
Abstract
Background
Circular RNAs (circRNAs) are a new class of noncoding RNAs that have gained increased attention in human tumor research. However, the identification and function of circRNAs are largely unknown in the context of gastric cancer (GC). This study aims to identify novel circRNAs and determine their action networks in GC.
Methods
A comprehensive strategy of data mining, reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and computational biology were conducted to discover novel circRNAs and to explore their potential mechanisms in GC. Promising therapeutic drugs for GC were determined by connectivity map (CMap) analysis.
Results
Six overlapped differentially expressed circRNAs (DECs) were screened from selected microarray and RNA-Seq datasets of GC, and the six DECs were then validated by sanger sequencing and RNase R treatment. Subsequent RT-qPCR analysis of GC samples confirmed decreased expressions of the six DECs (hsa_circ_0000390, hsa_circ_0000615, hsa_circ_0001438, hsa_circ_0002190, hsa_circ_0002449 and hsa_circ_0003120), all of which accumulated preferentially in the cytoplasm. MiRNA binding sites and AGO2 occupation of the six circRNAs were predicted using online databases, and circRNA–miRNA interactions including the six circRNAs and 33 miRNAs were determined. Then, 5320 target genes of the above 33 miRNAs and 1492 differently expressed genes (DEGs) from The Cancer Genome Atlas (TCGA) database were identified. After intersecting the miRNA target genes and the 889 downregulated DEGs, 320 overlapped target genes were acquired. The Kyoto Encyclopedia of Genes and Genomes enrichment analysis indicated that these target genes were related to two critical tumor-associated signaling pathways. A protein–protein interaction network with the 320 target genes was constructed using STRING, and fifteen hubgenes (ATF3, BTG2, DUSP1, EGR1, FGF2, FOSB, GNAO1, GNAI1, GNAZ, GNG7, ITPR1, ITPKB, JUND, NR4A3, PRKCB) in the network were identified. Finally, bioactive chemicals (including vorinostat, trichostatin A and astemizole) based on the fifteen hubgenes were identifed as therapeutic agents for GC through the CMap analysis.
Conclusions
This study provides a novel insight for further exploration of the pathogenesis and therapy of GC from the circRNA-miRNA-mRNA network perspective.
Similar content being viewed by others

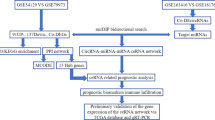
Background
Circular RNAs (circRNAs) are a newly identified class of noncoding RNAs (ncRNAs) that have recently elicited increased attention [1, 2]. CircRNAs are formed by back-splicing of a downstream splice donor site to an upstream splice acceptor site, thus producing a covalently closed RNA molecule. Lacking 5′ caps and 3′ poly (A) tails, circRNAs have not received significant attention for a long time; recently, with the application of high-throughput sequencing, a growing number of circRNAs have been unveiled [3]. Studies on circRNAs have revealed that they are structurally stable, presumably because their lack of free ends is resistant to exonuclease activity, which enables circRNAs to serve as a new class of diagnostic or prognostic biomarkers of diseases. Furthermore, emerging lines of studies have revealed that some circRNAs play important roles in physiological and pathological conditions including malignant tumors, and they may provide new potential therapeutic targets [4].
Gastric cancer (GC) is the fifth most common cancer and the fourth-leading cause of cancer-related deaths worldwide [5] and is thus a global cancer burden. Although diagnosis and treatment have improved over the last decades, the prognosis remains poor and the 5-year survival rate remains low in patients with GC [6]. Therefore, the discovery of effective biomarkers and therapeutic targets is of great importance. Competing endogenous RNA (ceRNA) refers to RNAs that sequester or sponge miRNAs to regulate mRNA transcripts containing common miRNA recognition elements (MREs) [7]. Recent studies reported that a large amount of conserved MREs present in circRNAs [8], making circRNAs a new research hotspot in the field of ceRNA. CircRNAs have been found to participate in various biological processes in GC by working as ceRNAs [9,10,11]. However, the identification and function of circRNAs in GC still require further investigation.
In the current study, data mining and bioinformatics analysis were combined to identify novel circRNAs and to investigate the underlying mechanism of circRNA-miRNA-mRNA network in GC (Additional file 2: Figure S1). First, we collected GC-related circRNA microarray and RNA-Seq datasets from the Gene Expression Omnibus (GEO) databases and PubMed publications and screened overlapped differentially expressed circRNAs (DECs). Then we validated these overlapped DECs by sanger sequencing and RNase R treatment, and verified their expression via reverse transcription-quantitative polymerase chain reaction (RT-qPCR) using GC samples. Further subcellular localization analysis and AGO2-binding sites mining indicated that these DECs function as ceRNAs. After predicting the sponge miRNAs of the DECs and miRNA target genes, we constructed circRNA-miRNA-mRNA networks; Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis on the target genes were conducted to investigate the potential pathogenesis of GC. Then, a protein–protein interaction (PPI) network with target genes was set up and hubgenes were determined. Moreover, we performed a connectivity map (CMap) analysis based on the hubgenes to identify potential bioactive compounds, which provides possible alternatives and chemotherapeutics for the treatment of GC patients.
Methods
Screening of DECs in GC from GEO database and PubMed publications
The microarray and RNA-Seq data was obtained from GEO databases (http://www.ncbi.nlm.nih.gov/gds/) [12] and PubMed publications. Three published microarray datasets that analyzed circRNA expression profile between GC tissues and matched nontumor tissues were retrieved from GEO database until June 2019 (Table 1), and the microarray dataset reported by Dang et al. was selected for further analysis since it has the largest number of identified circRNAs among the three datasets [13]. And the RNA-Seq dataset reported by Chen et al. [11] was selected for the same reason that it has the largest number of identified circRNAs among RNA-Seq datasets until June 2019. Limma [14], a Bioconductor package, was used to screen DECs, and “|Log2 (fold change) |> 1 and P-value < 0.05” were defined as the criteria for the DECs screening. The overlapped DECs were identified by intersecting the DECs from the aforementioned two datasets.
Cell culture and tissue specimens
GES-1-T cells were constructed in the previous study [15], and were cultured in Dulbecco′s modified Eagle′s medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. Human GC cell lines HGC-27 and MGC-803 were cultured in RPMI 1640 medium with 10% FBS and 1% penicillin–streptomycin. All cells were incubated in a humidified atmosphere containing 5% CO2 at 37 °C. Eight pairs of fresh frozen GC tissues and corresponding adjacent normal tissues were obtained from patients who underwent surgery at the Fifth Affiliated Hospital of Guangzhou Medical University. All samples used in this study were collected with patients’ consent. The present study was approved by the Ethics Committee of the Fifth Affiliated Hospital of Guangzhou Medical University.
RNA isolation, nuclear-cytoplasmic fractionation and RNase R treatment
Total RNA was extracted from cells and tissues using TRIzol (Invitrogen) according to the manufacturer’s instructions. Nuclear and cytoplasmic RNAs were isolated with PARIS Kit (Invitrogen, AM1921) following the manufacturer’s instruction. For RNase R treatment, total 10 ug RNA was incubated for 20 min at 37 °C with or without 2 units of RNase R (Epicentre, RNR07250); the resulting RNA was then purified with an RNeasy MinElute Cleanup Kit (Qiagen, 74,204).
RT-qPCR
RNA was reversely transcribed into cDNA using PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa, RR047A) in the presence of random hexamers (TaKaRa). qPCR was conducted using a TB Green® Premix Ex Taq™ II Kit (TaKaRa, RR820A) on the Applied Biosystems 7500 Real Time PCR System. β-actin served as an internal control. The specific primers of circRNAs were listed in Table 2.
Prediction of MREs
MiRNA binding sites in DECs were predicted using web tools CircInteractome (https://circinteractome.nia.nih.gov/) [16] and Circbank (www.circbank.cn) [17] respectively. Only MREs predicted consistently by both Circbank and CircInteractome were used for further analysis.
AGO2-binding sites from cross-linking immunoprecipitation (CLIP) data sets
The evidence for AGO2-binding sites was acquired from published online CLIP data sets on doRiNA database (http://dorina.mdc-berlin.de) [18]. These data sets contain AGO2 PAR-CLIP or HITS-CLIP data from several cell lines. After downloading the available data sets, AGO2-binding sites of circRNA genomic region were acquired.
Prediction of miRNA target genes
MiRNAs targeted genes were predicted using miRWalk 2.0 [19], which involves 12 algorithms (miRWalk, Microt4, miRanda, mirbridge, miRDB, miRMap, miRNAMap, Pictar2, PITA, RNA22, RNAhybrid and Targetscan). Target genes identified by at least eight algorithms were chosen.
Collection of differently expressed genes (DEGs) of GC from The Cancer Genome Atlas (TCGA)
RNA-Seq data of 415 gastric adenocarcinoma samples and 35 normal tissues was downloaded from the TCGA database. DEGs were determined by DEseq [20], a Bioconductor package; with the cutoff of gene reads > 1, total 12,408 genes were inclusive in all samples, and 1492 genes were differentially expressed with the criteria of |log2 (fold change) |> 1 and P-value < 0.05. Among the 1492 genes, 603 genes were upregulated and 889 genes were downregulated.
Construction of circRNA–miRNA–mRNA networks
The predicted miRNA target genes and the downregulated DEGs from TCGA were intersected to obtain the overlapped genes. Then, circRNA–miRNA–mRNA networks were constructed and visualized using the Cytoscape3.6.1 software [21].
Establishment of a PPI network and identification of hubgenes
The PPI network directly indicate the protein interactions established using STRING (version 11.0) [22]. Seven active interaction sources were selected: textmining, experiment, database, co-expression, neighborhood, gene fusion, and co-occurrence. Confident score was set to medium (score 0.400). The results were visualized by the Cytoscape3.6.1 software. Next, the “Molecular Complex Detection” (MCODE), a plugin of Cytoscape, was employed to find the highly interacted hubgene clusters.
Bioinformatic analysis tools
The KEGG Pathway based data integration and visualization was carried out using Pathview Web server (https://pathview.uncc.edu/) [23]: expression data of 320 target genes in ENSEMBL ID type of GC tissues (as sample) and normal tissues (as control) were loaded into the Pathview input tool, and the output suffix was set as KEGG pathview. Venn diagrams were generated using an online analysis platform (http://bioinfogp.cnb.csic.es/tools/venny/index.html).
CMap analysis
CMap (https://portals.broadinstitute.org/CMap/) [24, 25] is a collection of gene expression profiles from cultured human cells treated with bioactive small molecules; CMap helps scientists to discover functional connections among diseases, genetic perturbation, and drug action. Downregulated tags of the fifteen hubgenes were loaded into the CMap online tool, searching against 6100 treatment-controls (instances) in which 1309 bioactive molecules were involved. A connectivity score ranging from − 1 to 1 was used to estimate the closeness between query signatures and compounds: a positive score indicates a promotive effect of compound on the query signatures, whereas a negative score denotes an inverse effect of compound on the query signatures.
CircRNA overexpression and cell transfection
CircRNA overexpression plasmids were cloned with the pCD2.1-ciR vector, and all the circRNA sequence was confirmed by Sanger sequencing. For transient transfection, GC cells were transfected with the reagents using Lipofectamine™ 3000 (Invitrogen) according to the manufacturer’s instructions.
Cell counting Kit-8 (CCK-8) assay
Cell proliferation was assayed by CCK-8 (Dojindo, Tokyo) according to the manufacturer’s protocols. GC cells in logarithmic growth were plated in each well of a 96-well plate. Then, on the indicated time, 10 μl of CCK-8 solution was added to each well. Following 1 h of incubation at 37 °C, the absorbance of each well was measured at 450 nM by Synergy 2 microplate reader (BioTek, Winooski, VT, USA).
Results
Identification of six downregulated DECs in GC
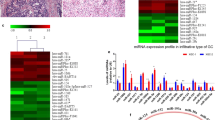
First, we collected GC-related circRNA microarray and RNA-Seq datasets from GEO databases and PubMed publications and screened overlapped DECs. As displayed in Table 1, one microarray dataset from GEO databases (GSE100170) [13] and one RNA-Seq dataset from PubMed publications (PMID: 27986464) [11] were selected for analysis in this study since these two datasets had the largest numbers of identified circRNAs respectively. A total of 180 DECs were identified in the study performed by Chen et al. [11], including 82 upregulated circRNAs and 98 downregulated circRNAs (Fig. 1A); a total of 713 DECs were identified in the study by Dang et al. [13], of which 191 circRNAs were upregulated and 522 circRNAs were downregulated (Fig. 1A).

Identification of differentially expressed circRNAs (DECs) in gastric cancer (GC) using microarray and RNA-Seq datasets. A Venn diagram showing overlapped circRNAs that were upregulated or downregulated in studies performed by Chen et al. and Dang et al., respectively. B, C Heatmap for the six DECs in individual microarray or RNA-Seq datasets. Red indicates a higher expression and blue indicates a lower expression
Further Venn diagram analysis showed that there were six overlapped downregulated circRNAs and none overlapped upregulated circRNAs in both studies (Fig. 1A). The six overlapped downregulated circRNAs were hsa_circ_0000390, hsa_circ_0000615, hsa_circ_0001438, hsa_circ_0002190, hsa_circ_0002449 and hsa_circ_0003120. The expression of the six DECs in individual microarray or RNA-Seq dataset was displayed: in the study by Chen et al. (Fig. 1B), six DECs was significantly lower with good consistency in GC tissues comparing to the matched nontumor tissues; downregulation of the six DECs in GC tissues was also observed in the study by Dang et al. (Fig. 1C).
Validation of the six DECs and verification of their downregulation with GC samples
We then analyzed the six DECs. The principal characteristics of the six DECs were listed in Table 3. Divergent primers (Table 2) were designed against the six DECs. By sanger sequencing of the RT-PCR product, head-to-tail splicing in the six DECs was confirmed (Fig. 2A). Moreover, after the treatment of RNase R, a processive 3′ to 5′ exonuclease, the six DECs was apparently enriched, which was consistent with the positive control CDR1as [26]; however, the abundance of linear GAPDH mRNA was apparently decreased (Fig. 2B). These results confirmed the circular characteristics of the six DECs.

Validation of the six DECs by Sanger sequencing and RNase R treatment. A Head-to-tail splicing in the RT-qPCR product of the six DECs by Sanger sequencing. Blue arrow indicates the back-splicing of DECs. B qRT-PCR analysis of the abundance of the six DECs in MGC-803 cells after treatment with RNase R. The amounts were normalized to the value measured in the untreated group
Then, RT-qPCR analysis was performed with 8 pairs of GC samples and adjacent nontumor tissues to verify the induced expression of the six DECs. As shown in Fig. 3A–F, expression of the six DECs were dramatically downregulated in GC tissues than adjacent normal tissues.

Expression of the six DECs in GC by RT-qPCR analysis: A hsa_circ_0000390, B hsa_circ_0000615, C hsa_circ_0001438, D hsa_circ_0002190, E hsa_circ_0002449, and F hsa_circ_0003012. The amounts of circRNAs were normalized to the value measured in adjacent normal tissues. ANT, adjacent normal tissues; GC, gastric cancer
Identification of circRNA-miRNA interactions and AGO2-binding sites
We subsequently determined the potential function of the six DECs in GC. Since the function of circRNAs was associated with their subcellular localization, RT-qPCR analysis of nuclear or cytoplasmic fractions of GC cell lines was performed to evaluate the subcellular distribution of the six DECs. As expected, U1 snRNA was enriched in the nuclear while the coding GAPDH mRNA was primarily localized in cytoplasm; as demonstrated, all the six DECs were found predominantly in the cytoplasm (Fig. 4A).

Subcellular distribution of the six DECs and identification of circRNA-miRNA interactions and AGO2 occupation. A qRT-PCR analysis of subcellular distribution of the six DECs. The amounts of circRNAs were normalized to the value measured in the nucleus. C/N ratio, the ratio of cytoplasm to nucleus. B Structural patterns of the six DECs with predicted MREs and AGO2-binding sites. Red dots represent MREs predicted by CircInteractome while blue dots and green dots represent MREs predicted by Circbank using miRanda and Targetscan, respectively. Black triangles represent AGO2-binding sites
Given that increasing circRNAs have been reported to play important roles in tumor through absorbing miRNA and that the six DECs accumulated preferentially in the cytoplasm, three online databases were used to determine whether the six DECs function as ceRNAs in GC. Herein CircInteractome and Circbank were utilized to predict potential target miRNAs of the six DECs, and only circRNA–miRNA interactions identified by both Circbank and CircInteractome were used (Fig. 4B), which are also shown in Additional file 2: Table S1. A total of 36 circRNA-miRNA interactions consisting of 6 circRNAs and 33 miRNAs (3 overlapped miRNAs) were identified. Moreover, online AGO2 PAR-CLIP or HITS-CLIP data was downloaded from doRiNA database [18] to ascertain whether AGO2 occupies in the region of the six DECs. The result demonstrated that all the six DECs contain AGO2 binding sites and 4 out of 6 circRNAs contain at least two AGO2 binding sites (Fig. 4B).
Taken together, the six DECs possibly function by sponging miRNAs.
Construction of circRNA-miRNA-mRNA networks and KEGG pathway analysis of the target genes
To further illustrate the potential ceRNA mechanisms of the six DECs in GC, prediction of miRNA target genes was performed by miRWalk with the criteria of that target genes identified by at least 8 out of 12 algorithms were chosen for further analysis, and a total of 5320 target genes of the above-mentioned 33 miRNAs were obtained. In addition, 1492 DEGs in GC were obtained from TCGA (Additional file 1), of which 603 DEGs were upregulated and 889 DEGs were downregulated (Fig. 5A). After intersecting the 5320 miRNA target genes and the 889 downregulated DEGs from TCGA, 320 overlapped target genes were acquired (Fig. 5B), which probably serve crucial roles in GC.

Identification of 320 overlapped genes that probably play critical roles in GC. A Volcano plot of the differentially expressed genes (DEGs) in GC based on the data from The Cancer Genome Atlas (TCGA). Significantly upregulated circRNAs are indicated in red and downregulated circRNAs are indicated in blue (FC ≥ 2.0 or FC ≤ 0.5 and p < 0.05). B Venn diagram for the intersection between downregulated DEGs and miRNA target genes
Then, circRNA-miRNA-mRNA networks were generated using a combination of data from circRNA-miRNA interactions and the overlapped 320 target genes, which indicated an overall perspective of regulation networks consisting of six circRNAs, 33 miRNAs and 320 target mRNAs. The ceRNA networks with six DECs were shown in Additional file 2: Figure S2–3.
Next, KEGG pathway enrichment analysis were performed using pathview [23] to illustrate the functional annotations of the 320 target genes. As displayed in Fig. 6, the top two enriched pathways were “MAPK signaling pathway” and “PI3K-AKT signaling pathway”, both of which were tumor-related pathways [27,28,29,30].

Pathway analysis and visualization of 320 target genes. The top two enriched pathways: A MAPK signaling pathway; B PI3K-AKT signaling pathway
Identification of fifteen hubgenes from the PPI network with MCODE and validation of their downregulation in GC and their regulation by circRNAs
Then, a PPI network was built to analyze protein interactions with the 320 target genes. After removing unconnected nodes and nodes with connecting frequency smaller than 5, 125 nodes and 305 edges were mapped in the PPI network (Fig. 7A). Considering the key role of hubgenes in biological process, MCODE, a plugin of Cytoscape, was utilized to screen hubgenes from the PPI network. Using the criteria of that the k-core is 2, nine subnetworks were identified, and one subnetwork with highest MCODE score 5.571 that contains 15 nodes and 39 edges was selected (Fig. 7B). There were fifteen genes (ATF3, BTG2, DUSP1, EGR1, FGF2, FOSB, GNAI1, GNAO1, GNAZ, GNG7, ITPKB, ITPR1, JUND, NR4A3, and PRKCB) in this subnetwork, and these fifteen genes were identified as hubgenes.

Construction of a protein–protein interaction (PPI) network with 320 target mRNAs and identification of hubgenes from the PPI network. A The PPI network with 320 target genes. B The subnetwork with 15 hubgenes that extracted from the PPI network with the “Molecular Complex Detection (MCODE)” algorithm. A, B The node color changes gradually from light blue to dark blue in ascending order according to the fold change of target genes; the thickness of line between nodes indicate the interacting proteins varies based on the combined score for the protein association
The expression levels of the fifteen genes in GC from TCGA were demonstrated in Fig. 8A–O, and results showed that all these fifteen hubgenes was downregulated in GC tissues compared with normal tissues. Then, RT-qPCR analysis was performed with the above-mentioned 8 pairs of GC samples and adjacent nontumor tissues to verify the induced expression of these hubgenes; the results showed that expression of the fifteen hubgenes were dramatically decreased in GC tissues than adjacent normal tissues (Additional file 2: Figure S4A–D).

Box-scatter plots for the 15 hubgenes expression in GC from TCGA and the effect of circRNAs overexpressions on hubgenes expressions. A–O Box scatter plots for the expression of 15 hubgenes in GC from TCGA: A ATF3, B BTG2, C DUSP1, D EGR1, E FGF2, F FOSB, G GNAI1, H GNAO1, I GNAZ, J GNG7, K ITPKB, L ITPR1, M JUND, N NR4A3, and OPRKCB. P–Q Overexpressing plasmids of the six circRNAs were constructed and GC cells were transfected with pooled circRNA vectors. P The overexpression efficiency of circRNA vectors on RNA level of circRNAs were measured by qRT-PCR analysis. Q The mRNA levels of 15 hubgenes in GC cells transfected with circRNA vectors by qRT-PCR analysis
In addition, to determine the effect of circRNAs on the expression of hubgenes with wet lab experiments, overexpressing plasmids of the six circRNAs were constructed and GC cells were transfected with pooled circRNA vectors. The respective increase of circRNA expressions induced by overexpressing plasmids was confirmed by qRT-PCR analysis (Fig. 8P); meanwhile, significant increases of almost all 15 hubgenes (except four of them) in mRNA level was observed (Fig. 8Q). Collectively, these results indicate that the hubgenes are modulated by circRNAs.
Identification of three therapeutic drugs for GC based on CMap analysis and validation of their effect on GC cell proliferation and hubgenes expressions
CMap can be used to predict compounds that may induce or reverse a given gene expression signature, and we employed CMap to find potential compounds that could disturb the gene expression pattern of the fifteen hubgenes. Downregulated tags of the fifteen hubgenes were submitted to the CMap online tool for analysis. After the signature query, three chemicals, vorinostat, astemizole, and trichostatin A (TSA), with the most significant negative enrichment score were identified as the potential therapeutic compounds for GC (Table 4). The chemical structures of the three drugs were shown in Fig. 9A–C. The instances for each compound according to the permuted results of CMap analysis were listed in Additional file 2: Table S2; as shown, all the three compounds with different doses or in different cell lines consistently exhibited negative correlations to the query signature.

Chemical structures of the three compounds identified by the connectivity map (CMap) analysis and the effect of these three compounds on GC cell proliferation and hubgenes expressions. A–C Chemical structures of the three compounds: A Vorinostat, B Astemizole, and C Trichostatin A. D, F, H GC cells were treated with increasing concentrations of D Vorinostat, F Astemizole, and H Trichostatin A, and GC cell proliferations were measured by CCK-8 assays. E, G, I GC cells were treated with E Vorinostat, G Astemizole, and I Trichostatin A, and the mRNA levels of 15 hubgenes were measured by qRT-PCR analysis
In addition, we validated whether the three bioactive drugs can alleviate GC progression and can modulate expression of hubgenes. Our results showed that treatment of GC cells with increasing concentrations of all the three drugs markedly reduced GC cell proliferation by CCK-8 assays in a dose-dependent manner (Fig. 9D, F and H), and these drugs treatments further upregulated the mRNA levels of almost all 15 hubgenes by qRT-PCR analysis (Fig. 9E, G and I). These results indicate that the three drugs predicted by CMap analysis could be used as therapeutic agents for GC.
Discussion
CircRNAs that were first discovered in eukaryotic cells in 1979 [31] has been recently re-recognized and elicited attention. Unlike the formation of traditional linear RNA, circRNAs display a unique covalently closed circular form, which is responsible for their high stability and resistance to exonucleases. In addition, previous studies have proved their sequence conservation, abundant presence in exosomes and plasma, and spatial and cell-type specificity [32]. Thereby, circRNAs have great potential to serve as a biomarker for early diagnosis and prognosis of many human diseases including cancers. With the development of high-throughput sequencing and bioinformatics analysis, mounting circRNAs were found and confirmed to be involved in the regulation of diversified biological processes in different cancer types [33, 34]. In GC, some circRNAs, such as circHECTD1 [35] and circFAT1 [36], have been reported to be involved in GC cell proliferation, migration and invasion, and thereby exert their tumor-promoting or tumor-suppressing effects. However, the identification and function of circRNAs in GC still require further exploration.
In this study, we collected microarray and RNA-Seq datasets of GC from GEO databases and PubMed publications and screened six downregulated circRNAs (hsa_circ_0000390, hsa_circ_0000615, hsa_circ_0001438, hsa_circ_0002190, hsa_circ_0002449 and hsa_circ_0003120). Following sanger sequencing, RNase R treatment and RT-qPCR analysis, the six DECs were validated and their decreased expression in GC tissues was confirmed. Previous studies have revealed that hsa_circ_0000615 acts as miRNA sponge to exert different roles in different solid tumors [37,38,39,40]; for example, hsa_circ_0000615 upregulates Sp1 expression by adsorbing miR-150-5p, and thereby promoting the proliferation and metastasis of nasopharyngeal cancer cells [38]. However, until now studies about the effect of hsa_circ_0000615 and the other five DECs on GC have been rarely reported. Further studies are imperative to determine their potential roles in GC.
As ceRNA, circRNA harboring MREs can sponge miRNAs, which suppress the miRNA activity and result in alteration of expression level of miRNA target genes [41, 42]. Subcellular localization analysis and AGO2-binding sites mining in this study indicate that the six DECs in the cytoplasm probably sponge miRNAs. To further determine whether the above six DEC act as ceRNAs in GC, their corresponding MREs were predicted with two online tools, CircInteractome and Circbank. The former web tool forecasts MREs via Targetscan algorithm, which predicts MREs by surveying for 7-mer or 8-mer complementarity to seed region and the 3′ end of each miRNA [16, 43]. The latter web tool predicts MREs based on two different algorithms including miRanda [44] and Targetscan [43]. We selected miRNAs predicted by both CircInteractome and Circbank as target miRNAs of the six DECs, and collectively, 36 circRNA-miRNA interactions composed of 6 circRNAs and 33 miRNAs were identified.
After intersecting the target genes of the aforementioned 33 miRNAs and DEGs in GC from TCGA, 320 overlapped target genes were acquired to establish circRNA-miRNA-mRNA regulatory networks, providing an evidence of the ceRNA functional mechanism of the six DECs in GC. The KEGG pathway analysis indicated that these target genes were related to two critical tumor-associated signaling pathways, “MAPK signaling pathway” [27, 28] and “PI3K-AKT signaling pathway” [29, 30]. To comprehensively reveal potential relationships of target genes, we developed a PPI network using the 320 target genes and extracted 15 hubgenes (ATF3, BTG2, DUSP1, EGR1, FGF2, FOSB, GNAI1, GNAO1, GNAZ, GNG7, ITPKB, ITPR1, JUNID, NR4A3, PRKCB); and the differential expressions of the 15 hubgenes were subsequently validated with GC tissues from TCGA and with our GC samples. As previously demonstrated, some of the fifteen genes serve crucial roles in GC [45,46,47]. For example, Tang et al. have demonstrated that FOSB was significantly decreased in GC tissues, consistent with the results of this study, and moreover, downregulated expression of FOSB was correlated with poor prognosis for GC patients [46].
With the development of novel agents in recent years, survival outcomes of GC patients have improved [48]. However, the overall prognosis of patients with advanced gastric cancer remains poor, and more effective drugs against GC are needed. Therefore, CMap analysis of the fifteen hubgenes was performed to explore available compounds for the treatment of GC. Based on the genome-wide expression profiling of gene transcripts technology, CMap presents a data-driven and reliable approach for identifying new drugs or repositioning existing drugs [49]. Three chemicals (vorinostat, TSA, and astemizole) were determined and validated as the therapeutic options for GC. As histone deacetylase (HDAC) inhibitor, vorinostat alters the level of histone and nonhistone protein acetylation and thereby regulates gene expression, cell proliferation, angiogenesis and cell survival [50]. Vorinostat is FDA-approved for the treatment of cutaneous T cell lymphoma [51] and has been investigated in diverse clinical trials as a potential mono- or combination-drug therapy for solid tumors including GC [52, 53]. TSA is another kind of typical HDAC inhibitor used mainly in laboratory experiments. TSA is known to induce cell cycle arrest and apoptosis in different cancer cell lines, and its antitumor effect in solid tumors including GC has been illuminated previously [54]. Astemizole is an old anti-histamine that can target important proteins involved in the cancer progression, namely, ether à-go-go (Eag1) and Eag-related gene potassium channels [55], thus inhibiting tumor cell proliferation [56]. Moreover, previous evidence has revealed that Eag1 is expressed in several human tumor cell lines, including those from GC [57]. However, its anti-GC effect have not been elucidated presently. In this study, we found that it potentially serves as therapeutic agent for GC.
This study provides a basis for exploring the pathogenesis and treatment strategy of GC from the circRNA-miRNA-mRNA network perspective and provides more evidence for three bioactive compounds (vorinostat, TSA, astemizole) as anti-GC agents. A recent study has presented perspectives on circRNA‐miRNA‐mRNA networks to explore the pathogenesis and therapy for pancreatic ductal adenocarcinoma (PDAC) [58], and another study reported similar insights into the pathogenesis and therapy of HCC from the circRNA–miRNA–mRNA network view [59]. However, these perspectives or insights needs further wet lab investigations. Nevertheless, combined with our findings, these studies further our understanding of tumors from the perspective of circRNA‐related ceRNA networks.
Conclusions
In conclusion, through an integrated analysis of microarray and RNA-Seq data, RT-qPCR and computational biology, six circRNA-miRNA-mRNA regulatory networks were established and these ceRNA networks uncovered six circRNAs (hsa_circ_0000390, hsa_circ_0000615, hsa_circ_0001438, hsa_circ_0002190, hsa_circ_0002449 and hsa_circ_0003120) that might function as ceRNA to play important roles in GC. Additionally, three bioactive compounds (vorinostat, TSA, astemizole) obtained from the CMap analysis were identified as therapeutic agents for GC. Our study provides a new insight for further exploration of the pathogenesis and therapy strategies of GC from the circRNA-miRNA-mRNA network perspective.
Availability of data and materials
All data generated or analyzed during this study are included in this article. The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Abbreviations
- CircRNA:
-
Circular RNA
- GC:
-
Gastric cancer
- GEO:
-
Gene Expression Omnibus
- RNA-seq:
-
RNA sequencing
- DECs:
-
Differentially expressed circRNAs
- RT-qPCR:
-
Reverse transcription-quantitative polymerase chain reaction
- DEGs:
-
Differently expressed genes
- TCGA:
-
The Cancer Genome Atlas
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- ceRNA:
-
Competing endogenous RNA
- MRE:
-
MiRNA response element
- PPI:
-
Protein–protein interaction
- CMap:
-
Connectivity map
- CLIP:
-
Cross-linking immunoprecipitation
- MCODE:
-
Molecular Complex Detection
- HDAC:
-
Histone deacetylase
- FDA:
-
Food and Drug Administration
References
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–8.
Vo JN, Cieslik M, Zhang Y, Shukla S, Xiao L, Wu YM, et al. The landscape of circular RNA in cancer. Cell. 2019;176(4):869-81 e13.
Wang PL, Bao Y, Yee MC, Barrett SP, Hogan GJ, Olsen MN, et al. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE. 2014;9(6):e90859.
Chen Y, Li C, Tan C, Liu X. Circular RNAs: a new frontier in the study of human diseases. J Med Genet. 2016;53(6):359–65.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Allemani C, Weir HK, Carreira H, Harewood R, Spika D, Wang XS, et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. 2015;385(9972):977–1010.
Qi X, Zhang DH, Wu N, Xiao JH, Wang X, Ma W. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015;52(10):710–8.
Zhong Y, Du Y, Yang X, Mo Y, Fan C, Xiong F, et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol Cancer. 2018;17(1):79.
Liu H, Liu Y, Bian Z, Zhang J, Zhang R, Chen X, et al. Circular RNA YAP1 inhibits the proliferation and invasion of gastric cancer cells by regulating the miR-367-5p/p27 (Kip1) axis. Mol Cancer. 2018;17(1):151.
Zhang J, Liu H, Hou L, Wang G, Zhang R, Huang Y, et al. Circular RNA_LARP4 inhibits cell proliferation and invasion of gastric cancer by sponging miR-424-5p and regulating LATS1 expression. Mol Cancer. 2017;16(1):151.
Chen J, Li Y, Zheng Q, Bao C, He J, Chen B, et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett. 2017;388:208–19.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013;41(Database issue):D991–5.
Dang Y, Ouyang X, Zhang F, Wang K, Lin Y, Sun B, et al. Circular RNAs expression profiles in human gastric cancer. Sci Rep. 2017;7(1):9060.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Yang Q, Xu E, Dai J, Wu J, Zhang S, Peng B, et al. miR-21 regulates N-methyl-N-nitro-N’-nitrosoguanidine-induced gastric tumorigenesis by targeting FASLG and BTG2. Toxicol Lett. 2014;228(3):147–56.
Dudekula DB, Panda AC, Grammatikakis I, De S, Abdelmohsen K, Gorospe M. CircInteractome: a web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016;13(1):34–42.
Liu M, Wang Q, Shen J, Yang BB, Ding X. Circbank: a comprehensive database for circRNA with standard nomenclature. RNA Biol. 2019;16(7):899–905.
Blin K, Dieterich C, Wurmus R, Rajewsky N, Landthaler M, Akalin A. DoRiNA 2.0—upgrading the doRiNA database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2015;43(Database issue):D160–7.
Dweep H, Gretz N, Sticht C. miRWalk database for miRNA-target interactions. Methods Mol Biol. 2014;1182:289–305.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
Su G, Morris JH, Demchak B, Bader GD. Biological network exploration with Cytoscape 3. Curr Protoc Bioinformatics. 2014;47:8–13.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–8.
Luo W, Pant G, Bhavnasi YK, Blanchard SG Jr, Brouwer C. Pathview Web: user friendly pathway visualization and data integration. Nucleic Acids Res. 2017;45(W1):W501–8.
Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171(6):1437-52 e17.
Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–35.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–8.
Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120(22):3446–56.
Drosten M, Barbacid M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell. 2020;37(4):543–50.
Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med. 2016;67:11–28.
Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18(1):26.
Hsu MT, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979;280(5720):339–40.
Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J, et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25(8):981–4.
Li P, Yang X, Yuan W, Yang C, Zhang X, Han J, et al. CircRNA-Cdr1as exerts anti-oncogenic functions in bladder cancer by sponging MicroRNA-135a. Cell Physiol Biochem. 2018;46(4):1606–16.
Hu X, Wu D, He X, Zhao H, He Z, Lin J, et al. circGSK3beta promotes metastasis in esophageal squamous cell carcinoma by augmenting beta-catenin signaling. Mol Cancer. 2019;18(1):160.
Cai J, Chen Z, Wang J, Wang J, Chen X, Liang L, et al. circHECTD1 facilitates glutaminolysis to promote gastric cancer progression by targeting miR-1256 and activating beta-catenin/c-Myc signaling. Cell Death Dis. 2019;10(8):576.
Fang J, Hong H, Xue X, Zhu X, Jiang L, Qin M, et al. A novel circular RNA, circFAT1(e2), inhibits gastric cancer progression by targeting miR-548g in the cytoplasm and interacting with YBX1 in the nucleus. Cancer Lett. 2019;442:222–32.
He Y, Huang H, Jin L, Zhang F, Zeng M, Wei L, et al. CircZNF609 enhances hepatocellular carcinoma cell proliferation, metastasis, and stemness by activating the Hedgehog pathway through the regulation of miR-15a-5p/15b-5p and GLI2 expressions. Cell Death Dis. 2020;11(5):358.
Zhu L, Liu Y, Yang Y, Mao XM, Yin ZD. CircRNA ZNF609 promotes growth and metastasis of nasopharyngeal carcinoma by competing with microRNA-150-5p. Eur Rev Med Pharmacol Sci. 2019;23(7):2817–26.
Zuo Y, Shen W, Wang C, Niu N, Pu J. Circular RNA Circ-ZNF609 promotes lung adenocarcinoma proliferation by modulating miR-1224-3p/ETV1 signaling. Cancer Manag Res. 2020;12:2471–9.
Du S, Zhang P, Ren W, Yang F, Du C. Circ-ZNF609 accelerates the radioresistance of prostate cancer cells by promoting the glycolytic metabolism through miR-501-3p/HK2 axis. Cancer Manag Res. 2020;12:7487–99.
Zhang Y, Liang W, Zhang P, Chen J, Qian H, Zhang X, et al. Circular RNAs: emerging cancer biomarkers and targets. J Exp Clin Cancer Res. 2017;36(1):152.
Bossi L, Figueroa-Bossi N. Competing endogenous RNAs: a target-centric view of small RNA regulation in bacteria. Nat Rev Microbiol. 2016;14(12):775–84.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20.
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1.
Liu HT, Liu S, Liu L, Ma RR, Gao P. EGR1-Mediated transcription of lncRNA-HNF1A-AS1 promotes cell-cycle progression in gastric cancer. Cancer Res. 2018;78(20):5877–90.
Tang C, Jiang Y, Shao W, Shi W, Gao X, Qin W, et al. Abnormal expression of FOSB correlates with tumor progression and poor survival in patients with gastric cancer. Int J Oncol. 2016;49(4):1489–96.
Yeh CM, Chang LY, Lin SH, Chou JL, Hsieh HY, Zeng LH, et al. Epigenetic silencing of the NR4A3 tumor suppressor, by aberrant JAK/STAT signaling, predicts prognosis in gastric cancer. Sci Rep. 2016;6:31690.
Lordick F, Shitara K, Janjigian YY. New agents on the horizon in gastric cancer. Ann Oncol. 2017;28(8):1767–75.
Qu XA, Rajpal DK. Applications of Connectivity Map in drug discovery and development. Drug Discov Today. 2012;17(23–24):1289–98.
Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5(9):769–84.
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109(1):31–9.
Deng R, Zhang P, Liu W, Zeng X, Ma X, Shi L, et al. HDAC is indispensable for IFN-gamma-induced B7–H1 expression in gastric cancer. Clin Epigenetics. 2018;10(1):153.
Yoo C, Ryu MH, Na YS, Ryoo BY, Lee CW, Maeng J, et al. Phase I and pharmacodynamic study of vorinostat combined with capecitabine and cisplatin as first-line chemotherapy in advanced gastric cancer. Invest New Drugs. 2014;32(2):271–8.
Meneses-Morales I, Izquierdo-Torres E, Flores-Peredo L, Rodriguez G, Hernandez-Oliveras A, Zarain-Herzberg A. Epigenetic regulation of the human ATP2A3 gene promoter in gastric and colon cancer cell lines. Mol Carcinog. 2019;58(6):887–97.
de Guadalupe C-L, Perez-Carreon JI, Zuniga-Garcia V, Diaz-Chavez J, Herrera LA, Caro-Sanchez CH, et al. Astemizole-based anticancer therapy for hepatocellular carcinoma (HCC), and Eag1 channels as potential early-stage markers of HCC. Tumour Biol. 2015;36(8):6149–58.
Diaz L, Ceja-Ochoa I, Restrepo-Angulo I, Larrea F, Avila-Chavez E, Garcia-Becerra R, et al. Estrogens and human papilloma virus oncogenes regulate human ether-a-go-go-1 potassium channel expression. Cancer Res. 2009;69(8):3300–7.
Ding XW, Luo HS, Jin X, Yan JJ, Ai YW. Aberrant expression of Eag1 potassium channels in gastric cancer patients and cell lines. Med Oncol. 2007;24(3):345–50.
Xiao Y. Construction of a circRNA-miRNA-mRNA network to explore the pathogenesis and treatment of pancreatic ductal adenocarcinoma. J Cell Biochem. 2020;121(1):394–406.
Xiong DD, Dang YW, Lin P, Wen DY, He RQ, Luo DZ, et al. A circRNA-miRNA-mRNA network identification for exploring underlying pathogenesis and therapy strategy of hepatocellular carcinoma. J Transl Med. 2018;16(1):220.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (81673206 and 31500142), the Science and Technology Program of Guangzhou (201902020001, 201905010004 and 20191A011090), the Project of Educational Commission of Guangdong Province (2019KTSCX142), and Guangdong Medical Science and Technology Research Fund Project (A2019466).
Author information
Authors and Affiliations
Contributions
WY, ZL, ML, ZD, and XZ contributed conception and design of the study; WY, ZD, and ML organized the data; ZD, ZL, and WY performed the statistical analysis; ZD and JP performed the experiments; ZD, JP, HL, and WL collected the clinical specimens. All authors contributed to manuscript revision, read and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The Ethical Committee of the Fifth Affiliated Hospital of Guangzhou Medical University approved the present study. Primary GC samples were from the Fifth Affiliated Hospital of Guangzhou Medical University with informed consent.
Consent for publication
The authors agree for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
1492 DEGs in GC obtained from TCGA.
Additional file 2: Table S1.
CircRNA–miRNA interactions identified by both CircInteractome and Circbankdatabases. Table S2. Permuted results of the three compounds by CMap analysis. Figure S1. Flow chart of thepresent study. Figure S2. The circRNA–miRNA–mRNA regulatory networks of hsa_circ_0000615,hsa_circ_0001438, hsa_circ_0002190 and hsa_circ_0002449 in GC. Figure S3. The circRNA–miRNA–mRNAregulatory networks of hsa_circ_0000390 and hsa_circ_0003012 in GC. Figure S4. Expression of fifiteenhubgenes in GC by RT-qPCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dong, Z., Liu, Z., Liang, M. et al. Identification of circRNA–miRNA–mRNA networks contributes to explore underlying pathogenesis and therapy strategy of gastric cancer. J Transl Med 19, 226 (2021). https://doi.org/10.1186/s12967-021-02903-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-021-02903-5