
Abstract
Background
Increasing evidence implicates circular RNAs (circRNAs) have been involved in human cancer progression. However, the mechanism remains unclear. In this study, we identified novel circRNAs related to gastric cancer and constructed a circRNA-miRNA-mRNA network.
Methods
Microarray datasets GSE83521 and GSE93541 were obtained from the Gene Expression Omnibus (GEO). Then, we used computational biology to identify circRNAs that were differentially expressed in both GC tissue and plasma compared to normal controls; then, we detected the expression of the selected circRNAs in gastric cell lines by quantitative real-time polymerase chain reaction (qRT-PCR). We also identified circRNA-related candidate miRNAs and their target genes with online tools. Combining the predicted miRNAs and target mRNAs, a competing endogenous RNA regulatory network was established. Functional and pathway enrichment analyses were performed, and interactions between proteins were predicted by using String and Cytoscape. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed to elucidate the possible functions of these differentially expressed circRNAs. The regulatory network constructed using the microarray datasets (GSE83521 and GSE93541) contained three differentially co-expressed circRNAs (DECs). A circRNA-miRNA-mRNA network was constructed based on 3 circRNAs, 43 miRNAs and 119 mRNAs.
Results
GO and KEGG analysis showed that the regulation of apoptotic signaling pathway and PI3K−Akt signaling pathway were highest degrees of enrichment respectively. We established a protein-protein interaction (PPI) network consisting of 165 nodes and 170 edges and identified hub genes by using MCODE plugin in Cytoscape. Furthermore, a core circRNA-miRNA-mRNA network was constructed based on hub genes. Hsa_circ_0001013 was finally determined to play an important role in the pathogenesis of GC according to the core circRNA-miRNA-mRNA network.
Conclusions
We propose a new circRNA-miRNA-mRNA network that is associated with the pathogenesis of GC. The network may become a new molecular biomarker and could be used to develop potential therapeutic strategies for gastric cancer.
Similar content being viewed by others

Introduction
Gastric cancer is one of the most common malignant tumors. Every year, there are nearly 1 million new cases of gastric cancer around the world, making it the third leading cause of cancer-related deaths and prompting the World Health Organization to declare it a public health problem [1]. The pathogenesis of gastric cancer is multifactorial and involves multiple steps, but these steps are currently unclear. It is widely believed that Helicobacter pylori is one of the main pathogenic factors of gastric cancer [2, 3]. Almost 90% of new cases of non-cardiac gastric cancer are related to Helicobacter pylori. Although the techniques for the detection and treatment of gastric cancer have dramatically improved, the prognosis is still very poor [4]. The 5-year survival rate of patients with advanced gastric cancer is approximately 18–29% [5]. Therefore, early diagnosis and treatment are critical to improving the curative effect and reducing the mortality rate.
In 1976, a new type of 3′-5′ head-to-tail covalently closed RNA called circular RNAs (circRNAs) were identified [6, 7]. However, in subsequent decades, circular RNAs were thought to be the product of mis-splicing [8]. In recent years, it has been recognized that circRNAs are normal co-products of numerous eukaryotic protein-coding genes [9]. CircRNAs have been a hotspot of research in the fields of life science and medicine and have been identified as critical regulators of a variety of diseases, including various malignant tumors [10,11,12]. CircRNAs can regulate variable splicing or the expression of their host genes by blocking transcriptional initiation sites, and can even be translated into proteins or peptides. However, the role of competitive endogenous RNA (ceRNA) in sponging miRNA is considered to be one of the main functions of circRNA in various cancers. CircRNA is also regarded as a potential biomarker for cancers because it is more stable than linear RNA [13]. There are many differentially expressed circRNAs associated with gastric cancer. For example, Wei et al. found that circHIPK3 promotes the proliferation and migration of gastric cancer cells by sponging miR-107 and regulating BDNF expression [14, 15]. He et al. confirmed that circular RNA circ_0006282 contributes to the progression of gastric cancer by sponging miR-155 to upregulate the expression of FBXO22 [16]. Pan et al. reported that circUBA1 promotes gastric cancer proliferation and metastasis by acting as a competitive endogenous RNA by sponging miR-375 and regulating TEAD4 [17]. Additionally, circRNAs have also been proposed as diagnostic or prognostic biomarkers. Wang et al. demonstrated that hsa_circ_0005654 might serve as a new and promising diagnostic biomarker for early gastric cancer screening. The AUC, sensitivity, and specificity of hsa_circ_0005654 are significantly higher than those of present gastric cancer associated-biomarkers [18]. Although related studies have emerged, the network involved in circRNA-mediated regulation of gastric cancer remains unclear.
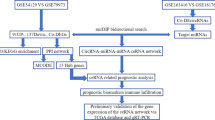
The aim of our study was to identify differentially co-expressed circRNAs in tissues and plasma of patients with gastric cancer. The expression profiles of circRNAs were obtained from the Gene Expression Omnibus (GEO). The bioinformatic data were analyzed and differentially expressed circRNAs (DECs) were screened. Next, the potential miRNAs sponged by DECs and their target genes were identified by bioinformatic analysis. Moreover, the core circRNA-miRNA-mRNA regulatory network was constructed. Gene enrichment analyses of the candidate miRNAs or mRNAs were performed with the gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases and R software, which resulted in the prediction of the signaling pathways involved in GC. A flow chart of the methods used in this study is provided in Fig. 1. The results of this study may help to clarify the potential mechanism of the pathogenesis of gastric cancer, provide new biomarkers for gastric cancer and facilitate future research in GC treatment and diagnosis.

Flow chart of the methods used in the present study. GO gene ontology, circRNA circular RNA, KEGG Kyoto Encyclopedia of Genes and Genome, mRNA messenger RNA, PPI protein-protein interaction
Methods
Microarray analysis of gene expression
Two circRNA expression profiles for human samples derived from patients with gastric cancer were obtained from GEO (www.ncbi.nlm.nih.gov/geo/). We chose the GSE83521 and GSE93541 circRNA expression profiles, both of which were generated from the Agilent-069978 Arraystar Human CircRNA microarray V1 GPL19978 platform. The GSE83521 dataset contained six gastric cancer tissues and six normal mucosa tissues, and the GSE93541 dataset included three plasma samples of gastric cancer patients and three healthy controls.
Identification of DECs
Differential expression of the circRNAs in the two datasets was analyzed by using the GEO2R online analysis tool. The absolute value of log fold change > 1.5 and p value < 0.05 were used as cut-off criteria. The significantly differentially expressed circRNAs in the two datasets were screened, and the co-expressed circRNAs were detected by Venn analysis. The basic structural features of the differentially expressed circRNAs were obtained from the Cancer-Specific CircRNA Database (http://gb.whu.edu.cn/CSCD/).
Prediction of circRNA-miRNA and miRNA-mRNA interactions
Online tools circBank (http://www.circbank.cn/) and Circular RNA Interactome (https://circinteractome.nia.nih.gov/index.html) were used to predict the possible interactions between circRNAs and miRNAs. The miRNAs predicted by both circBank and circInteractome were selected as candidate miRNAs. mirTarBase (http.//mirtarba se.mbc.nctu.edu.tw/php/index.php) was used to obtain experimentally strongly supported target genes of these miRNAs. Only candidate genes that could be verified by reporter assays as well as Western blot or qPCR experiments were selected.
GO and KEGG functional enrichment analysis
FunRich software (version 3.1.1) was used to conduct GO analysis for candidate miRNAs. GO annotation and KEGG pathway analyses were conducted with the R (version 3.6) package (http://www.bioconductor.org/) clusterProfiler to explore the potential biological roles of candidate genes. The analysis results were visualized with the ggplot2 package of R software. Both a p value and q value < 0.05 were considered significant for GO annotation, while a p value < 0.05 and q value < 1 were considered significant for KEGG pathway analysis.
Construction of the protein-protein interaction and circRNA-miRNA-mRNA network
Candidate target genes of the candidate miRNAs were put into the Search Tool for the Retrieval of Interacting Genes database (STRING, https://string-db.org/), and an interaction network chart with a combined score > 0.4 was saved and exported. Then the protein-protein interaction (PPI) network was visualized using Cytoscape software (version 3.6.1; http://cytoscape.org/). The MCODE plugin of Cytoscape software was used to identify hub genes among candidate targets. The circRNA–miRNA–mRNA network was also visualized by Cytoscape software.
Cell culture
The gastric cancer cell line SGC-7901 and human gastric mucosal epithelial cell line GES-1 were purchased from the Cell Resource Center, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Science. All of the cell lines were maintained under the recommended culture conditions and incubated at 37 °C in a humidified environment with 5% CO2.
RNA extraction and quantitative real-time polymerase chain reaction
Total RNA was isolated from the cell lines with TRIzol reagent (Invitrogen, CA, USA) following the manufacturer’s instructions. The concentration and purity of the total RNA samples were assessed using a NanoDrop spectrophotometer (Thermo, Wilmington, DE, USA). Total RNA was reverse transcribed using HiScript Q RT SuperMix for qPCR with gDNA wiper (Vazyme Biotech, Nanjing, China), and qPCR assays were performed in triplicate using the AceQ qPCR SYBR Green Master Mix kit (Vazyme Biotech, Nanjing, China) on a 7500 real-time PCR system (ABI). The divergent primers used for detecting circRNAs were synthesized from Shanghai Generay Biotech (Shanghai, China), and β-actin was used as an internal control. The following primer pairs were used for qPCR: β-actin forward, 5′-AGAAAATCTGGCACCACACC-3′ and reverse, 5′-CAGAGGCGTACAGGGATAGC-3′; hsa_circ_0001013 forward, 5′-GTCAAAGGAAGCAAAAGAAAGTCT-3′ and reverse, 5′-GATCGCACCTCTACACTCCA-3′; hsa_circ_0007376 forward, 5′-ATCGACTCCATGGCCAACTC-3′ and reverse, 5′-AAGCCCCGGAGAACAGC-3′; hsa_circ_0043947 forward, 5′-CAATTGTGGTTGTGCAGCC-3′ and reverse, 5′-ACACAAACTCAGCATCATGGA-3′. The expression of circRNAs was normalized to that of the internal control β-actin by using the 2-ΔΔC method [19].
Statistical analysis
All computations were carried out using GraphPad Prism 8 (GraphPad Software, CA, USA). Data are expressed as mean ± SEM. Student’s t test was conducted to compare the differences in circRNA expression between GES-1 and SGC-7901 cells. P < 0.05 was considered statistically significant.
Results
Identification of DECs
Two circRNA expression profiles, GSE83521 and GSE93541, were obtained from GEO, and the GEO2R method was applied to analyze DECs. The GSE83521 dataset was derived from gastric cancer tissues, and the GSE93541 dataset was derived from plasma samples. CircRNAs that were differentially expressed both in the tissues and the plasmas of gastric cancer patients were selected as our target circRNAs. We found that 53 circRNAs were differentially expressed in GSE83521, including 39 upregulated and 14 downregulated circRNAs, while 267 differentially expressed circRNAs were identified in GSE93541, including 138 upregulated and 129 downregulated circRNAs. Among them, 3 common upregulated and 0 common downregulated circRNAs were observed in the two circRNA expression profiles. A Venn diagram of the results is shown in Fig. 2A and B. The upregulated circRNAs that overlapped in the two datasets (hsa_circ_0001013, hsa_circ_0007376, hsa_circ_0043947) were selected for further analysis. Details of the overlapping upregulated circRNAs are listed in Table 1, and the basic structural features of the three selected circRNAs are shown in Fig. 2C.

Differentially co-expressed circRNAs in gastric cancer tissues and plasma. A, B Venn diagram used to select the three overlapping differentially expressed circRNAs detected by analysis of the GSE89143 and GSE93541 datasets. C The essential characteristics and basic structural patterns of DECs were analyzed by the cancer-specific circRNA database. MRE miRNA response element, RBP RNA binding protein, ORF open reading frame
Expression of circRNAs in datasets and cell lines
As shown in Fig. 3A and B, the expression patterns of the three selected circRNAs were upregulated in both tissues and plasma samples according to the datasets. We also detected the expression of selected circRNAs in the gastric cancer cell line SGC-7901 and the human gastric epithelial cell line GES-1 by qRT-PCR. The results showed that all three selected circRNAs had higher expression levels in SGC-7901 than in GES-1 as shown in Fig. 3C.

Expression of DECs in gastric cancer samples and cell lines. A The expression of three selected circRNAs in six gastric cancer tissues and six normal mucosa tissues. B The expression of three selected circRNAs in three gastric cancer plasmas and three healthy controls. C The relative expression of three selected circRNAs in gastric cancer cell line SGC-7901 and human gastric epithelial cell line GES-1. **< 0.05
Prediction of circRNA-miRNA and their functional analysis
An increasing amount of evidence demonstrates that circRNAs might function as competing endogenous RNAs (ceRNAs) that operate by competitively binding common microRNAs (miRNAs) and increase the expression of the target genes of these miRNAs. Target miRNAs of the three selected circRNAs were predicted by two online tools, circBank and circInteractome. A total of 43 consensus miRNAs from both prediction tools were identified and DECs potentially binding to these miRNAs are presented in Table 2. The results showed that one specific circRNA might bind to more miRNAs, while different circRNAs could interact with one specific miRNA. FunRich software was used for GO analysis of the 43 miRNAs. The top five enriched terms are shown in Fig. 4: “regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolism,” “regulation of cell growth,” “Cell cycle,” “regulation of enzyme activity”: and “cell-cell adhesion’ for biological progress (BP),” “cytoplasm and nucleus,” “nucleus,” “lysosome,” “actin cytoskeleton” and “endosome” for cellular component (CC), and “transcription factor activity,” “receptor signaling complex scaffold activity”, “translation regulator activity”, “protein binding” and “RNA binding” for molecular function (MF). All of these results indicated that circRNAs might impact on GC progression by modulating various miRNAs.

GO analysis for 43 miRNAs by RichFun software. A–C Top five enrichment items for BP, CC, and MF respectively. BP biological progress, CC cellular component, MF molecular function
Construction of the ceRNA network
We identified 119 experimentally strongly supported target genes of 43 miRNAs by using the mirTarBase online tool (Table 2). Then, we used 3 circRNAs, 43 miRNAs, and 119 mRNAs in Cytoscape 3.6.1 to construct a circRNA-miRNA-mRNA visualization network (Fig. 5).

The circRNA-miRNA-mRNA network was constructed based on 3 circRNAs, 43 miRNAs, and 119 mRNAs
Functional and pathway enrichment analysis and PPI network
GO analysis indicated that the 119 mRNAs were mainly enriched in “regulation of apoptotic signaling pathway,” “autophagy,” and “process utilizing autophagic mechanism” (BPs); “glutamatergic synapse,” “nuclear chromatin” and “external side of plasma membrane” (CCs); and “DNA-binding transcription activator activity, RNA polymeraseII-specific” (MFs) (Fig. 6A). KEGG pathway analysis revealed strong enrichment in the “PI3K-Akt signaling pathway” (Fig. 6B). After obtaining the target genes of candidate miRNAs, we created a PPI network composed of 165 nodes and 170 edges (Fig. 7A). Following the identification of the vital functions of hub genes in the network, 18 hub genes (CCND2, STAT3, TP53, MCL1, MYC, FOXO1, FOXO3, BCL2L11, PTEN, MTOR, CDH1, CASP3, IL6, GSK3B, CDKN1A, MAPK1, SMAD4, CDC42) were identified in GC using the MCODE plugin, MCODE_Score = 13.76. These hub genes were predicted target genes for hsa-miR-197-3p, hsa-miR-451a, hsa-miR-136-5p, hsa-miR-337-3p, hsa-miR-654-3p, hsa-miR-182-5p, hsa-miR-1228-3p, hsa-miR-942-5p, hsa-miR-488-3p and hsa-miR-876-3p, and all 10 miRNAs were predicted miRNAs for hsa_circ_0001013. The core circRNA–miRNA–mRNA network based on hub genes is displayed in Fig. 7B.

GO and KEEG pathway analysis for 119 mRNAs. A Top 10 enriched gene ontology (GO) terms. B Top 30 significant KEGG pathways. KEEG Kyoto Encyclopedia of Genes and Genomes, BP biological process, CC cellular component, MF molecular function

PPI network and core circRNA-miRNA-mRNA network. A PPI network composed of 165 nodes and 170 edges, and hub genes identified from the PPI network by Cytoscape. B The core circRNA-miRNA-mRNA network based on 1circRNA, 10 miRNAs, and the 18 hub genes. circRNA, circular RNA; miRNA, microRNA; mRNA, messenger RNA; PPI, protein-protein interaction
Discussion
Emerging evidence indicates that circRNAs are frequently dysregulated in various cancers and may play a vital role in cancer progression. Moreover, the increased stability of circRNAs compared with that of linear RNAs in the serum, makes circRNAs vital biomarkers for cancer diagnosis and prognosis. However, the mechanism of circRNA in cancer progression has not been clearly elucidated. Current evidence demonstrates that circRNAs can target miRNAs, often referred to as “miRNA sponges,” to reduce the level of miRNAs and release their targeting inhibition to mRNAs. These studies have shown that the circRNA-miRNA-mRNA axis can play a role as a wide range of gene expression regulatory networks and can be used as a biomarker for cancer diagnosis and prognosis. Gastric cancer is one of the most common malignant tumors of the digestive tract. At present, radical resection is the main treatment for gastric cancer, but the prognosis of patients is still not satisfactory. Previous studies have confirmed that circRNAs are involved in the tumorigenesis and progression of gastric cancer. Wu et al. investigated the functions of circRNA ring finger protein 111 (circ-RNF111) in GC and found that Circ-RNF111 was higher expressed in GC tissues. Silencing of circ-RNF111 restrained cell viability, colony formation, migration, invasion, cell cycle process, and glycolysis and induced apoptosis in GC cells by regulating KLF12 expression via absorbing miR-876-3p [20]. Liu et al. found that circ-PVT1 contributes to paclitaxel resistance in gastric cancer cells through the regulation of ZEB1 expression by sponging miR-124-3p [21]. Xie et al. showed that the downregulated expression of hsa_circ_0074362 in gastric cancer is related to lymph node metastasis and has diagnostic value for gastric cancer [22]. The expression of hsa_circ_0000190 in gastric cancer tissue and serum is downregulated compared to that in normal samples, suggesting that it may be a more potential biomarker of gastric cancer than the common tumor markers CEA and CA19-9 [23]. Liu et al. attempted to construct the regulatory network of circRNA-miRNA-mRNA in gastric cancer. Their study focused on three downregulated circRNAs (hsa_circ_0001190, hsa_circ_0036287 and hsa_circ_0048607) in gastric cancer tissues and plasma and successfully established the circRNA-miRNA-hub gene network through bioinformatics analysis [24]. However, biomarkers with relatively low abundance are less sensitive to detection than those with high abundance. Therefore, based on previous studies, we attempted to find highly expressed circRNAs in gastric cancer tissues and plasma and to further improve the circRNA-miRNA-mRNA regulatory network to provide a theoretical basis for the study of gastric cancer. In our study, we screened the circRNA expression profiles in the GSE89143 and GSE93541 GEO datasets for gastric cancer tissue and plasma ssample data to identify differentially expressed circRNAs, with the significance threshold set as P < 0.05 and |log2FC| > 1.5. Three upregulated circRNAs were selected for further analysis, namely hsa_circ_0001013, hsa_circ_0007376, and hsa_circ_0043947. They have not been reported until now.
Currently, it is generally believed that circRNAs have miRNA response elements (MREs) and can interact with miRNA through a “sponging” action. CiRS-7 was the first circRNA to be reported to act as a ceRNA [25] and circHECTD1 has been shown to act as a ceRNA to promote gastric cancer proliferation by sponging miR-1256 [26]. We also screened 43 miRNAs that may interact with the three selected circRNAs through bioinformatics analyis, and the GO analysis showed that these 43 miRNAs were involved in the regulation of nucleobase, regulation of cell growth, etc. These biological processes are also very active in the development and progression of tumors. We further predicted the downstream target genes of these 43 miRNAs by an online tool and a total of 119 target mRNAs were selected. Next, we analyzed these target genes by using GO and KEGG pathway analysis to gain an understanding of the function of the target genes. The GO analysis showed that the target genes mainly participated in the regulation of apoptotic signaling pathway for BP, glutamatergic synapse for CC and DNA-binding transcription activator activity, RNA polymerase II-specific for MF. The KEGG pathway analysis indicated that the most enriched term was the PI3K-Akt signaling pathway, which is one of the most frequently activated downstream signal transduction pathways in human cancer. The PI3K-Akt signaling pathway plays an important role in regulating cell proliferation, growth, and apoptosis. Peng et al. reported that hsa_circ_0010882 promotes the progression of gastric cancer via regulation of the PI3K/Akt/mTOR signaling pathway [27]. We established a protein-protein interaction (PPI) network consisting of 165 nodes and 170 edges and identified 18 hub genes by using the MCODE plugin in Cytoscape. The 18 hub genes have been reported to be associated with gastric cancer, which are CCND2 [28], STAT3 [29], TP53 [30], MCL1 [31], MYC [32], FOXO1 [33], FOXO3 [34], BCL2L11 [35], GSK3B [36], CDKN1A [37], CDH1 [38], PTEN [39], MTOR [40], MAPK1 [41], CASP3, CDC42 [42], SMAD4 [43], and IL6 [44]. All of them were predicted target genes for hsa-miR-197-3p, hsa-miR-451a, hsa-miR-136-5p, hsa-miR-337-3p, hsa-miR-654-3p, hsa-miR-182-5p, hsa-miR-1228-3p, hsa-miR-942-5p, hsa-miR-488-3p, and hsa-miR-876-3p, and all 10 miRNAs were predicted miRNAs for hsa_circ_0001013. Therefore, a core circRNA-miRNA-mRNA regulatory network was constructed based on 1 circRNA, 10 miRNAs, and 18 hub genes which are called gastric cancer-related genes. Finally, hsa_circ_0001013 was determined to play a key role in the pathogenesis of GC. Although the exact mechanisms of circRNAs in gastric cancer are not clear, our results provide insights into the underlying mechanisms of gastric cancer pathogenesis. The results of this study are based solely on bioinformatics models. This is a pilot study and further studies are needed to verify the biological role of these circRNAs in gastric cancer.
Conclusions
We obtained circRNA expression profiles in gastric cancer tissue and plasma from the GEO database. Three circRNAs that were upregulated in gastric cancer tissue and plasma compared to normal controls were identified as potential regulators. A core circRNA-miRNA-mRNA network was constructed by using bioinformatics methods. We found that hsa_circ_0001013 may act as a ceRNA and play a critical role in carcinogenesis-related pathways. These findings provide a new pathway for mechanistic studies and offer potential biomarkers for GC. Further studies are needed to examine the role of regulatory modules in GC carcinogenesis.
Availability of data and materials
We declare that the data and materials in this study will be provided free of charge to scientists for noncommercial purposes.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Pormohammad A, Ghotaslou R, Leylabadlo HE, Nasiri MJ, Dabiri H, Hashemi A. Risk of gastric cancer in association with Helicobacter pylori different virulence factors: A systematic review and meta-analysis. Microb Pathog. 2018;118:214–9.
Sugano K. Effect of Helicobacter pylori eradication on the incidence of gastric cancer: a systematic review and meta-analysis. Gastric Cancer. 2019;22:435–45.
Eusebi LH, Telese A, Marasco G, Bazzoli F, Zagari RM. Gastric cancer prevention strategies: A global perspective. J Gastroenterol Hepatol. 2020;35:1495–502.
Banks M, Graham D, Jansen M, Gotoda T, Coda S, di Pietro M, et al. British Society of Gastroenterology guidelines on the diagnosis and management of patients at risk of gastric adenocarcinoma. Gut. 2019;68:1545–75.
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976;73:3852–6.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–8.
Salzman J. Circular RNA expression: its potential regulation and function. Trends Genet. 2016;32:309–16.
Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675–91.
Cui C, Yang J, Li X, Liu D, Fu L, Wang X. Functions and mechanisms of circular RNAs in cancer radiotherapy and chemotherapy resistance. Mol Cancer. 2020;19:58.
Li J, Sun D, Pu W, Wang J, Peng Y. Circular RNAs in cancer: biogenesis, function, and clinical significance. Trends Cancer. 2020;6:319–36.
Tang Q, Hann SS. Biological roles and mechanisms of circular RNA in human cancers. Onco Targets Ther. 2020;13:2067–92.
Chaichian S, Shafabakhsh R, Mirhashemi SM, Moazzami B, Asemi Z. Circular RNAs: A novel biomarker for cervical cancer. J Cell Physiol. 2020;235:718–24.
Wei J, Xu H, Wei W, Wang Z, Zhang Q, De W, et al. circHIPK3 promotes cell proliferation and migration of gastric cancer by sponging miR-107 and regulating BDNF expression. Onco Targets Ther. 2020;13:1613–24.
Jahani S, Nazeri E, Majidzadeh AK, Jahani M, Esmaeili R. Circular RNA; a new biomarker for breast cancer: A systematic review. J Cell Physiol. 2020;235:5501–10.
He Y, Wang Y, Liu L, Liu S, Liang L, Chen Y, et al. Circular RNA circ_0006282 contributes to the progression of gastric cancer by sponging miR-155 to upregulate the expression of FBXO22. Onco Targets Ther. 2020;13:1001–10.
Pan H, Pan J, Chen P, Gao J, Guo D, Yang Z, et al. WITHDRAWN: Circular RNA circUBA1 promotes gastric cancer proliferation and metastasis by acting as a competitive endogenous RNA through sponging miR-375 and regulating TEAD4. Cancer Lett. 2020;19:S0304-3835(20)30085-9.
Wang Y, Xu S, Chen Y, Zheng X, Li T, Guo J. Identification of hsa_circ_0005654 as a new early biomarker of gastric cancer. Cancer Biomark. 2019;26:403–10.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8.
Wu G, Zhang A, Yang Y, Wu D. Circ-RNF111 aggravates the malignancy of gastric cancer through miR-876-3p-dependent regulation of KLF12. World J Surg Oncol. 2021;19:259.
Liu YY, Zhang LY, Du WZ. Circular RNA circ-PVT1 contributes to paclitaxel resistance of gastric cancer cells through the regulation of ZEB1 expression by sponging miR-124-3p. Biosci Rep. 2019;39:BSR20193045.
Xie Y, Shao Y, Sun W, Ye G, Zhang X, Xiao B, et al. Downregulated expression of hsa_circ_0074362 in gastric cancer and its potential diagnostic values. Biomark Med. 2018;12:11–20.
Chen S, Li T, Zhao Q, Xiao B, Guo J. Using circular RNA hsa_circ_0000190 as a new biomarker in the diagnosis of gastric cancer. Clin Chim Acta. 2017;466:167–71.
Liu J, Li Z, Teng W, Ye X. Identification of downregulated circRNAs from tissue and plasma of patients with gastric cancer and construction of a circRNA-miRNA-mRNA network. J Cell Biochem. 2020;121:4590-600.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–8.
Cai J, Chen Z, Wang J, Wang J, Chen X, Liang L, et al. circHECTD1 facilitates glutaminolysis to promote gastric cancer progression by targeting miR-1256 and activating beta-catenin/c-Myc signaling. Cell Death Dis. 2019;10:576.
Peng YK, Pu K, Su HX, Zhang J, Zheng Y, Ji R, et al. Circular RNA hsa_circ_0010882 promotes the progression of gastric cancer via regulation of the PI3K/Akt/mTOR signaling pathway. Eur Rev Med Pharmacol Sci. 2020;24:1142–51.
Shi H, Han J, Yue S, Zhang T, Zhu W, Zhang D. Prognostic significance of combined microRNA-206 and CyclinD2 in gastric cancer patients after curative surgery: A retrospective cohort study. Biomed Pharmacother. 2015;71:210–5.
Huang Z, Cai Y, Yang C, Chen Z, Sun H, Xu Y, et al. Knockdown of RNF6 inhibits gastric cancer cell growth by suppressing STAT3 signaling. Onco Targets Ther. 2018;11:6579–87.
Ha TMT, Le TNU, Nguyen VN, Tran VH. Association of TP53 gene codon 72 polymorphism with Helicobacter pylori-positive non-cardia gastric cancer in Vietnam. J Infect Dev Ctries. 2019;13:984–91.
Han Y, Wu N, Jiang M, Chu Y, Wang Z, Liu H, et al. Long non-coding RNA MYOSLID functions as a competing endogenous RNA to regulate MCL-1 expression by sponging miR-29c-3p in gastric cancer. Cell Prolif. 2019;52:e12678.
Yang DD, Chen ZH, Yu K, Lu JH, Wu QN, Wang Y, et al. METTL3 promotes the progression of gastric cancer via targeting the MYC pathway. Front Oncol. 2020;10:115.
Xie C, Guo Y, Lou S. LncRNA ANCR promotes invasion and migration of gastric cancer by regulating foxo1 expression to inhibit macrophage M1 polarization. Dig Dis Sci. 2020;65:2863-72.
He X, Zou K. MiRNA-96-5p contributed to the proliferation of gastric cancer cells by targeting FOXO3. J Biochem. 2020;167:101–8.
Zhang H, Duan J, Qu Y, Deng T, Liu R, Zhang L, et al. Onco-miR-24 regulates cell growth and apoptosis by targeting BCL2L11 in gastric cancer. Protein Cell. 2016;7:141–51.
Lin JX, Xie XS, Weng XF, Qiu SL, Xie JW, Wang JB, et al. Overexpression of IC53d promotes the proliferation of gastric cancer cells by activating the AKT/GSK3beta/cyclin D1 signaling pathway. Oncol Rep. 2019;41:2739–52.
Ma JX, Yang YL, He XY, Pan XM, Wang Z, Qian YW. Long noncoding RNA MNX1-AS1 overexpression promotes the invasion and metastasis of gastric cancer through repressing CDKN1A. Eur Rev Med Pharmacol Sci. 2019;23:4756–62.
Shenoy S. CDH1 (E-Cadherin) Mutation and gastric cancer: genetics, molecular mechanisms and guidelines for management. Cancer Manag Res. 2019;11:10477–86.
He JQ, Zhang SR, Li DF, Tang JY, Wang YQ, He X, et al. Experimental study on the effect of a weifufang on human gastric adenocarcinoma cell line BGC-823 xenografts and PTEN gene expression in nude mice. Cancer Biother Radiopharm. 2020;35:199-207.
Xiang L, Wang W, Zhou Z, Lv M, Tao L, Ni T, et al. COX-2 promotes metastasis and predicts prognosis on gastric cancer via regulating mTOR. Biomark Med. 2020;14:421-32.
Diao L, Wang S, Sun Z. Long noncoding RNA GAPLINC promotes gastric cancer cell proliferation by acting as a molecular sponge of miR-378 to modulate MAPK1 expression. Onco Targets Ther. 2018;11:2797–804.
Hirano T, Shinsato Y, Tanabe K, Higa N, Kamil M, Kawahara K, et al. FARP1 boosts CDC42 activity from integrin alphavbeta5 signaling and correlates with poor prognosis of advanced gastric cancer. Oncogenesis. 2020;9:13.
Yang T, Huang T, Zhang D, Wang M, Wu B, Shang Y, et al. TGF-beta receptor inhibitor LY2109761 enhances the radiosensitivity of gastric cancer by inactivating the TGF-beta/SMAD4 signaling pathway. Aging (Albany NY). 2019;11:8892–910.
Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X, et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget. 2017;8:20741–50.
Acknowledgments
We thank the reviewers for their constructive comments.
Funding
This work was supported by the Young Science & Technology Project of Changzhou Health Commission (QN202033); Changzhou Science & Technology Project (CJ20200097); the Young Scientists Foundation of Changzhou No.2 People’s Hospital (2018K010).
Author information
Authors and Affiliations
Contributions
YG, YJ, and XQ collected related data from GEO; identified differentially expressed circRNAs; collected and analyzed information about miRNAs and mRNAs; conducted validation of qRT-PCR; constructed the PPI network; conducted GO and KEGG analyses; and wrote the main manuscript. TL designed the entire experiment. JF and JQ helped to sort out data from datasets. JZ and HY checked all of the data used in the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Consent for publication have been obtained from participants.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gong, Y., Jiao, Y., Qi, X. et al. Construction of a circRNA-miRNA-mRNA network based on differentially co-expressed circular RNA in gastric cancer tissue and plasma by bioinformatics analysis. World J Surg Onc 20, 34 (2022). https://doi.org/10.1186/s12957-022-02503-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-022-02503-7