
Abstract
Background
Endometrial carcinoma (EC) is the most common gynecological malignant tumors which poses a serious threat to women health. This study aimed to screen the candidate genes differentially expressed in EC by bioinformatics analysis.
Methods
GEO database and GEO2R online tool were applied to screen the differentially expressed genes (DEGs) of EC from the microarray datasets. Protein-protein interaction (PPI) network for the DEGs was constructed to further explore the relationships among these genes and identify hub DEGs. Gene ontology and KEGG enrichment analyses were performed to investigate the biological role of DEGs. Besides, correlation analysis, genetic alteration, expression profile, and survival analysis of these hub DEGs were also investigated to further explore the roles of these hub gene in mechanism of EC tumorigenesis. qRT-PCR analysis was also performed to verify the expression of identified hub DEGs.
Results
A total of 40 DEGs were screened out as the DEGs with 3 upregulated and 37 downregulated in EC. The gene ontology analysis showed that these genes were significantly enriched in cell adhesion, response to estradiol, and growth factor activity, etc. The KEGG pathway analysis showed that DEGs were enriched in focal adhesion, leukocyte transendothelial migration, PI3K-Akt signaling pathway, and ECM-receptor interaction pathway. More importantly, COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 were identified as the hub genes of EC. The genetic alteration analysis showed that hub genes were mainly altered in mutation and deep deletion. Expression validation by bioinformatic analysis and qRT-PCR also proved the expression of these six hub genes were differentially expressed in EC. Additionally, significantly better overall survival and disease-free survival were observed with six hub genes altered, and survival outcome in high expression of COL1A1, IGF1, and PTEN patients was also significantly better than low expression patients.
Conclusions
COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 involved in the pathogenesis of EC and might be candidate genes for diagnosis of EC.
Similar content being viewed by others

Introduction
Endometrial carcinoma (EC) is an epithelial malignant tumor of the endometrium and regarded as the most common cancers in female reproductive system [1]. There are about 319,600 new cases diagnosed around the world annually, ranking first in gynecological malignant tumors in high-income countries [2]. In recent years, due to the changes of people's living habits, abusing of informal hormone and sex hormone replacement therapy, the incidence of EC increases significantly, and the population tends to be younger, which poses a serious threat to women health [3, 4]. According to the clinicopathological features, EC can be divided into type I and type II, of which type I (hormone dependent) accounts for about 80% of the EC [5]. Surgical treatment is the main means for EC at present, and appropriate adjuvant therapy is also applied according to the pathological and clinical stages of the tumors. However, about 15–20% of the tumors still relapse after surgical treatment, and the curative effect of the systematic therapy is limited [6]. The lack of early diagnosis and timely treatment to prevent the tumorigenesis or progression particularly held responsible for the most deaths of EC. Therefore, exploring the suitable biomarkers and potential targets for the accurate prediction or diagnosis of EC and to seek the possibilities to improve the therapeutic effect and clinical prognosis of EC patients is urgently needed.
The oncogenesis is an extremely complicated pathophysiological process and regulated by various factors and pathways. Recently, high-throughput sequencing technology is widely used in clinical research, with the pronounced advantages relying on its ability to simultaneously determine the expression information of massive genes one time [7]. This is an ideal method to quickly and effectively screen differentially expressed genes (DEGs) related to certain disease. Based on the massive microarray datasets information stored in a large number of public databases such as The Cancer Genome Atlas (TCGA) database and Gene Expression Omnibus (GEO) database, the target data could be mined, integrated, and reanalyzed by bioinformatics method, which provide useful clues for the research of various diseases [8,9,10]. It has important value in the discovery of disease related biomarkers, disease mechanisms, the prediction of disease prognosis, and the discovery of targeted drugs. In recent years, many microarray profiling studies also have been performed in EC, but most of the results are limited and usually generated from a single cohort study.
In the present study, we applied integrated bioinformatics analysis for the identification of DEGs as potential biomarkers of EC based on the two mRNA expression profiles from the Gene Expression Omnibus (GEO) database. These analysis may provide possible perspectives for the progression and development of EC as well as identification potential candidate genes for EC diagnose or treatment.
Material and methods
Microarray data information
The microarray data used in this study were acquired from GEO database (http://www.ncbi.nlm.nih.gov/gds/), which is a public repository containing the high-throughput gene expression data submitted by research institutions around the world. We chose the mRNA expression profiles of GSE115810 and GSE36389 from the GEO database. The array data of GSE115810 was consisted with 24 EC tissues and 3 normal adjacent mucosa tissues based on the platform of GPL96 [HG-U133A] Affymetrix Human Genome U133A Array. The array data of GSE36389 was consisted with 13 EC tissues and 7 normal adjacent mucosa tissues based on the platform of GPL6480 Agilent-014850 Whole Human Genome Microarray 4x44K G4112F (Probe Name version).
Differential expression analysis of EC
The two mRNA expression microarray datasets obtained from GEO database were screened by an interactive online tool, GEO2R (www.ncbi.nlm.nih.gov/geo/geo2r). The raw data of microarray datasets were pre-processed via background correction and normalization, then |log2fold change (FC)| ≥ 1.5 and P value < 0.05 were conducted as the cutoff criteria for the DE mRNAs mining. Then, DE mRNAs from GSE115810 and GSE36389 were intersected and the overlapped mRNAs were identified as the DEGs of EC.
Integration of protein-protein interaction (PPI) network and hub gene identification
To investigate the interaction associations of DEGs, we applied the DEGs to the Search Tool for the Retrieval of Interacting Genes (STRING, http://string-db.org/), an online tool to explore and analyze PPI interactions of the genes. The DEGs of EC were mapped and the interactions with combined score ≥ 0.4 meet the criterion. Afterwards, a PPI network was constructed and visualized by Cytoscape software (version 3.4.0, http://www.cytoscape.org/). Subsequently, the topological properties including node degree of the DEGs in the PPI network were calculated to further analyze the candidate hub genes from the PPI network.
Gene functional enrichment analysis of DEGs
Based on the identified DEGs, GO analysis and KEGG pathway analysis of the selected DEGs were carried out by DAVID database (http://www.david.abcc.ncifcrf.gov/). This database can integrate biological data and analytical tools to provide systematic comprehensive biologic annotation information for large scale genes or protein lists (hundreds of ID or protein ID lists) to understand the biological meaning behind numerous genes. GO terms and KEGG pathways with P < 0.05 as the cutoff criterion, and the DEGs enriched GO terms and KEGG pathway were ranked by enrichment score (− log10 (P value)).
Correlation analysis of the DEGs
Based on raw data of the gene expression profiles from the two microarray datasets we download, correlations between the identified DEGs were also analyzed. Pearson’s correlation analysis was used to conduct the correlation analysis; P < 0.05 was considered to be statistically significant.
Exploring cancer genomics data of hub genes
The cBio Portal for Cancer Genomics (http://cbioportal.org) provides visualization, analysis, and download of large-scale cancer genomics data sets; users can select specific samples to form a dataset and chose certain genes to analyze the copy number aberration frequency and mutation frequency of these genes, as well as the co-expression genes and survival curves. In this study, we used the cBio Cancer Portal to investigate the cancer genomics data of the candidate hub genes. The result of the genomics datasets was shown through a concise and compact genomic alterations graphical summary of the inputted genes list in EC. In addition, overall survival and disease/progression-free survival analyses were also performed by grouping EC data alterations using input of the hub genes.
Hub gene expression and survival analysis
Online tool UALCAN (http://ualcan.path.uab.edu/index.html) was applied to evaluate the expression profiles of the hub DEGs in EC. The EC expression data was based on The Cancer Genome Atlas (TCGA) database, which generates comprehensive, multi-dimensional maps of the key genomic changes over 30 types of cancer. The Human Protein Atlas (https://www.proteinatlas.org/) online tool was also used to obtain the immunohistochemistry-based map of the hub genes protein expression profiles in normal and EC tissues, and the survival analysis in EC was also performed based on 541 EC patients.
qRT-PCR validation
To further verify the expression of hub DEGs in EC, we performed qRT-PCR to detect the mRNA level of identified hub DEGs in 13 EC clinical samples and 13 normal endometrial clinical samples from Zhejiang Cancer Hospital, China. This experiment was approved by the Ethical Committee of Zhejiang Cancer Hospital, China, and all participated patients are informed consent and agreement for the research use of the clinical endometrial samples. Total RNA was extracted by the TRIzol Reagent (Sangon Biotech, Shanghai, China), and 1 μg RNA was used to synthesize DNA using SuperScript III Reverse Transcriptase (Invitrogen). Real-time PCR was further performed with SYBR Green Master Mix (Takara, Japan) according to the manufacturer’s protocols. The primers were listed as follows: COL1A1, Forward-5′-AGTGGTTTGGATGGTGCCAA -3′; Reverse-5′-GCACCATCATTTCCACGAGC -3′; IGF1, Forward-5′-AGTGCTGCTTTTGTGATTTCTTGA-3′; Reverse-5′-ACCCTGTGGGCTTGTTGAAA -3′; COL5A1, Forward-5′-CTGACAAGAAGTCCGAAGGG -3′; Reverse-5′-CCACATAGGAGAGCAGTTTCC-3′; CXCL12, Forward-5′-GAAAGCCATGTTGCCAGAGC-3′; Reverse-5′-CTCTCACATCTTGAACCTCTTGT-3′; PTEN, Forward-5′- TCCCAGACATGACAGCCATC-3′; Reverse-5′-TGCTTTGAATCCAAAAACCTTACT-3′; SPP1, Forward-5′-AGCAGCTTTACAACAAATACCCAG- 3′; Reverse-5′-TTACTTGGAAGGGTCTGTGGG- 3′; GAPDH, Forward-5′-AATGGGCAGCCGTTAGGAAA-3′; Reverse-5′-GCCCAATACGACCAAATCAGAG-3′. GAPDH were used as an internal control and relative gene expressions were calculated with the 2−ΔΔCt method.
Statistical analysis
Statistical analysis was performed using SPSS Statistics software (version 22.0). Two-tailed Student’s t test was used for calculating the difference between two groups. P value < 0.05 was considered statistically significant.
Results
DEGs identification
Based on the GEO database and GEO2R online analysis tool, a total of 160 DE mRNAs were detected from GSE115810, of which 13 were upregulated mRNAs and 137 were downregulated; a total of 67 DE mRNAs were detected from GSE36389, of which 15 were upregulated and 52 were downregulated (Fig. 1). Subsequently, these two groups of DE mRNAs were intersected and there were 40 mRNAs which overlapped between GSE115810 and GSE36389. Finally, these 40 overlapped mRNAs, with 3 upregulated and 37 downregulated, were filtered out and identified as the DEGs of EC (Fig. 2a).

Volcano plot (a) and heat map (b) of GSE115810 and GSE36389. In the volcano plot, red plot represents upregulated gene, green plot represents downregulated gene, and black plot represents gene without significant expression in microarray dataset

Identification of the DEGs. a Venn diagrams for the identification of DEGs in endometrial carcinoma (EC); 40 genes were overlapped in GSE115810 and GSE36389, and then were identified as DEGs. b Protein-protein interaction (PPI) network for the DEGs
PPI network construction and hub genes identification
A PPI network of the DEGs was constructed by STRING and visualized by Cytoscape to further explore the relationships among these genes. According to the node pair combing score ≥ 0.4, a total of 26 DEGs were filtered into the PPI network that interacted with other DEG(s) (Fig. 2b). Furthermore, according to the topological properties analysis of the PPI network, several nodes have higher connectivity degree, with degree ≥ 5 set as the criterion. These nodes were Collagen alpha-1(I) chain (COL1A1, degree = 11), insulin-like growth factor 1 (IGF1, degree = 9), secreted protein acidic and rich in cysteine (SPARC, degree = 8), collagen alpha-1(V) chain (COL5A1, degree = 6), fibrillin-1 (FBN1, degree = 6), CXC chemokine ligand 12 (CXCL12, degree = 6), phosphatase and tensin homolog (PTEN, degree = 5), secreted phosphoprotein 1 (SPP1, degree = 5), and Wilms tumor 1 (WT1, degree = 5). Besides, most of these nodes could interact with each other in the PPI network.
GO term and KEGG pathway enrichment analysis of DEGs
GO and KEGG enrichment analysis were performed to gain further insight of the bioinformatics information of the 40 DEGs (Table 1). The each enriched GO term of DEGs was ranked by − log10 (P value) (Fig. 3). GO terms includes biological processes, cellular components, and molecular function. The significant enriched biological process terms were extracellular matrix (ECM) organization, response to estradiol, collagen fibril organization, negative regulation of cell proliferation, cell adhesion, blood vessel development, positive regulation of epidermal cell differentiation/apoptotic process, cell migration, etc.; the significantly enriched cellular component terms were proteinaceous ECM, extracellular region, ECM, basement membrane, extracellular exosome, platelet alpha granule lumen, and cell projection; and the significantly enriched molecular function terms were integrin/heparin binding, ECM structural constituent, ECM binding, calcium ion binding, insulin-like growth factor receptor binding, etc. KEGG pathways enrichment analysis of the DEGs suggests that focal adhesion, ECM-receptor interaction, PI3K-Akt signaling pathway, and pathways in cancer were found to be significantly enrichment pathways (Fig. 4a). Moreover, for the multiple genes involved in the identified KEGG pathways, the DEG-pathway network showed that some of the DEGs exhibited active participations, including COL1A1, IGF1, COL5A1, PTEN, CXCL12, and SPP1 (Fig. 4b). Former PPI analysis also identified these genes with higher node degree, then these six DEGs were considered as hub DEGs and utilized for further analysis.

The gene ontology (GO) enrichment analysis of the DEGs in EC. GO were divided into three categories: biological process, cellular component, molecular function. Each enriched GO terms of DEGs were ranked by − log10 (P value)

Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the DEGs. a KEGG pathway of the DEGs. b DEG-KEGG pathway network
Genetic alterations of the hub genes in EC
cBio Portal online database was applied for a further investigation of these hub genes in EC oncogenesis. Genetic alterations of hub genes and their cancer clinical profiles in EC samples were observed from the TCGA database based on cBio Portal platform. As shown in Fig. 5a, tumor samples with sequencing and CNA data for each gene also implied that 562 cases (52%) of 1076 sequenced cases had alterations in at least one of the hub DEGs queried. For COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1, the genetic alteration rate were 6%, 1.9%, 9%, 1.4%, and 1.8%, respectively. When it comes to PTEN, it showed that half of the cases were altered (50%) and the alterations were classified as truncating mutation, missense mutation, deep deletion, frame mutation, and amplification.

Genetic alterations and survival analysis of the total six hub genes in EC by cBio portal. a A visual summary of alteration across a set of EC samples (data taken from the TCGA Data Portal) based on a query of the six genes (COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1). Distinct genomic alterations are summarized and color-coded, presented by % changes in particular affected genes in individual tumor samples. Each row represents a gene and each column represents a tumor sample. b Overall survival and the disease-free survival rates of the EC patients with or without COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 mutations. The red curves in the Kaplan-Meier plots include cases with gene alteration, and the blue curves include cases without gene alteration
The survival results indicated that these total six DEGs were also obtained from the cBio Portal platform. Overall survival and disease-free survival were compared between cancer cases that have alteration of these queried genes and without alteration of these genes in EC samples (Fig. 5b). For the overall survival, cases without alteration in the hub genes have worse survival than cases with alteration, and significant difference was found with a P value of 6.862E−05 < 0.05. For the disease-free survival, the outcome consists of the overall survival, with a P value of 1.445E−03 < 0.05.
Correlations between COL1A1 and IGF1, COL5A1, PTEN, CXCL12, and SPP1
According to the raw data of the gene expression profiles in GSE115810 and GSE36389, the correlations between COL1A1 and IGF1, COL5A1, PTEN, CXCL12, and SPP1 were analyzed. As shown in Fig. 6a, the results indicated that there were significant correlations between COL1A1 and IGF1 (R = 0.566, P < 0.01), COL5A1 (R = 0.510, P < 0.01), PTEN (R = 0.807, P < 0.01), CXCL12 (R = 0.504, P < 0.01), and SPP1 (R = 0.44, P < 0.01), respectively.

a Correlation analysis between COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1. b Expression profiles of COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 in EC patients based on TCGA samples. The blue box represents normal samples; red box represents tumor samples
Expression validation of COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 in EC
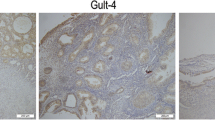
Due to the former analysis found that hub DEGs were frequently mutated in EC patients and related with EC survival, we made further analysis on these genes. Based on TCGA platform and online tool UALCAN, the expression profiles of hub DEGs in EC were further validated. As shown in Fig. 6b, it indicated that the COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 were significantly differently expressed in EC tumor compared with those in normal samples (P = 4.93E−02, P = 5.24E−03, P = 2.83E−04, P = 1.58E−06, P = 2.11E−12, and P = 6.91E−13, respectively). Furthermore, the immunohistochemistry map of the hub genes protein expression in normal and EC tissues was also shown in Fig. 7a, and different expressions of these hub genes in normal and EC tissues also could be observed. The mRNA expression of these hub DEGs were also verified using qRT-PCR in 13 pairs of samples form EC patients and adjacent normal samples. qRT-PCR results showed that the mRNA expression of COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 were also significantly differently expressed in normal samples and tumor samples (Fig. 7b). The relative expression of COL1A1, IGF1, COL5A1, CXCL12, and PTEN in tumor samples was significantly lower compared to those in adjacent normal samples (P < 0.05), while SPP1 expression was significantly higher in tumor samples compared to the adjacent normal samples (P < 0.05).

a Immunohistochemistry map of the hub genes protein expression in normal and EC tissues. b qRT-PCR validation of the expression of COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 in 13 pairs of samples form EC patients and adjacent normal samples. *P < 0.05, **P < 0.01
Prognosis values of the COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 in EC
Prognosis value of the each hub gene was also analyzed with the survival analysis. As shown in Fig. 8, the Kaplan-Meier survival analysis showed that the survival outcome in high expression of COL1A1, IGF1, and PTEN patients was significantly better than low expression patients (P = 0.042, P < 0.001, P = 0.015, respectively).

Survival analysis of COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 in EC. P < 0.05 was considered as statistically significant. The blue line represents the low expression of DEG; red line represents the high expression of DEG
Discussion
EC, as common gynecological malignant tumors posing serious threat to the women health worldwide, its pathophysiological process remains elusive and complicated. In present study, bioinformatics analysis was performed based on the gene expression microarray dataset of GSE115810 and GSE36389 from the GEO database. A total of 40 genes were screened out as the EC related DEGs. Functional enrichment analysis showed that these DGEs were significantly enriched in biological processes including extracellular matrix (ECM) organization, response to estradiol, collagen fibril organization, negative regulation of cell proliferation, cell adhesion, blood vessel development, positive regulation of epidermal cell differentiation/apoptotic process, cell migration, etc.; KEGG pathways include focal adhesion, ECM-receptor interaction, PI3K-Akt signaling pathway, and pathways in cancer. The functional enrichment evidence strongly shored up the supposition that these DEGs can affect the proliferation, differentiation, migration, and apoptosis of EC cells through the aforementioned biological processes and pathways and thus regulate the onset and progression of EC. More importantly, COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1 were identified as hub DEGs with potential values in EC.
In this study, the expression of COL1A1 and COL5A1 were downregulated in EC samples compared with normal samples in microarray datasets and further expression validation. Survival analysis also found that the survival outcome of EC patients in high expression of COL1A1 was significantly better than that in low COL1A1 expression patients (P = 0.042). And in 1076 sequenced EC cases, the genetic alteration rate of COL1A1 was 6% and COL5A1 was 9%; the major genetic alterations were amplification, deep deletion, and missense mutation. COL1A1 and COL5A1 all belong to the collagen family; this family is the major component of the tumor microenvironment, it interacts with substances in the ECM (such as matrix metalloproteinases), integrins, tyrosine kinase receptors, and some signaling pathways to influence tumor cell behavior and activity [11, 12]. Evidencing studies of collagen in diverse cancers illustrate the dual roles of collagen. Study found that COL1A1 gene expression was markedly decreased in hepatocellular carcinoma tumor tissues (log2 ratio − 1.1) with a poor overall survival rate (P = 0.013) [13]. Tumorigenesis of EC is a complex process, with multiple oncogenes and anti-oncogenes critically involved, forming a tight connected network, affecting multiple signaling pathways and metabolism or biological processes in the organs, and thus inducing the progression of tumor. PPI network in the present study showed that these genes could mostly interact with each other; correlation analysis also revealed that the expression of COL1A1 and the rest of hub genes IGF1, COL5A1, CXCL12, PTEN, and SPP1 was significantly correlated in EC. This indicated that these genes have tight correlations with others, and these correlations might amplify their roles in EC.
Periodic changes in IGF1 expression play a key role in regulating the transition of premenopausal endometrium through procreation, secretion, and menstrual cycle. In this study, IGF1 was screened as a hub DEG of EC, expression validation in TCGA database and clinical samples proved that IGF1 was downregulated in EC samples, and survival analysis also indicated that the EC patients with high IGF1 expression have better survival outcome than those low expression group patients (P < 0.001). IGF1 is thought to be a potential mediator for the effects of estradiol on uterine growth. Epidemiological, clinical, and experimental data also shore up the evidence that IGF1 is an important players in general gynecological cancers, and particularly in endometrial tumors [14,15,16]. The downregulation of IGF-1 was also found in endometrial cancer specimens compared to the adjacent normal specimens in Soufla’s study, which seems to comprise the main features of endometrial carcinogenesis [17]. CXCL12 expression is also associated with the survival of cancer patients, but the role is dual. High CXCL12 expression was associated with reduced overall survival in patients with esophagogastric (P = 0.002), pancreatic (P = 0.0005), and lung cancer (P = 0.01), whereas in breast cancer patients, high CXCL12 expression conferred an overall survival advantage (P < 0.001) [18]. Felix et al. suggest that the positive CXCL12 expression was associated with longer overall survival (log-rank P = 0.006) and longer recurrence-free survival (log-rank p = 0.01) in estrogen receptor (ER) negative EC patients, but not in ER positive EC patients [19]. Another study found that the expression of CXCR4 was predominant (P = 0.035) whereas CXCL12 was low-expressed (P = 0.002) in EC [20].
It is proved that PTEN produces tumor suppressor effect mainly through blocking the PI3K/Akt signaling pathway based on its lipid phosphatase activity, thus inhibiting the activity of Akt and inducing the apoptosis of tumor cells [21]. This is consistent with our KEGG enrichment results that PTEN and other hub DEGs were mainly involved in the PI3K/Akt signaling pathway. Hyperactivation of PI3K/Akt signaling is the risk factors for carcinogenesis of cells, and it is regulated by a variety of factors, whose negative feedbacks are mainly regulated including PTEN [22]. As a tumor suppressor gene, the lost or mutation of PTEN was revealed and regarded as diagnostic biomarker in multiple types of cancers, like prostate cancer [23], breast cancer [24], gastric cancer [25], cervical carcinoma [26], and EC [27, 28]. In our study, microarray analysis and expression validation found that PTEN was downregulated in EC samples; survival analysis also indicated that the EC patients with high PTEN expression have better survival outcome than those low expression group patients (P < 0.015). SPP1, also called osteopontin, is involved in various cellular processes like ECM binding, osteoblast differentiation, cell adhesion, immune regulation, and ECM receptor interaction acting through interaction with multiple cell surface receptors including CD44 [29, 30]. Except for the critical role in wound healing and chronic inflammatory diseases, the expression of SPP1 also has strong relationship with numerous tumors. Abnormal expression of SPP1 was found in numerous cancers and also has potential correlation with cancer prognosis [31, 32]. Our study showed that in 1076 sequenced EC cases, the genetic alteration rate of SPP1 was 1.8%, and the major alteration was missense mutation. Tu et al.’s study also observed that mutations in SPP1 primarily occurred in cutaneous melanoma and endometrial cancer, with the ratio of alteration which ranged from 1.03 to 9.23% [33].
Genetic alterations results in this study implied that half of the EC cases (52%) had alterations in at least one of the hub DEGs queried, with the genetic alteration rate varied from 1.4 to 50% for hub genes, especially for PTEN alteration. It showed that half of the cases were altered (50%) and the alterations were classified as truncating mutation, missense mutation, deep deletion, frame mutation, and amplification. Gene mutation is responsible for the abnormal functions of the code protein and might further cause abnormal phenotype, including tumor occurrence. Mutations of anti-oncogene were often observed in variety of tumors, and this may correlated to the acceleration of tumorigenesis [34]. PTEN is the most commonly and frequently mutated gene in EC, and the loss of PTEN is considered to be an early event of endometrial tumorigenesis. In this study, further overall and disease-free survival analysis observed statistically different outcomes with these DEG alterations, and individual survival analysis of hub genes in EC patients also demonstrated that low expression of COL1A1, IGF1, and PTEN were related with poor survival outcomes in EC patients, which indicated that these DEGs might critically correlate with the EC prognosis.
Conclusion
In conclusion, the present study identified six genes (COL1A1, IGF1, COL5A1, CXCL12, PTEN, and SPP1) with crucial role in tumorigenesis and progression in EC; our results suggested these genes could add a new dimension to our understanding of the EC and might be served as potential biomarkers that will be assisting cancer biologists and clinical oncologists in developing novel therapeutic strategies for EC patients. However, there are some limitations in this study. Further larger clinical sample size and in-depth experiment studies in vivo and in vitro are needed to clarify the clear mechanism and warrant the prognostic value of these DEGs in EC.
Availability of data and materials
The datasets supporting the conclusion of this article are included within the article.
Abbreviations
- EC:
-
Endometrial carcinoma
- DEGs:
-
Differentially expressed genes
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPI:
-
Protein-protein interaction network
- TCGA:
-
The Cancer Genome Atlas
- COL1A1:
-
Collagen alpha-1(I) chain
- IGF1:
-
Insulin-like growth factor 1
- SPARC:
-
Secreted protein acidic and rich in cysteine
- COL5A1:
-
Collagen alpha-1(V) chain
- FBN1:
-
Fibrillin-1
- CXCL12:
-
CXC chemokine ligand 12
- PTEN:
-
Phosphatase and tensin homolog
- SPP1:
-
Secreted phosphoprotein 1
- WT1:
-
Wilms tumor 1
- ECM:
-
Extracellular matrix
- IRS1:
-
Insulin receptor substrate 1
- MAPK:
-
Mitogen-activated protein kinases
- CXCR4:
-
CXC motif chemokine receptor 4
- SHIP2:
-
SH2-containing inositol 5-phosphatase
- PIP3:
-
Plasma membrane intrinsic protein 3
References
Salvesen HB, Haldorsen IS, Trovik J. Markers for individualised therapy in endometrial carcinoma. Lancet Oncology. 2012;13(8):e353.
Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. Ca A Cancer Journal for Clinicians. 2016;66(4):271–89.
Chen M, Jin Y, Li Y, et al. Oncologic and reproductive outcomes after fertility-sparing management with oral progestin for women with complex endometrial hyperplasia and endometrial cancer. Int J Gynaecol Obstet. 2015;132(1):34–8.
Buhtoiarova TN, Brenner CA, Singh M. Endometrial carcinoma: role of current and emerging biomarkers in resolving persistent clinical dilemmas. American Journal of Clinical Pathology. 2016;145(1):8–21.
Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecologic Oncology. 1983;15(1):10–7.
Group A S, Blake P, Swart AM, et al. Adjuvant external beam radiotherapy in the treatment of endometrial cancer (MRC ASTEC and NCIC CTG EN.5 randomised trials): pooled trial results, systematic review, and meta-analysis. Lancet. 2009;373(9658):137–46.
Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum. mol. genet. 2003;12(12. Spec):R145–52.
Wang XG, Peng Y, Song XL, et al. Identification potential biomarkers and therapeutic agents in multiple myeloma based on bioinformatics analysis. Eur Rev Med Pharmacol Sci. 2016;20(5):810–7.
Liang B, Li C, Zhao J. Identification of key pathways and genes in colorectal cancer using bioinformatics analysis. Medical Oncology. 2016;33(10):111.
Bijnens AP, Lutgens E, Ayoubi T, et al. Genome-wide expression studies of atherosclerosis: critical issues in methodology, analysis, interpretation of transcriptomics data. Arteriosclerosis Thrombosis & Vascular Biology. 2006;26(6):1226–35.
Jabłońska-Trypuć A, Matejczyk M, Rosochacki S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. Journal of Enzyme Inhibition and Medicinal Chemistry. 2016;31(sup1):177–83.
Xu S, Xu H, Wang W, et al. The role of collagen in cancer: from bench to bedside. J Transl Med. 2019;17(1):309.
Hayashi M, Nomoto S, Hishida M, et al. Identification of the collagen type 1 alpha 1 gene (COL1A1) as a candidate survival-related factor associated with hepatocellular carcinoma. BMC Cancer. 2014;14(1):108.
Liu L, Wang X, Li X, et al. Upregulation of IGF1 by tumor-associated macrophages promotes the proliferation and migration of epithelial ovarian cancer cells. Oncology Reports. 2018;39(2):818–26.
Merritt MA, Strickler HD, Einstein MH, et al. Insulin/IGF and sex hormone axes in human endometrium and associations with endometrial cancer risk factors. Cancer Causes & Control. 2016;27(6):737–48.
Shafiee MN, Seedhouse C, Mongan N, et al. Up-regulation of genes involved in the insulin signalling pathway (IGF1, PTEN and IGFBP1) in the endometrium may link polycystic ovarian syndrome and endometrial cancer. Molecular & Cellular Endocrinology. 2016;424(C):94–101.
Soufla G, Sifakis S, Spandidos DA. FGF2 transcript levels are positively correlated with EGF and IGF-1 in the malignant endometrium. Cancer Letters. 2008;259(2):146–55.
Samarendra H, Jones K, Petrinic T, et al. A meta-analysis of CXCL12 expression for cancer prognosis. British Journal of Cancer. 2017;117(1):124–35.
Felix AS, Stone RA, Chivukula M, et al. Survival outcomes in endometrial cancer patients are associated with CXCL12 and estrogen receptor expression. International Journal of Cancer Journal International Du Cancer. 2012;131(2):114–21.
Gelmini S, Mangoni M, Castiglione F, Beltrami C, et al. The CXCR4/CXCL12 axis in endometrial cancer. Clinical & Experimental Metastasis. 2009;26(3):261–8.
Haddadi N, Lin Y, Travis G, et al. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Molecular Cancer. 2018;17(1):37.
Fresno Vara JA, Casado E, De CJ, et al. PI3K/Akt signalling pathway and cancer. Cancer Treatment Reviews. 2004;30(2):193–204.
Su Q, Zhang B, Zhang L, et al. Jagged1 upregulation in prostate epithelial cells promotes formation of reactive stroma in the PTEN null mouse model for prostate cancer. Oncogene. 2017;36(5):618–27.
Wang LL, Hao S, Zhang S, et al. PTEN/PI3K/Akt protein expression is related to clinicopathological features and prognosis in breast cancer with axillary lymph node metastases. Human Pathology. 2017;61:49–57.
Ma J, Guo X, Zhang J, et al. PTEN gene induces cell invasion and migration via regulating Akt/GSK-3β/β-catenin signaling pathway in human gastric cancer. Digestive Diseases & Sciences. 2017;62(12):1–11.
Feng Y, Zou W, Hu C, et al. Modulation of CASC2/miR-21/PTEN pathway sensitizes cervical cancer to cisplatin. Archives of Biochemistry & Biophysics. 2017:623–4.
Yang HP, Meeker A, Guido R, et al. PTEN expression in benign human endometrial tissue and cancer in relation to endometrial cancer risk factors. Cancer Causes & Control. 2015;26(12):1729–36.
Ju Z, Westin SN, Broaddus R, et al. PTEN loss as a context-dependent determinant of patient outcomes in obese and non-obese endometrioid endometrial cancer patients. Genetics Selection Evolution Gse. 2013;38(3):313–20.
Ahmed M, Behera R, Chakraborty G, et al. Osteopontin: a potentially important therapeutic target in cancer. Expert Opinion on Therapeutic Targets. 2011;15(9):1113–26.
Chen Q, Shou P, Zhang L, et al. An osteopontin-integrin interaction plays a critical role in directing adipogenesis and osteogenesis by mesenchymal stem cells. Stem Cells. 2014;32(2):327–37.
Zhuo C, Li X, Zhuang H, et al. Elevated THBS2, COL1A2, and SPP1 expression levels as predictors of gastric cancer prognosis. Cellular Physiology & Biochemistry International Journal of Experimental Cellular Physiology Biochemistry & Pharmacology. 2016;40(6):1316–24.
Zeng B, Zhou M, Wu H, et al. SPP1 promotes ovarian cancer progression via integrin β1/FAK/Akt signaling pathway. Oncotargets & Therapy. 2018;11:1333–43.
Tu Y, Chen C, Fan G, et al. Association between the expression of secreted phosphoprotein-related genes and prognosis of human cancer. BMC Cancer. 2019;19(1):1230.
Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature genetics. 1997;15(4):356–62.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China [Grant no. 81602267], the Natural Science Foundation of Zhejiang Province, China [Grant no. LYY19H310001], and Science and Technology of traditional Chinese Medicine Project of Zhejiang Province, China [Grant no. 2017ZA031].
Author information
Authors and Affiliations
Contributions
The conception and design of the study: Yiming Zhu, Tao Zhu. Acquisition of data, analysis and interpretation of data: Yiming Zhu, Liang Shi, Ping Chen. Drafting the article and revising it critically for important intellectual content: Yiming Zhu, Yingli Zhang, Tao Zhu. Final approval of the version to be submitted: all the authors. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, Y., Shi, L., Chen, P. et al. Identification of six candidate genes for endometrial carcinoma by bioinformatics analysis. World J Surg Onc 18, 161 (2020). https://doi.org/10.1186/s12957-020-01920-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-020-01920-w