other extracellular signaling proteins. Science. 1994;264:1415–21.
Siveen KS, Sikka S, Surana R, Dai XY, Zhang JW, Kumar AP, Tan BKH, Sethi G, Bishayee A. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845:136–54.
Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao YX, Pestell RG, Albanese C, Darnell JE. Stat3 as an oncogene. Cell. 1999;98:295–303.
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736–46.
Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19:2607–11.
Mitsuyama K, Matsumot S, Masuda J, Yamasaki H, Kuwaki K, Takedatsu H, Sata M. Therapeutic strategies for targeting the IL-6/STAT3 cytokine signaling pathway in inflammatory bowel disease. Anticancer Res. 2007;27:3749–56.
Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia WY, Wei YK, Bartholomeusz G, Shih JY, Hung MC. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–89.
Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol. 2015;34:75–82.
Hashemi V, Masjedi A, Hazhir-karzar B, Tanomand A, Shotorbani SS, Hojjat-Farsangi M, Ghalamfarsa G, Azizi G, Anvari E, Baradaran B, Jadidi-Niaragh F. The role of DEAD-box RNA helicase p68 (DDX5) in the development and treatment of breast cancer. J Cell Physiol. 2019;234:5478–87.
Tawara K, Scott H, Emathinger J, Wolf C, LaJoie D, Hedeen D, Bond L, Montgomery P, Jorcyk C. HIGH expression of OSM and IL-6 are associated with decreased breast cancer survival: synergistic induction of IL-6 secretion by OSM and IL-1beta. Oncotarget. 2019;10:2068–85.
Tawara K, Scott H, Emathinger J, Ide A, Fox R, Greiner D, LaJoie D, Hedeen D, Nandakumar M, Oler AJ, et al. Co-expression of VEGF and IL-6 family cytokines is associated with decreased survival in HER2 negative breast cancer patients: subtype-specific IL-6 family cytokine-mediated VEGF secretion. Transl Oncol. 2019;12:245–55.
Hao SN, Chen X, Wang F, Shao QQ, Liu J, Zhao H, Yuan C, Ren HX, Mao HT. Breast cancer cell-derived IL-35 promotes tumor progression via induction of IL-35-producing induced regulatory T cells. Carcinogenesis. 2018;39:1488–96.
Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, Zhong H, Schneider RJ. Inflammatory breast cancer promotes development of M2 tumor-associated macrophages and cancer mesenchymal cells through a complex chemokine network. Cancer Res. 2019;79:3360–71.
Ma M, Huang W, Kong DH. IL-17 inhibits the accumulation of myeloid-derived suppressor cells in breast cancer via activating STAT3. Int Immunopharmacol. 2018;59:148–56.
Li A, Chen P, Leng Y, Kang JH. Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3-COX2-dependent pathway. Oncogene. 2018;37:5952–66.
Li L, Zhou JX, Calvet JP, Godwin AK, Jensen RA, Li XG. Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis. 2018;9:17.
Pang Y, Wu J, Li X, Wang C, Wang M, Liu J, Yang G. NEAT1/miR-124/STAT3 feedback loop promotes breast cancer progression. Int J Oncol. 2019;55(3):745–54.
Hosea R, Hardiany NS, Ohneda O, Wanandi SI. Glucosamine decreases the stemness of human ALDH(+) breast cancer stem cells by inactivating STAT3. Oncol Lett. 2018;16:4737–44.
Veenstra C, Karlsson E, Mirwani SM, Nordenskjold B, Fornander T, Perez-Tenorio G, Stal O. The effects of PTPN2 loss on cell signalling and clinical outcome in relation to breast cancer subtype. J Cancer Res Clin Oncol. 2019;145:1845–56.
Sun YQ, Li XQ, Zhang LL, Liu X, Jiang BH, Long ZG, Jiang YY. Cell permeable NBD peptide-modified liposomes by hyaluronic acid coating for the synergistic targeted therapy of metastatic inflammatory breast cancer. Mol Pharm. 2019;16:1140–55.
Kim SL, Choi HS, Kim JH, Jeong DK, Kim KS, Lee DS. Dihydrotanshinone-induced NOX5 activation inhibits breast cancer stem cell through the ROS/Stat3 signaling pathway. Oxidative Med Cell Longev. 2019;16:9296439. https://doi.org/10.1155/2019/9296439.
Woosley AN, Dalton AC, Hussey GS, Howley BV, Mohanty BK, Grelet S, Dincman T, Bloos S, Olsen SK, Howe PH. TGF beta promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene. 2019;38:3794–811.
Egusquiaguirre SP, Yeh JE, Walker SR, Liu SH, Frank DA. The STAT3 target gene TNFRSF1A modulates the NF-kappa B pathway in breast cancer cells. Neoplasia. 2018;20:489–98.
Abrhale T, Brodie A, Sabnis G, Macedo L, Tian CS, Yue BB, Serrero G. GP88 (PC-cell derived growth factor, progranulin) stimulates proliferation and confers letrozole resistance to aromatase overexpressing breast cancer cells. BMC Cancer. 2011;11:10.
Wang WG, Hayashi J, Serrero G. PC cell-derived growth factor confers resistance to dexamethasone and promotes tumorigenesis in human multiple myeloma. Clin Cancer Res. 2006;12:49–56.
Laudisi F, Cherubini F, Di Grazia A, Dinallo V, Di Fusco D, Frenz E, Ortenzi A, Salvatori I, Scaricamazza S, Monteleone I, et al. Progranulin sustains STAT3 hyper-activation and oncogenic function in colorectal cancer cells. Mol Oncol. 2019;13:2142–59.
Wang JB, Wang LW, Li Y, Huang CQ, Zheng CH, Li P, Xie JW, Lin JX, Lu J, Chen QY, et al. CDK5RAP3 acts as a tumor suppressor in gastric cancer through inhibition of beta-catenin signaling. Cancer Lett. 2017;385:188–97.
Egusquiaguirre SP, Liu SH, Tosic I, Jiang K, Walker SR, Nicolais M, Saw TY, Xiang M, Bartel K, Nelson EA, Frank DA. CDK5RAP3 is a co-factor for the oncogenic transcription factor STAT3. Neoplasia. 2020;22:47–59.
Lin W-h, Dai W-g, Xu X-d, Yu Q-h. Zhang B, Li J, Li H-p: Downregulation of DPF3 promotes the proliferation and motility of breast cancer cells through activating JAK2/STAT3 signaling. Biochem Biophys Res Commun. 2019;514:639–44.
Chun J, Song K, Kim YS. Sesquiterpene lactones-enriched fraction of Inula helenium L. induces apoptosis through inhibition of signal transducers and activators of transcription 3 signaling pathway in MDA-MB-231 breast cancer cells. Phytother Res. 2018;32:2501–9.
Xie Q, Yang ZJ, Huang XM, Zhang ZK, Li JB, Ju JH, Zhang H, Ma JY. Ilamycin C induces apoptosis and inhibits migration and invasion in triple-negative breast cancer by suppressing IL-6/STAT3 pathway. J Hematol Oncol. 2019;12:14.
Chang RX, Song LL, Xu Y, Wu YJ, Dai C, Wang XY, Sun X, Hou YY, Li W, Zhan XB, Zhan LX. Loss of Wwox drives metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat Commun. 2018;9:12.
Pham TH, Bak Y, Kwon T, Kwon SB, Oh JW, Park JH, Choi YK, Hong JT, Yoon DY. Interleukin-32 theta inhibits tumor-promoting effects of macrophage-secreted CCL18 in breast cancer. Cell Commun Signal. 2019;17:14.
Park Y, Kim J. Regulation of IL-6 signaling by miR-125a and let-7e in endothelial cells controls vasculogenic mimicry formation of breast cancer cells. BMB Rep. 2019;52:214–9.
Shi PF, Chen C, Li X, Wei ZJ, Liu ZM, Liu YJ. MicroRNA-124 suppresses cell proliferation and invasion of triple negative breast cancer cells by targeting STAT3. Mol Med Rep. 2019;19:3667–75.
Zhang Y, Dai GQ. Overexpression of MicroRNA-9 inhibits proliferation of human breast cancer cells by targeting STAT3. Trop J Pharm Res. 2018;17:1753–8.
Li JP, Xiang Y, Fan LJ, Yao A, Li H, Liao XH. Long noncoding RNA H19 competitively binds miR-93-5p to regulate STAT3 expression in breast cancer. J Cell Biochem. 2019;120:3137–48.
Li ZM, He F, Yang ZJ, Cao XM, Dai SY, Zou J, Xu PS, Zhou Z. Exosomal miR-25-3p derived from hypoxia tumor mediates IL-6 secretion and stimulates cell viability and migration in breast cancer. RSC Adv. 2019;9:1451–9.
Yao A, Xiang Y, Si YR, Fan LJ, Li JP, Li H, Guo W, He HX, Liang XJ, Tan Y, et al. PKM2 promotes glucose metabolism through a let-7a-5p/Stat3/hnRNP-A1 regulatory feedback loop in breast cancer cells. J Cell Biochem. 2019;120:6542–54.
Wang H, Yao F, Luo SY, Ma K, Liu M, Bai LC, Chen S, Song C, Wang TY, Du Q, et al. A mutual activation loop between the Ca2+−activated chloride channel TMEM16A and EGFR/STAT3 signaling promotes breast cancer tumorigenesis. Cancer Lett. 2019;455:48–59.
Hu GW, Pen W, Wang M. TRIM14 promotes breast cancer cell proliferation by inhibiting apoptosis. Oncol Res. 2019;27:439–47.
Guan MX, Tong YN, Guan MH, Liu XB, Wang M, Niu RF, Zhang F, Dong D, Shao J, Zhou YL. Lapatinib inhibits breast cancer cell proliferation by influencing PKM2 expression. Technol Cancer Res Treat. 2018;17:12.
Wang LP, Shi HM, Zhang XY, Zhang XL, Liu Y, Kang WY, Shi XY, Wang TX. I157172, a novel inhibitor of cystathionine -lyase, inhibits growth and migration of breast cancer cells via SIRT1-mediated deacetylation of STAT3. Oncol Rep. 2019;41:427–36.
Kamran MZ, Patil P, Gude RP. Role of STAT3 in cancer metastasis and translational advances. Biomed Res Int. 2013;15:421821. https://doi.org/10.1155/2013/421821.
Zhang F, Yin GD, Han XF, Jiang XQ, Bao ZS. Chlorogenic acid inhibits osteosarcoma carcinogenesis via suppressing the STAT3/Snail pathway. J Cell Biochem. 2019;120:10342–50.
Li ZZ, Chen YD, An TT, Liu PF, Zhu JY, Yang HC, Zhang W, Dong TX, Jiang J, Zhang Y, et al. Nuciferine inhibits the progression of glioblastoma by suppressing the SOX2-AKT/STAT3-Slug signaling pathway. J Exp Clin Cancer Res. 2019;38:15.
Ma Q, Gao F-F, He X, Li K, Gao Y, Xu X-L, Jiang N-H, Ding L, Song W-J, He Y-Q, et al. Antitumor effects of saikosaponin b2 on breast cancer cell proliferation and migration. Mol Med Rep. 2019;20:1943–51.
Kim MS, Lee HS, Kim YJ, Lee DY, Kang SG, Jin W. MEST induces Twist-1-mediated EMT through STAT3 activation in breast cancers. Cell Death Differ. 2019;26(12):2594–606.
Khanna P, Lee JS, Sereemaspun A, Lee H, Baeg GH. GRAMD1B regulates cell migration in breast cancer cells through JAK/STAT and Akt signaling. Sci Rep. 2018;8:10.
Doherty MR, Parvani JG, Tamagno I, Junk DJ, Bryson BL, Cheon HJ, Stark GR, Jackson MW. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019;21:12.
Han ML, Wang YM, Guo GC, Li L, Dou DW, Ge X, Lv PW, Wang F. Gu YT: microRNA-30d mediated breast cancer invasion, migration, and EMT by targeting KLF11 and activating STAT3 pathway. J Cell Biochem. 2018;119:8138–45.
Liang MM, Ma QY, Ding N, Luo F, Bai Y, Kang F, Gong XS, Dong R, Dai JJ, Dai QJ, et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019;10:12.
Gao X, Liu X, Lu Y, Wang Y, Cao W, Liu X, Hu H, Wang H. PIM1 is responsible for IL-6-induced breast cancer cell EMT and stemness via c-myc activation. Breast Cancer (Tokyo, Japan). 2019;26(5):663–71.
Hata T, Rajabi H, Yamamoto M, Jin C, Ahmad R, Zhang Y, Kui L, Li W, Yasumitzu Y, Hong D, et al. Targeting MUC1-C inhibits TWIST1 signaling in triple-negative breast cancer. Mol Cancer Ther. 2019;18(10):1744–54. https://doi.org/10.1158/1535-7163.MCT-19-0156.
Qu JK, Zhao XX, Liu X, Sung YC, Wang JZ, Liu L, Wang JS, Zhang J. Natriuretic peptide receptor a promotes breast cancer development by upregulating MMP9. Am J Cancer Res. 2019;9:1415–28.
Monteleone E, Orecchia V, Corrieri P, Schiavone D, Avalle L, Moiso E, Savino A, Molineris I, Provero P, Poli V. SP1 and STAT3 functionally synergize to induce the RhoU small GTPase and a subclass of non-canonical WNT responsive genes correlating with poor prognosis in breast cancer. Cancers. 2019;11:17.
Dai XP, Geng F, Dai JL, Li MS, Liu M. Rho GTPase activating protein 24 (ARHGAP24) regulates the anti-cancer activity of Sorafenib against breast cancer MDA-MB-231 cells via the signal transducer and activator of transcription 3 (STAT3) signaling pathway. Med Sci Monit. 2018;24:8669–77.
Zhao LM, Pang AX, Li YC. Function of GCN5 in the TGF-1-induced epithelial-to-mesenchymal transition in breast cancer. Oncol Lett. 2018;16:3955–63.
Abyaneh HS, Gupta N, Alshareef A, Gopal K, Lavasanifar A, Lai R. Hypoxia induces the acquisition of cancer stem-like phenotype via upregulation and activation of signal transducer and activator of transcription-3 (STAT3) in MDA-MB-231, a triple negative breast cancer cell line. Cancer Microenviron. 2018;11:141–52.
Ma J-H, Qi J, Lin S-Q, Zhang C-Y, Liu F-Y, Xie W-D, Li X. STAT3 targets ERR-alpha to promote epithelial-mesenchymal transition, migration and invasion in triple negative breast cancer cells. Mol Cancer Res. 2019;17(11):2184–95.
Tzeng YDT, Liu PF, Li JY, Liu LF, Kuo SY, Hsieh CW, Lee CH, Wu CH, Hsiao M, Chang HT, Shu CW. Kinome-wide siRNA screening identifies Src-enhanced resistance of chemotherapeutic drugs in triple-negative breast cancer cells. Front Pharmacol. 2018;9:11.
Castellaro AM, Rodriguez-Baili MC, Di Tada CE, Gil GA. Tumor-associated macrophages induce endocrine therapy resistance in ER plus breast cancer cells. Cancers. 2019;11:29.
Wang TY, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue CY, Zhang CY, Lifshitz V, Song J, Yuan Y, et al. JAK/STAT3-regulated fatty acid beta-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018;27:136–+.
Wang SQ, Yao YN, Yao MY, Fu PF, Wang WL. Interleukin-22 promotes triple negative breast cancer cells migration and paclitaxel resistance through JAK-STAT3/MAPKs/AKT signaling pathways. Biochem Biophys Res Commun. 2018;503:1605–9.
Liu C, Xing H, Guo C, Yang Z, Wang Y, Wang Y. MiR-124 reversed the doxorubicin resistance of breast cancer stem cells through STAT3/HIF-1 signaling pathways. Cell Cycle (Georgetown, Tex). 2019;18(18):2215–27.
Cheng CC, Shi LH, Wang XJ, Wang SX, Wan XQ, Liu SR, Wang YF, Lu Z, Wang LH, Ding Y. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int J Oncol. 2018;53:339–48.
Kim JY, Kim JC, Lee JY, Park MJ. Oct4 suppresses IR-induced premature senescence in breast cancer cells through STAT3-and NF-B-mediated IL-24 production. Int J Oncol. 2018;53:47–58.
Xiang S, Dauchy RT, Hoffman AE, Pointer D, Frasch T, Blask DE, Hill SM. Epigenetic inhibition of the tumor suppressor ARHI by light at night-induced circadian melatonin disruption mediates STAT3-driven paclitaxel resistance in breast cancer. J Pineal Res. 2019;67(2):e12586.
Tseng C-C, Zhang P, Lee AS. The COOH-terminal proline-rich region of GRP78 is a key regulator of its cell surface expression and viability of tamoxifen-resistant breast cancer cells. Neoplasia (New York, NY). 2019;21:837–48.
Wang L, Wang QR, Gao MZ, Fu L, Li Y, Quan HT, Lou LG. STAT3 activation confers trastuzumab-emtansine (T-DM1) resistance in HER2-positive breast cancer. Cancer Sci. 2018;109:3305–15.
Feng F, Zhu XL, Wang CY, Chen L, Cao WP, Liu YQ, Chen Q, Xu WL. Downregulation of hypermethylated in cancer-1 by miR-4532 promotes adriamycin resistance in breast cancer cells. Cancer Cell Int. 2018;18:12.
Chen D, Ma YM, Li PQ, Liu M, Fang Y, Zhang JJ, Zhang BL, Hui YY, Yin Y. Piperlongumine induces apoptosis and synergizes with doxorubicin by inhibiting the JAK2-STAT3 pathway in triple-negative breast cancer. Molecules. 2019;24:15.
Kettner NM, Vijayaraghavan S, Durak MG, Bui TY, Kohansal M, Ha MJ, Liu B, Rao XY, Wang J, Yi M, et al. Combined inhibition of STAT3 and DNA repair in palbociclib-resistant ER-positive breast cancer. Clin Cancer Res. 2019;25:3996–4013.
Tian JL, Chen X, Fu SL, Zhang RJ, Pan L, Cao Y, Wu XJ, Xiao H, Lin HJ, Lo HW, et al. Bazedoxifene is a novel IL-6/GP130 inhibitor for treating triple-negative breast cancer. Breast Cancer Res Treat. 2019;175:553–66.
Liu CL, Dong LH, Sun Z, Wang L, Wang QP, Li HY, Zhang J, Wang XJ. Esculentoside A suppresses breast cancer stem cell growth through stemness attenuation and apoptosis induction by blocking IL-6/STAT3 signaling pathway. Phytother Res. 2018;32:2299–311.
Choi HS, Kim JH, Kim SL, Deng HY, Lee D, Kim CS, Yun BS, Lee DS. Catechol derived from aronia juice through lactic acid bacteria fermentation inhibits breast cancer stem cell formation via modulation Stat3/IL-6 signaling pathway. Mol Carcinog. 2018;57:1467–79.
Al-Asmari AK, Riyasdeen A, Islam M. Scorpion venom causes upregulation of p53 and downregulation of Bcl-x(L) and BID protein expression by modulating signaling proteins Erk(1/2) and STAT3, and DNA damage in breast and colorectal cancer cell lines. Integr Cancer Ther. 2018;17:271–81.
He JY, Wei XH, Li SJ, Quan XP, Li RM, Du HZ, Yuan ST, Sun L. DT-13 suppresses breast cancer metastasis by modulating PLOD2 in the adipocytes microenvironment. Phytomedicine. 2019;59:9.
Yang YG, Zhou HF, Liu WM, Wu J, Yue XL, Wang JC, Quan LN, Liu H, Guo L, Wang ZP, et al. Ganoderic acid A exerts antitumor activity against MDA-MB-231 human breast cancer cells by inhibiting the Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway. Oncol Lett. 2018;16:6515–21.
Qiu CW, Zhang T, Zhu XY, Qiu JX, Jiang KF, Zhao G, Wu HC, Deng GZ. Methylseleninic acid suppresses breast cancer growth via the JAK2/STAT3 pathway. Reprod Sci. 2019;26:829–38.
Jang H, Ko H, Song K, Kim YS. A Sesquiterpenoid from Farfarae Flos induces apoptosis of MDA-MB-231 human breast cancer cells through inhibition of JAK-STAT3 signaling. Biomolecules. 2019;9(7):278. https://doi.org/10.3390/biom9070278.
Hu YH, Yague E, Zhao J, Wang LY, Bai JC, Yang QX, Pan T, Zhao H, Liu JJ, Zhang J. Sabutoclax, pan-active BCL-2 protein family antagonist, overcomes drug resistance and eliminates cancer stem cells in breast cancer. Cancer Lett. 2018;423:47–59.
Zhang XH, Yang Y, Liu JJ, Shen L, Shi Z, Wu J. Tagalide A and tagalol A, naturally occurring 5/6/6/6-and 5/6/6-fused cyclic dolabrane-type diterpenes: a new insight into the anti-breast cancer activity of the dolabrane scaffold. Org Chem Front. 2018;5:1176–83.
Ma X, Ning SL. Cyanidin-3-glucoside attenuates the angiogenesis of breast cancer via inhibiting STAT3/VEGF pathway. Phytother Res. 2019;33:81–9.
Huang Q, Li S, Zhang LW, Qiao XF, Zhang YY, Zhao XY, Xiao GJ, Li ZB. CAPE-pNO(2) inhibited the growth and metastasis of triple-negative breast cancer via the EGFR/STAT3/Akt/E-cadherin signaling pathway. Front Oncol. 2019;9:14.
Esparza-Lopez J, Alvarado-Munoz JF, Escobar-Arriaga E, Ulloa-Aguirre A, de Jesus I-SM. Metformin reverses mesenchymal phenotype of primary breast cancer cells through STAT3/NF-kappaB pathways. BMC Cancer. 2019;19:728.
Cui L, Bu WQ, Song J, Feng L, Xu TT, Liu D, Ding WB, Wang JH, Li CY, Ma BE, et al. Apoptosis induction by alantolactone in breast cancer MDA-MB-231 cells through reactive oxygen species-mediated mitochondrion-dependent pathway. Arch Pharm Res. 2018;41:299–313.
Chen W, Wang H, Cheng M, Ni L, Zou L, Yang Q, Cai XH, Jiao BW. Isoharringtonine inhibits breast cancer stem-like properties and STAT3 signaling. Biomed Pharmacother. 2018;103:435–42.
Ko H, Lee JH, Kim HS, Kim T, Han YT, Suh YG, Chun J, Kim YS, Ahn KS. Novel Galiellalactone analogues can target STAT3 phosphorylation and cause apoptosis in triple-negative breast cancer. Biomolecules. 2019;9:16.
Zhang ZL, Jiang QC, Wang SR. Schisandrin A reverses doxorubicin-resistant human breast cancer cell line by the inhibition of P65 and Stat3 phosphorylation. Breast Cancer. 2018;25:233–42.
Kim JW, Gautam J, Kim JE, Kim JA, Kang KW. Inhibition of tumor growth and angiogenesis of tamoxifen-resistant breast cancer cells by ruxolitinib, a selective JAK2 inhibitor. Oncol Lett. 2019;17:3981–9.
Khan MW, Saadalla A, Ewida AH, Al-Katranji K, Al-Saoudi G, Giaccone ZT, Gounari F, Zhang M, Frank DA, Khazaie K. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol Immunother. 2018;67:13–23.
Khaki-khatib F, Ghorbani M, Sabzichi M, Ramezani F, Mohammadian J. Adjuvant therapy with stattic enriches the anti-proliferative effect of doxorubicin in human ZR-75-1 breast cancer cells via arresting cell cycle and inducing apoptosis. Biomed Pharmacother. 2019;109:1240–8.
Lu L, Dong JL, Wang LL, Xia Q, Zhang D, Kim HJ, Yin T, Fan SJ, Shen Q. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene. 2018;37:5292–304.
Oh E, Kim YJ, An H, Sung D, Cho TM, Farrand L, Jang S, Seo JH, Kim JY. Flubendazole elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Int J Cancer. 2018;143:1978–93.
Yang B, Shen JW, Zhou DH, Zhao YP, Wang WQ, Zhu Y, Zhao HJ. Precise discovery of a STAT3 inhibitor from Eupatorium lindleyanum and evaluation of its activity of anti-triple-negative breast cancer. Nat Prod Res. 2019;33:477–85.
Zeng AQ, Yu Y, Yao YQ, Yang FF, Liao M, Song LJ, Li YL, Yu Y, Li YJ, Deng YL, et al. Betulinic acid impairs metastasis and reduces immunosuppressive cells in breast cancer models. Oncotarget. 2018;9:3794–804.
Zhang WD, Yu WY, Cai GP, Zhu JW, Zhang C, Li SS, Guo JP, Yin GP, Chen C, Kong LY. A new synthetic derivative of cryptotanshinone KYZ3 as STAT3 inhibitor for triple-negative breast cancer therapy. Cell Death Dis. 2018;9:11.
Locken H, Clamor C, Muller K. Napabucasin and related Heterocycle-fused Naphthoquinones as STAT3 inhibitors with Antiproliferative activity against cancer cells. J Nat Prod. 2018;81:1636–44.
Cai GP, Yu WY, Song DM, Zhang WD, Guo JP, Zhu JW, Ren YH, Kong LY. Discovery of fluorescent coumarin-benzo b thiophene 1, 1-dioxide conjugates as mitochondria-targeting antitumor STAT3 inhibitors. Eur J Med Chem. 2019;174:236–51.
Lee EJ, Kim SJ, Hahn YI, Yoon HJ, Han B, Kim K, Lee S, Kim KP, Suh YG, Na HK, Surh YJ. 15-Keto prostaglandin E-2 suppresses STAT3 signaling and inhibits breast cancer cell growth and progression. Redox Biol. 2019;23:17.
Sravanthi VS, Palaka BK, Vel G, Venkatesan R, Ampasala DR, Periyasamy L. Identification of novel inhibitors of signal transducer and activator of transcription 3 over signal transducer and activator of transcription 1 for the treatment of breast cancer by in-silico and in-vitro approach. Process Biochem. 2019;82:153–66.
Dai XX, Yin CT, Zhang Y, Guo GL, Zhao CG, Wang OC, Xiang YQ, Zhang XH, Liang G. Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J Exp Clin Cancer Res. 2018;37:11.
Vundavilli H, Datta A, Sima C, Hua J, Lopes R, Bittner ML. Bayesian inference identifies combination therapeutic targets in breast cancer. IEEE Trans Biomed Eng. 2019;66(9):2684–92.
Qin JJ, Yan L, Zhang J, Zhang WD. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res. 2019;38:16.
Acknowledgements
Not applicable
Funding
This work was supported in part by grants from Shandong Provincial Natural Science Foundation (No. ZR2019MH001), the National Natural Science Foundation of China (No.81803562 and No. 81273532), the Fundamental Research Funds for the Central Universities (No.2019ZRJC004).
Author information
Authors and Affiliations
Contributions
Dr. Jia-hui Ma and Dr. Qin Li did literature jobs and wrote this paper. Prof. Xia Li received research supports. The authors declare no conflict of interest. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
All authors agreed to consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, Jh., Qin, L. & Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun Signal 18, 33 (2020). https://doi.org/10.1186/s12964-020-0527-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-020-0527-z