
Abstract
The ADIPOR1 and ADIPOR2 proteins (ADIPORs) are generally considered as adiponectin receptors with anti-diabetic properties. However, studies on the yeast and C. elegans homologs of the mammalian ADIPORs, and of the ADIPORs themselves in various mammalian cell models, support an updated/different view. Based on findings in these experimental models, the ADIPORs are now emerging as evolutionarily conserved regulators of membrane homeostasis that do not require adiponectin to act as membrane fluidity sensors and regulate phospholipid composition. More specifically, membrane rigidification activates ADIPOR signaling to promote fatty acid desaturation and incorporation of polyunsaturated fatty acids into membrane phospholipids until fluidity is restored. The present review summarizes the evidence supporting this new view of the ADIPORs, and briefly examines physiological consequences.
Similar content being viewed by others


Background
The ADIPOR1 and ADIPOR2 proteins (ADIPORs) have been the subject of several high-profile articles suggesting that they are adiponectin receptors with anti-diabetic properties [1,2,3,4,5]. Separately, the yeast and C. elegans homologs of the mammalian ADIPORs have now been studied for over ten years and this is a brief account of what has been learned during that time.
The ADIPORs are PAQR proteins
The ADIPORs are members of the PAQR (progestin and adipoQ receptors) protein family, named after two of its founding members [6]. There are eleven PAQR proteins in the human genome (PAQR1-11), and ADIPOR1 and ADIPOR2 correspond to PAQR1 and PAQR2, respectively. PAQR proteins have three important characteristics: (1) seven transmembrane domains; (2) an orientation inverse that of GPCRs (G protein-coupled receptors), i.e. with their N-terminus being cytoplasmic and the C-terminus extracellular; and (3) they are part of the larger CREST protein family of hydrolases [7]. ADIPOR1 and ADIPOR2 were initially identified as putative receptors for the primarily adipocyte-secreted protein adiponectin (other tissues can produce adiponectin but adipocytes are by far the main source [8]), which was done by screening a cDNA expression library to identify proteins that bound fluorescent-tagged bacterially expressed recombinant adiponectin [3]. Several publications have since reported adiponectin-dependent ADIPOR signaling or physiological roles [1, 2, 4, 5, 9, 10]. Cautiously however, one study suggest that the ADIPORs may not be adiponectin receptors [11], and adiponectin itself is now emerging as a lipid carrier protein, which would explain its high concentration in plasma [12]. The crystal structure of the ADIPORs has been solved and consists of a barrel-shaped conformation open towards the cytoplasm with a cavity capable of accommodating fatty acids (FAs), or FA-like substrates, and a zinc-coordination site that may either stabilize the structure and/or participate in a hydrolytic reaction, such as the proposed ceramidase activity first suggested by studies of the yeast homologs and consistent with observations with the mammalian proteins [5, 13, 14].
Lessons from yeast
The yeast Izh1, -2, -3 and − 4 (implicated in zinc homeostasis) genes encode PAQR proteins similar to the ADIPORs. A 2004 study by Lyons et al. showed that the Izh genes regulate the levels of structural membrane sterols and, in this indirect way, may affect the permeability of certain ions, such as zinc [15]. Importantly, expression of Izh genes is regulated by an oleate response element (ORE) [16], and is inhibited by the presence of oleic acid (C18:1) or linoleic acid (C18:2), two unsaturated fatty acids (UFAs) that promote membrane fluidity when present in phospholipids [17]. Conversely, Izh gene expression is stimulated by the presence of saturated fatty acids (SFAs) that promote membrane rigidification when incorporated into phospholipids [15, 17]. In 2009, the Lyons group showed that IZH2 is a ceramidase, i.e. can hydrolyze ceramides to generate free FAs and sphingoid bases that act as second messengers [18]. The same group showed that the human ADIPOR1 and ADIPOR2 proteins can functionally mimic IZH proteins [19], and are functional as ceramidases when heterologously expressed in yeast cells [20]. The activity of the ADIPORs was enhanced by addition of adiponectin in the media during those experiments [20]; unfortunately, the source of adiponectin was not specified, which is an important point given that active recombinant adiponectin is difficult to produce and may contain biologically active impurities [21]. A separate study further strengthened the connection to membrane homeostasis by identifying genes mis-regulated in the Izh2 mutant: down-regulated genes included several lipid metabolism genes such as Ino1 (inositol-3-phosphate synthase), Cho1 (phosphatidylserine synthase), Tsc (important for sphingosine synthesis) and Erg28 (ergosterol biosynthesis); over-expressed genes included genes important for phosphate transport (Pho84, Pho89 and Pic2) [22]. Izh2 is also upregulated under condition of increased membrane rigidity in a yeast model with varying UFA content [23], and the Izh2 and Izh4 genes are continuously repressed under conditions of high UFAs, i.e. when membranes are not challenged by SFA-driven rigidification [24]. In summary, the yeast homologs of the ADIPORs regulate membrane composition (structural lipids), are up-regulated by membrane rigidification (e.g. excess SFAs), are inhibited by the presence of membrane-fluidizing UFAs, carry a ceramidase activity, signal via sphingoid bases and can be functionally replaced, at least partially, by the human ADIPORs.
Lessons from C. elegans
There are 5 PAQR proteins encoded by the C. elegans genome, two of which are clear ADIPOR homologs and are named PAQR-1 and PAQR-2 (there are no adiponectin homologs in C. elegans) [25]. Mutants lacking PAQR-1 have no obvious phenotypes, though they enhance the phenotypes of worms lacking PAQR-2, indicating redundancy [25, 26]. Worms lacking PAQR-2 have a deformed tail tip, impaired autophagy, reduced life span, brood size and locomotion, and are cold- and SFA-intolerant because both conditions promote membrane rigidification [25, 27,28,29,30,31]. Importantly, the primary defect in mutants lacking PAQR-2 is an excess of SFAs in their phospholipids accompanied by debilitating membrane rigidity, which we documented through lipidomics analysis and verified in vivo using fluorescence recovery after photobleaching (FRAP) [27, 28, 32,33,34,35]. A forward genetics screen led to the identification of the protein IGLR-2 as an obligate partner for PAQR-2 function: both proteins must be present in the same cell for systemic maintenance of membrane fluidity when worms are challenged with either cold temperatures or SFA-rich diets [28, 35]. IGLR-2 has a large extracellular N-terminal region containing one immunoglobulin domain and several leucine-rich repeats, one transmembrane alpha helix and a large cytoplasmic C-terminal domain [28]. IGLR-2 is loosely homologous to mammalian LRIG proteins, a family with nearly 40 members [36], though a functional IGLR-2 ortholog has not yet been identified. Bifluorescence complementation (BiFC) and fluorescence resonance energy transfer (FRET) studies have shown that IGLR-2 localizes to the plasma membrane where it interacts in a membrane rigidity-dependent manner with PAQR-2 [28, 37]. Additionally, structure-function studies indicate that the two proteins interact via their transmembrane domains and, more speculatively, that IGLR-2 may help displace the cytoplasmic PAQR-2 domain to facilitate access of substrates to the active site [26, 37]. Note also that IGLR-2 is not required for PAQR-1 function, suggesting that PAQR-1 either has a basal constitutive activity or is regulated via a separate mechanism [26].
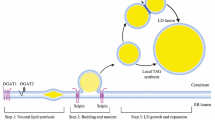
Remarkably, an unbiased genetic screen to identify mutants intolerant of dietary palmitic acid (a C16:0 SFA) showed that PAQR-2 and IGLR-2 are the only two C. elegans proteins specifically essential to prevent lethal membrane rigidification by SFA-rich diets [37]. Additionally, screens for secondary mutations that compensate for the lack of PAQR-2 revealed the existence of two independent downstream branches in the PAQR-2/IGLR-2 pathway: “Branch 1” acts by upregulating the expression of desaturases, and its function can be replaced by gain-of-function alleles of NHR-49 (a homolog of the mammalian PPARs), MDT-15 (a homolog of the mammalian mediator subunit MED15) or SBP-1 (a homolog of the mammalian SREBPs)[29]; “Branch 2” acts by promoting PUFA production and/or their incorporation into phospholipids, and its function can be replaced by loss-of-function mutations in FLD-1 (a homolog of the mammalian TLCD1 and TLCD2 proteins) or ACS-13 (a homolog of the mammalian ACSL1)[32, 33]. Only by simultaneously providing mutations that replace both branches can one achieve complete suppression of all phenotypes found in worms lacking PAQR-2 [32, 33](Fig. 1). In summary, work in C. elegans showed that the essential function of PAQR-2 is to respond to membrane rigidification by low temperature or SFA-rich diets by promoting a compensatory increase in phospholipids that contain UFAs.

The role of PAQR-2 in membrane homeostasis. Conditions that promote membrane rigidification, such as SFA-rich diets or low temperature, stimulate the formation, hence activation, of the PAQR-2/IGLR-2 complex. While PAQR-2 is definitely an ADIPOR ortholog, no functional homolog of IGLR-2 has yet been identified in mammals. Genetic studies reveal that two downstream branches mediate the effects of the PAQR-2/IGLR-2 complex: (1) Branch 1 stimulates the transcription of fatty acid desaturase genes and can be replaced by gain-of-function mutations in NHR-49 (homologous to the mammalian PPARs), MDT-15 or SBP-1; and (2) Branch 2 stimulates the production/incorporation of PUFAs into phospholipids and can be replaced by loss-of-function mutations in FLD-1 (homologous to the mammalian TLCD1/2 proteins) or ACS-13. The ultimate output of PAQR-2 signaling is to increase the UFA content in phospholipids, which promotes fluidity
Translation to mammalian cells
The findings concerning C. elegans PAQR-2 have been extended to the ADIPORs in mammalian cells using siRNA and CRISPR/Cas9 to silence or knock out target genes, RNA sequencing to monitor the transcriptome, lipidomics to monitor the FA composition of phospholipids, and three different methods to measure membrane fluidity, namely FRAP, Laurdan dye staining and atomic force microscopy. We found that ADIPOR1 and/or ADIPOR2 depletion causes excess SFAs in phospholipids accompanied by membrane rigidification in all cell lines studied (embryonic kidney cells-derived HEK293, hepatocyte-derived HepG2, astrocyte-like 1321N1) and in primary human umbilical vein endothelial cells (HUVECs), with the most potent effects occurring with dual silencing of ADIPOR1 and ADIPOR2 [27, 32, 33, 38, 39]. qPCR and an exhaustive RNAseq analysis also showed that ADIPOR2 is required for a normal transcriptional response to SFA challenges: thousands of genes become mis-regulated in ADIPOR2-deficient cells challenged with 200 µM palmitate [39]. Among the most significantly downregulated genes in ADIPOR2-KO cells are the desaturases SCD, FADS1 and FADS2 while genes of the UPR (unfolded protein response) are among the most upregulated genes, which is likely a secondary consequence of the membrane homeostasis failure [39]. Not surprisingly, the ADIPOR-deficient cells exhibit mitochondrial respiration defects, poor viability and abnormal morphology when challenged with SFAs [39]. Most phenotypes found in cells lacking one or both ADIPORs can be abrogated by supplementing the cultures with membrane-fluidizing FAs such as oleic acid (a C18:1 UFA), eicosapentaenoic acid (a C20:5 PUFA) or docosahexaenoic acid (a C22:6 PUFA), which indicates that membrane rigidification is the primary defect in these cells [33, 38, 39]. Importantly, adiponectin is not required for the membrane homeostasis function of ADIPOR1 and ADIPOR2 in the cultured cell types tested, and the addition of a commercially purchased recombinant adiponectin produced from a mammalian expression system had no effect on the membrane fluidity of HEK293 cells [38]. This is somewhat confusing given the many studies documenting ADIPOR-dependent in vivo effects of adiponectin, including its effect on ceramide levels [5, 10]. Unfortunately, no comprehensive analysis of phospholipid composition has been reported for ADIPOR1/2 or adiponectin mutant mice, though it is known that ADIPOR1 mutations cause a severe depletion of long-chain PUFAs in the phosphatidylcholines of the retina accompanied by impaired vision both in mice and humans [40,41,42,43]. In terms of downstream events, it is likely that the two pathway branches mediating the effects of C. elegans PAQR-2/IGLR-2 are conserved in the human ADIPORs since they too regulate desaturase gene expression (“Branch 1”) [38], and their activity can at least partially be replaced by silencing TLCD1/TLCD2 or ACSL1, the homologs of C. elegans FLD-1 and ACS-13, respectively (“Branch 2”) [32, 33].
Recently, genome-wide CRISPR/Cas9 screens by the groups of Kivanc Birsoy (Rockefeller University) and Vamsi Mootha (Harvard Medical School) have provided further incontrovertible evidence that the ADIPORs are crucial for membrane homeostasis. In particular, ADIPOR2 ranked 25th out of ~ 3 000 metabolism genes tested for their ability to prevent palmitate toxicity in T-cell-derived Jurkat cells [44], and ranked 4th out of ~ 20 000 genes tested for their ability to suppress membrane rigidification in K562 cells challenged with hypoxia (a condition that inhibits oxygen-dependent desaturase reactions)[45]. Adiponectin itself was not picked by either screen, which may be due to the fact that it does not regulate ADIPOR2 or because it belongs to a family of secreted protein that may have partially redundant functions [46,47,48,49]. In conclusion, the results of ours and other’s studies are unambiguous: the function of ADIPOR1 and ADIPOR2 is conserved with that of C. elegans PAQR-2, and regulation of membrane homeostasis in most cell types is likely the ancestral function of these proteins, possibly extending as far back as the common ancestor between yeasts and animals. It is not known at present if there is a functional homolog to IGLR-2 in mammalian cells. Speculatively, functionally equivalent mechanisms (i.e. fluidity-dependent interactions) could involve other types of documented protein-protein interactions (e.g. APPL1; [50]) or homo/heterodimerization of the ADIPORs themselves [51, 52]. Also, future research will elucidate the precise nature of the two “branches” downstream of the ADIPORs, which likely involves their ceramidase activity.
An updated view of the ADIPORs
So far, ADIPOR1 and ADIPOR2 have been primarily considered as adiponectin receptors that act via PPARα, AMPK, and/or ceramide depletion to improve insulin function and protect from the metabolic syndrome [3, 5, 9, 10, 53, 54]. The literature just now summarized suggests a different view: the ADIPORs are evolutionarily conserved regulators of membrane homeostasis that do not require adiponectin to act as membrane fluidity sensors and regulate phospholipid composition. More specifically, membrane rigidification activates ADIPOR signaling to promote fatty acid desaturation and incorporation of PUFAs into membrane phospholipids. In this view, impaired ADIPOR function primarily leads to increased intracellular SFA levels and membrane rigidity and, secondarily, to a panoply of side effects including elevated ceramide levels (a likely consequence of increased intracellular palmitate [5, 10, 55,56,57,58]), altered signaling by numerous receptors and increased susceptibility to several diseases.
Relevance for human health
Membrane homeostasis is essential for most cellular processes, including membrane trafficking and fusion, organelle function, cytokinesis, etc. In particular, many receptors, transporters and channels require specific lipid interactions for their activity [59]. For example, SFAs induce c-SRC clustering, leading to the activation of JNK that in turns inhibit the insulin pathway and contributes to insulin resistance [60, 61]. Conversely, membrane fluidity increases insulin receptor signaling [62]. TRPV channels, which sense and regulate blood pressure in response to shear stress [63], are sensitive to membrane composition, requiring PUFA-containing phospholipids for their activation [64,65,66]. On the other hand, SFAs reduce membrane fluidity and promote the accumulation of TLR4 receptors in specialized membrane domains called lipid rafts, leading to homodimerization and proinflammatory signaling [67, 68]. Cell membrane tension and membrane fluidity also affect conformational dynamics of GPCRs and their ability to sense shear stress in endothelial cells [69]. And as a last example, phase separation of signaling molecules, which is influenced by membrane fluidity, promotes T cell receptor signal transduction [70]. This, merely the tip of a large iceberg, should suffice to convince any reader that membrane composition/property homeostasis is an important parameter in the regulation of vital signaling pathways. There are also numerous conditions where membrane fluidity defects are likely implicated: retinitis pigmentosa [41,42,43, 71], male sterility (in mice) [72,73,74], diabetes [75,76,77,78,79,80,81,82], X-linked adrenoleukodystrophy [83, 84], Gaucher disease [85], glycogen storage diseases [86], hypertension [87,88,89,90,91,92], thromboembolic disorders [93], cancer [94,95,96,97,98,99,100,101,102,103], Niemann-Pick disease type C [104], Parkinson’s disease [105,106,107,108], Alzheimer’s disease [109,110,111,112,113], Huntington’s disease [114, 115], Batten disease [116], polycystic kidney disease [117], inflammation [67, 68, 118, 119] and aging [120,121,122,123,124]. Note that the ADIPORs are ubiquitously expressed and that their roles in membrane homeostasis are cell non-autonomous due to lipid exchange among cells/tissues [35], which may complicate therapeutic approaches targeting specific organs. Nevertheless, pharmacological modulation of the ADIPOR pathway, possibly using agonists such as AdipoRon [125, 126], could have wide-ranging medical usefulness, and continued research to further elucidate the fluidity-sensing mechanisms and the pathways downstream of the ADIPORs will contribute to this development.
Study strengths and limitations
A strength of this review is that it emphasizes recent work on the cell and molecular function of the ADIPORs with a focus on model systems such as yeast, the nematode C. elegans and cultured mammalian cells. Conversely, the review does not cover in details the much more complex in vivo roles where the physiology of the whole organism makes it difficult to deduce specific molecular functions.
Conclusion, application and future perspective
The primary and evolutionarily conserved function of the ADIPORs is to regulate cell membrane composition to maintain its fluidity; other effects of the ADIPORs are likely secondary to this primary function. In the future, it will be interesting to elucidate the precise fluidity-sensing mechanism and the precise nature of the molecular events downstream of the ADIPORs by which they regulate the fatty acid composition of phospholipids.
Availability of data and materials
Not applicable.
Abbreviations
- ADIPOR:
-
Adiponectin receptor
- BiFC:
-
Bifluorescence complementation
- FA:
-
Fatty acid
- FRAP:
-
Fluorescence recovery after photobleaching
- FRET:
-
Fluorescence resonance energy transfer
- PAQR:
-
Progestin and adipoQ receptor
- SFA:
-
Saturated fatty acid
- UFA:
-
Unsaturated fatty acid
References
Awazawa M, Ueki K, Inabe K, Yamauchi T, Kaneko K, Okazaki Y, Bardeesy N, Ohnishi S, Nagai R, Kadowaki T. Adiponectin suppresses hepatic SREBP1c expression in an AdipoR1/LKB1/AMPK dependent pathway. Biochem Biophys Res Commun. 2009;382:51–6.
Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464:1313–9.
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–9.
Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–9.
Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55–63.
Tang YT, Hu T, Arterburn M, Boyle B, Bright JM, Emtage PC, Funk WD. PAQR proteins: a novel membrane receptor family defined by an ancient 7-transmembrane pass motif. J Mol Evol. 2005;61:372–80.
Pei J, Millay DP, Olson EN, Grishin NV. CREST–a large and diverse superfamily of putative transmembrane hydrolases. Biol Direct. 2011;6:37.
Nishida M, Funahashi T, Shimomura I. Pathophysiological significance of adiponectin. Med Mol Morphol. 2007;40:55–67.
Ishikawa M, Kitayama J, Yamauchi T, Kadowaki T, Maki T, Miyato H, Yamashita H, Nagawa H. Adiponectin inhibits the growth and peritoneal metastasis of gastric cancer through its specific membrane receptors AdipoR1 and AdipoR2. Cancer Sci. 2007;98:1120–7.
Holland WL, Xia JY, Johnson JA, Sun K, Pearson MJ, Sharma AX, Quittner-Strom E, Tippetts TS, Gordillo R, Scherer PE. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol Metab. 2017;6:267–75.
Kita S, Fukuda S, Maeda N, Shimomura I. Native adiponectin in serum binds to mammalian cells expressing T-cadherin, but not AdipoRs or calreticulin. Elife. 2019;8:e48675.
Ye JJ, Bian X, Lim J, Medzhitov R. Adiponectin and related C1q/TNF-related proteins bind selectively to anionic phospholipids and sphingolipids. Proc Natl Acad Sci U S A. 2020;117:17381–8.
Tanabe H, Fujii Y, Okada-Iwabu M, Iwabu M, Nakamura Y, Hosaka T, Motoyama K, Ikeda M, Wakiyama M, Terada T, et al. Crystal structures of the human adiponectin receptors. Nature. 2015;520:312–6.
Vasiliauskaite-Brooks I, Sounier R, Rochaix P, Bellot G, Fortier M, Hoh F, De Colibus L, Bechara C, Saied EM, Arenz C, et al. Structural insights into adiponectin receptors suggest ceramidase activity. Nature. 2017;544:120–3.
Lyons TJ, Villa NY, Regalla LM, Kupchak BR, Vagstad A, Eide DJ. Metalloregulation of yeast membrane steroid receptor homologs. Proc Natl Acad Sci U S A. 2004;101:5506–11.
Karpichev IV, Small GM. Global regulatory functions of Oaf1p and Pip2p (Oaf2p), transcription factors that regulate genes encoding peroxisomal proteins in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:6560–70.
Karpichev IV, Cornivelli L, Small GM. Multiple regulatory roles of a novel Saccharomyces cerevisiae protein, encoded by YOL002c, in lipid and phosphate metabolism. J Biol Chem. 2002;277:19609–17.
Villa NY, Kupchak BR, Garitaonandia I, Smith JL, Alonso E, Alford C, Cowart LA, Hannun YA, Lyons TJ. Sphingolipids function as downstream effectors of a fungal PAQR. Mol Pharmacol. 2009;75:866–75.
Kupchak BR, Garitaonandia I, Villa NY, Mullen MB, Weaver MG, Regalla LM, Kendall EA, Lyons TJ. Probing the mechanism of FET3 repression by Izh2p overexpression. Biochim Biophys Acta. 2007;1773:1124–32.
Kupchak BR, Garitaonandia I, Villa NY, Smith JL, Lyons TJ. Antagonism of human adiponectin receptors and their membrane progesterone receptor paralogs by TNFalpha and a ceramidase inhibitor. Biochemistry. 2009;48:5504–6.
Tullin S, Sams A, Brandt J, Dahl K, Gong W, Jeppesen CB, Krogh TN, Olsen GS, Liu Y, Pedersen AA, et al. Recombinant adiponectin does not lower plasma glucose in animal models of type 2 diabetes. PLoS One. 2012;7:e44270.
Mattiazzi Usaj M, Prelec M, Brloznik M, Primo C, Curk T, Scancar J, Yenush L, Petrovic U. Yeast Saccharomyces cerevisiae adiponectin receptor homolog Izh2 is involved in the regulation of zinc, phospholipid and pH homeostasis. Metallomics. 2015;7:1338–51.
Degreif D, de Rond T, Bertl A, Keasling JD, Budin I. Lipid engineering reveals regulatory roles for membrane fluidity in yeast flocculation and oxygen-limited growth. Metab Eng. 2017;41:46–56.
Yan GL, Duan LL, Liu PT, Duan CQ. Transcriptional Comparison Investigating the Influence of the Addition of Unsaturated Fatty Acids on Aroma Compounds During Alcoholic Fermentation. Front Microbiol. 2019;10:1115.
Svensson E, Olsen L, Morck C, Brackmann C, Enejder A, Faergeman NJ, Pilon M. The adiponectin receptor homologs in C. elegans promote energy utilization and homeostasis. PLoS One. 2011;6:e21343.
Busayavalasa K, Ruiz M, Devkota R, Stahlman M, Bodhicharla R, Svensk E, Hermansson NO, Boren J, Pilon M. Leveraging a gain-of-function allele of Caenorhabditis elegans paqr-1 to elucidate membrane homeostasis by PAQR proteins. PLoS Genet. 2020;16:e1008975.
Devkota R, Svensk E, Ruiz M, Stahlman M, Boren J, Pilon M. The adiponectin receptor AdipoR2 and its Caenorhabditis elegans homolog PAQR-2 prevent membrane rigidification by exogenous saturated fatty acids. PLoS Genet. 2017;13:e1007004.
Svensk E, Devkota R, Stahlman M, Ranji P, Rauthan M, Magnusson F, Hammarsten S, Johansson M, Boren J, Pilon M. Caenorhabditis elegans PAQR-2 and IGLR-2 Protect against Glucose Toxicity by Modulating Membrane Lipid Composition. PLoS Genet. 2016;12:e1005982.
Svensk E, Stahlman M, Andersson CH, Johansson M, Boren J, Pilon M. PAQR-2 regulates fatty acid desaturation during cold adaptation in C. elegans. PLoS Genet. 2013;9:e1003801.
Chen YL, Tao J, Zhao PJ, Tang W, Xu JP, Zhang KQ, Zou CG. Adiponectin receptor PAQR-2 signaling senses low temperature to promote C. elegans longevity by regulating autophagy. Nat Commun. 2019;10:2602.
Lee D, An SWA, Jung Y, Yamaoka Y, Ryu Y, Goh GYS, Beigi A, Yang JS, Jung GY, Ma DK, et al. MDT-15/MED15 permits longevity at low temperature via enhancing lipidostasis and proteostasis. PLoS Biol. 2019;17:e3000415.
Ruiz M, Bodhicharla R, Stahlman M, Svensk E, Busayavalasa K, Palmgren H, Ruhanen H, Boren J, Pilon M. Evolutionarily conserved long-chain Acyl-CoA synthetases regulate membrane composition and fluidity. Elife. 2019;8:e47733.
Ruiz M, Bodhicharla R, Svensk E, Devkota R, Busayavalasa K, Palmgren H, Stahlman M, Boren J, Pilon M. Membrane fluidity is regulated by the C. elegans transmembrane protein FLD-1 and its human homologs TLCD1/2. Elife. 2018;7:e40686.
Devkota R, Pilon M. FRAP: a powerful method to evaluate membrane fluidity in Caenorhabditis elegans. BIO-Protocol. 2018;8:e2913.
Bodhicharla R, Devkota R, Ruiz M, Pilon M. Membrane Fluidity Is Regulated Cell Nonautonomously by Caenorhabditis elegans PAQR-2 and Its Mammalian Homolog AdipoR2. Genetics. 2018;210:189–201.
Homma S, Shimada T, Hikake T, Yaginuma H. Expression pattern of LRR and Ig domain-containing protein (LRRIG protein) in the early mouse embryo. Gene Expr Patterns. 2009;9:1–26.
Devkota R, Henricsson M, Borén J, Pilon M. The C. elegans PAQR-2 and IGLR-2 membrane homeostasis proteins are uniquely essential for tolerating dietary saturated fats. BBA Mol Cell Biol Lipids. 2021;1866:158883.
Ruiz M, Stahlman M, Boren J, Pilon M. AdipoR1 and AdipoR2 Maintain Membrane Fluidity in Most Human Cell Types and Independently of Adiponectin. J Lipid Res. 2019;60:995–1004.
Ruiz M, Palmgren H, Henricsson M, Devkota R, Jaiswal H, Maresca M, Bohlooly YM, Peng XR, Boren J, Pilon M. Extensive transcription mis-regulation and membrane defects in AdipoR2-deficient cells challenged with saturated fatty acids. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866:158884.
Rice DS, Calandria JM, Gordon WC, Jun B, Zhou Y, Gelfman CM, Li S, Jin M, Knott EJ, Chang B, et al. Adiponectin receptor 1 conserves docosahexaenoic acid and promotes photoreceptor cell survival. Nat Commun. 2015;6:6228.
Sluch VM, Banks A, Li H, Crowley MA, Davis V, Xiang C, Yang J, Demirs JT, Vrouvlianis J, Leehy B, et al. ADIPOR1 is essential for vision and its RPE expression is lost in the Mfrp(rd6) mouse. Sci Rep. 2018;8:14339.
Xu M, Eblimit A, Wang J, Li J, Wang F, Zhao L, Wang X, Xiao N, Li Y, Wong LJ, et al. ADIPOR1 Is Mutated in Syndromic Retinitis Pigmentosa. Hum Mutat. 2016;37:246–9.
Zhang J, Wang C, Shen Y, Chen N, Wang L, Liang L, Guo T, Yin X, Ma Z, Zhang B, Yang L. A mutation in ADIPOR1 causes nonsyndromic autosomal dominant retinitis pigmentosa. Hum Genet. 2016;135:1375–87.
Zhu XG, Puthenveedu SN, Shen YH, La K, Ozlu C, Wang T, Klompstra D, Gultekin Y, Chi JY, Fidelin J, et al. CHP1 Regulates Compartmentalized Glycerolipid Synthesis by Activating GPAT4. Mol Cell. 2019;74:45–58.
Jain IH, Calvo SE, Markhard AL, Skinner OS, To TL, Ast T, Mootha VK. Genetic Screen for Cell Fitness in High or Low Oxygen Highlights Mitochondrial and Lipid Metabolism. Cell. 2020;181:716–27.
Seldin MM, Tan SY, Wong GW. Metabolic function of the CTRP family of hormones. Rev Endocr Metab Disord. 2014;15:111–23.
Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Revett T, Gimeno R, Lodish HF. Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-gamma agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem J. 2008;416:161–77.
Wong GW, Wang J, Hug C, Tsao TS, Lodish HF. A family of Acrp30/adiponectin structural and functional paralogs. Proc Natl Acad Sci U S A. 2004;101:10302–7.
Davis KE, Scherer PE. Adiponectin: no longer the lone soul in the fight against insulin resistance? Biochem J. 2008;416:e7–9.
Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:516–23.
Kosel D, Heiker JT, Juhl C, Wottawah CM, Bluher M, Morl K, Beck-Sickinger AG. Dimerization of adiponectin receptor 1 is inhibited by adiponectin. J Cell Sci. 2010;123:1320–8.
Almabouada F, Diaz-Ruiz A, Rabanal-Ruiz Y, Peinado JR, Vazquez-Martinez R, Malagon MM. Adiponectin receptors form homomers and heteromers exhibiting distinct ligand binding and intracellular signaling properties. J Biol Chem. 2013;288:3112–25.
Sharma AX, Holland WL. Adiponectin and its Hydrolase-Activated Receptors. J Nat Sci. 2017;3:e396.
Yamauchi T, Kadowaki T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013;17:185–96.
Hanada K, Hara T, Nishijima M. Purification of the serine palmitoyltransferase complex responsible for sphingoid base synthesis by using affinity peptide chromatography techniques. J Biol Chem. 2000;275:8409–15.
Mandon EC, Ehses I, Rother J, van Echten G, Sandhoff K. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J Biol Chem. 1992;267:11144–8.
Merrill AH Jr. Characterization of serine palmitoyltransferase activity in Chinese hamster ovary cells. Biochim Biophys Acta. 1983;754:284–91.
Williams RD, Wang E, Merrill AH Jr. Enzymology of long-chain base synthesis by liver: characterization of serine palmitoyltransferase in rat liver microsomes. Arch Biochem Biophys. 1984;228:282–91.
Hedger G, Sansom MSP. Lipid interaction sites on channels, transporters and receptors: Recent insights from molecular dynamics simulations. Biochim Biophys Acta. 2016;1858:2390–400.
Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, Karin M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–84.
Solinas G, Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol Metab. 2017;6:174–84.
Morigny P, Houssier M, Mairal A, Ghilain A, Mouisel E, Benhamed F, Masri B. Interaction between hormone-sensitive lipase and ChREBP in fat cells controls insulin sensitivity. Nature Metabolism. 2019;1:133–46.
Baylie RL, Brayden JE. TRPV channels and vascular function. Acta Physiol (Oxf). 2011;203:99–116.
Kahn-Kirby AH, Dantzker JL, Apicella AJ, Schafer WR, Browse J, Bargmann CI, Watts JL. Specific polyunsaturated fatty acids drive TRPV-dependent sensory signaling in vivo. Cell. 2004;119:889–900.
Vasquez V, Krieg M, Lockhead D, Goodman MB. Phospholipids that contain polyunsaturated fatty acids enhance neuronal cell mechanics and touch sensation. Cell Rep. 2014;6:70–80.
Caires R, Sierra-Valdez FJ, Millet JRM, Herwig JD, Roan E, Vasquez V, Cordero-Morales JF. Omega-3 Fatty Acids Modulate TRPV4 Function through Plasma Membrane Remodeling. Cell Rep. 2017;21:246–58.
Wong SW, Kwon MJ, Choi AM, Kim HP, Nakahira K, Hwang DH. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009;284:27384–92.
Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife. 2015;4:e08009.
Chachisvilis M, Zhang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci U S A. 2006;103:15463–8.
Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, Vale RD. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science. 2016;352:595–9.
Kautzmann MI, Gordon WC, Jun B, Do KV, Matherne BJ, Fang Z, Bazan NG. Membrane-type frizzled-related protein regulates lipidome and transcription for photoreceptor function. FASEB J. 2020;34:912–29.
Bjursell M, Ahnmark A, Bohlooly YM, William-Olsson L, Rhedin M, Peng XR, Ploj K, Gerdin AK, Arnerup G, Elmgren A, et al. Opposing effects of adiponectin receptors 1 and 2 on energy metabolism. Diabetes. 2007;56:583–93.
Stoffel W, Holz B, Jenke B, Binczek E, Gunter RH, Kiss C, Karakesisoglou I, Thevis M, Weber AA, Arnhold S, Addicks K. Delta6-desaturase (FADS2) deficiency unveils the role of omega3- and omega6-polyunsaturated fatty acids. EMBO J. 2008;27:2281–92.
Roqueta-Rivera M, Stroud CK, Haschek WM, Akare SJ, Segre M, Brush RS, Agbaga MP, Anderson RE, Hess RA, Nakamura MT. Docosahexaenoic acid supplementation fully restores fertility and spermatogenesis in male delta-6 desaturase-null mice. J Lipid Res. 2010;51:360–7.
Dobrzyn P, Jazurek M, Dobrzyn A. Stearoyl-CoA desaturase and insulin signaling–what is the molecular switch? Biochim Biophys Acta. 2010;1797:1189–94.
Elmendorf JS. Fluidity of insulin action. Mol Biotechnol. 2004;27:127–38.
Hulbert AJ, Turner N, Storlien LH, Else PL. Dietary fats and membrane function: implications for metabolism and disease. Biol Rev Camb Philos Soc. 2005;80:155–69.
Murphy MG. Dietary fatty acids and membrane protein function. J Nutr Biochem. 1990;1:68–79.
Storlien LH, Hulbert AJ, Else PL. Polyunsaturated fatty acids, membrane function and metabolic diseases such as diabetes and obesity. Curr Opin Clin Nutr Metab Care. 1998;1:559–63.
Weijers RN. Lipid composition of cell membranes and its relevance in type 2 diabetes mellitus. Curr Diabetes Rev. 2012;8:390–400.
Gianfrancesco MA, Paquot N, Piette J, Legrand-Poels S. Lipid bilayer stress in obesity-linked inflammatory and metabolic disorders. Biochem Pharmacol. 2018;153:168–83.
Pilon M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis. 2016;15:167.
Berger J, Gartner J. X-linked adrenoleukodystrophy: clinical, biochemical and pathogenetic aspects. Biochim Biophys Acta. 2006;1763:1721–32.
Berger J, Forss-Petter S, Eichler FS. Pathophysiology of X-linked adrenoleukodystrophy. Biochimie. 2014;98:135–42.
Pavicevic A, Lakocevic M, Popovic M, Popovic-Bijelic A, Dakovic M, Mojovic M. Changes of the peripheral blood mononuclear cells membrane fluidity from type 1 Gaucher disease patients: an electron paramagnetic resonance study. Biol Chem. 2018;399:447–52.
Keddad K, Therond P, Motta C, Baussan C, Legrand A. Alterations in erythrocyte membrane fluidity and fatty acid composition in glycogen storage disease. Biochim Biophys Acta. 1996;1315:61–5.
Tsuda K. Electron paramagnetic resonance investigation on modulatory effect of benidipine on membrane fluidity of erythrocytes in essential hypertension. Heart Vessels. 2008;23:134–9.
Tsuda K, Nishio I. Membrane fluidity and hypertension. Am J Hypertens. 2003;16:259–61.
Tsuda K, Kinoshita Y, Nishio I, Masuyama Y. Hyperinsulinemia is a determinant of membrane fluidity of erythrocytes in essential hypertension. Am J Hypertens. 2001;14:419–23.
Tsuda K, Kinoshita Y, Nishio I, Masuyama Y. Role of insulin in the regulation of membrane fluidity of erythrocytes in essential hypertension: an electron paramagnetic resonance investigation. Am J Hypertens. 2000;13:376–82.
Miyajima T, Tsujino T, Saito K, Yokoyama M. Effects of eicosapentaenoic acid on blood pressure, cell membrane fatty acids, and intracellular sodium concentration in essential hypertension. Hypertens Res. 2001;24:537–42.
Tsuda K, Yoshikawa A, Kimura K, Nishio I. Effects of mild aerobic physical exercise on membrane fluidity of erythrocytes in essential hypertension. Clin Exp Pharmacol Physiol. 2003;30:382–6.
Ambrus JL, Ambrus CM, Dembinsky W, Sykes D, Kulaylat MN, Patel R, Akhter S, Islam A. Thromboembolic disease susceptibility related to red cell membrane fluidity in patients with polycythemia vera and effect of phlebotomies. J Med. 1999;30:299–304.
Mounier C, Bouraoui L, Rassart E. Lipogenesis in cancer progression (review). Int J Oncol. 2014;45:485–92.
Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech. 2013;6:1353–63.
Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, Dehairs J, Escalona-Noguero C, Schmieder R, Cornfield T, et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature. 2019;566:403–6.
Lin L, Ding Y, Wang Y, Wang Z, Yin X, Yan G, Zhang L, Yang P, Shen H. Functional lipidomics: Palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology. 2017;66:432–48.
Sok M, Sentjurc M, Schara M, Stare J, Rott T. Cell membrane fluidity and prognosis of lung cancer. Ann Thorac Surg. 2002;73:1567–71.
Peck B, Schug ZT, Zhang Q, Dankworth B, Jones DT, Smethurst E, Patel R, Mason S, Jiang M, Saunders R, et al. Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab. 2016;4:6.
Peck B, Schulze A. Lipid desaturation - the next step in targeting lipogenesis in cancer? FEBS J. 2016;283:2767–78.
Kopecka J, Trouillas P, Gasparovic AC, Gazzano E, Assaraf YG, Riganti C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist Updat. 2019;49:100670.
Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniels VW, Machiels J, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70:8117–26.
Escriba PV. Membrane-lipid therapy: A historical perspective of membrane-targeted therapies - From lipid bilayer structure to the pathophysiological regulation of cells. Biochim Biophys Acta Biomembr. 2017;1859:1493–506.
Koike T, Ishida G, Taniguchi M, Higaki K, Ayaki Y, Saito M, Sakakihara Y, Iwamori M, Ohno K. Decreased membrane fluidity and unsaturated fatty acids in Niemann-Pick disease type C fibroblasts. Biochim Biophys Acta. 1998;1406:327–35.
Fecchio C, Palazzi L, de Laureto PP. alpha-Synuclein and Polyunsaturated Fatty Acids: Molecular Basis of the Interaction and Implication in Neurodegeneration. Molecules. 2018;23:1531.
O’Leary EI, Jiang Z, Strub MP, Lee JC. Effects of phosphatidylcholine membrane fluidity on the conformation and aggregation of N-terminally acetylated alpha-synuclein. J Biol Chem. 2018;293:11195–205.
Fanning S, Haque A, Imberdis T, Baru V, Barrasa MI, Nuber S, Termine D, Ramalingam N, Ho GPH, Noble T, et al. Lipidomic Analysis of alpha-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol Cell. 2018;73:1001–14.
Vincent BM, Tardiff DF, Piotrowski JS, Aron R, Lucas MC, Chung CY, Bacherman H, Chen Y, Pires M, Subramaniam R, et al. Inhibiting Stearoyl-CoA Desaturase Ameliorates alpha-Synuclein Cytotoxicity. Cell Rep. 2018;25:2742–54.
Tang K, Hynan LS, Baskin F, Rosenberg RN. Platelet amyloid precursor protein processing: a bio-marker for Alzheimer’s disease. J Neurol Sci. 2006;240:53–8.
Zainaghi IA, Forlenza OV, Gattaz WF. Abnormal APP processing in platelets of patients with Alzheimer’s disease: correlations with membrane fluidity and cognitive decline. Psychopharmacology. 2007;192:547–53.
Wood WG, Igbavboa U. Cholesterol trafficking and amyloid beta peptides. Pharmacopsychiatry. 2003;36(Suppl 2):144–8.
Muller WE, Koch S, Eckert A, Hartmann H, Scheuer K. beta-Amyloid peptide decreases membrane fluidity. Brain Res. 1995;674:133–6.
Eckert GP, Cairns NJ, Maras A, Gattaz WF, Muller WE. Cholesterol modulates the membrane-disordering effects of beta-amyloid peptides in the hippocampus: specific changes in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2000;11:181–6.
Eckmann J, Clemens LE, Eckert SH, Hagl S, Yu-Taeger L, Bordet T, Pruss RM, Muller WE, Leuner K, Nguyen HP, Eckert GP. Mitochondrial membrane fluidity is consistently increased in different models of Huntington disease: restorative effects of olesoxime. Mol Neurobiol. 2014;50:107–18.
Sameni S, Malacrida L, Tan Z, Digman MA. Alteration in Fluidity of Cell Plasma Membrane in Huntington Disease Revealed by Spectral Phasor Analysis. Sci Rep. 2018;8:734.
Schultz ML, Tecedor L, Lysenko E, Ramachandran S, Stein CS, Davidson BL. Modulating membrane fluidity corrects Batten disease phenotypes in vitro and in vivo. Neurobiol Dis. 2018;115:182–93.
Cohen EP. Membrane fluidity and polycystic kidney disease: a hypothesis. Nephron Physiol. 2008;108:8–10.
Schumann J. It is all about fluidity: Fatty acids and macrophage phagocytosis. Eur J Pharmacol. 2016;785:18–23.
Heinrichsdorff J, Olefsky JM. Fetuin-A: the missing link in lipid-induced inflammation. Nat Med. 2012;18:1182–3.
Fabelo N, Martin V, Santpere G, Marin R, Torrent L, Ferrer I, Diaz M. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol Med. 2011;17:1107–18.
Giusto NM, Salvador GA, Castagnet PI, Pasquare SJ, Ilincheta de Boschero MG. Age-associated changes in central nervous system glycerolipid composition and metabolism. Neurochem Res. 2002;27:1513–23.
Levi M, Wilson P, Nguyen S, Iorio E, Sapora O, Parasassi T. In K562 and HL60 cells membrane ageing during cell growth is associated with changes in cholesterol concentration. Mech Ageing Dev. 1997;97:109–19.
Pamplona R. Membrane phospholipids, lipoxidative damage and molecular integrity: a causal role in aging and longevity. Biochim Biophys Acta. 2008;1777:1249–62.
Tamburini I, Quartacci MF, Izzo R, Bergamini E. Effects of dietary restriction on age-related changes in the phospholipid fatty acid composition of various rat tissues. Aging Clin Exp Res. 2004;16:425–31.
Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, Yamaguchi M, Tanabe H, Kimura-Someya T, Shirouzu M, et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 2013;503:493–9.
Iwabu M, Okada-Iwabu M, Tanabe H, Ohuchi N, Miyata K, Kobori T, Odawara S, Kadowaki Y, Yokoyama S, Yamauchi T, Kadowaki T. AdipoR agonist increases insulin sensitivity and exercise endurance in AdipoR-humanized mice. Commun Biol. 2021;4:45.
Acknowledgements
I extend my fondest gratitude to the members of the Pilon group who for many years have made possible our advances in the understanding of the ADIPOR pathway either in worms or in mammalian cells. In particular, I thank: Catarina Mörck, Emma Svensk, Manish Rauthan, Parmida Ranji, Ranjan Devkota, Rakesh Bodhicharla, Mario Ruiz, Kiran Busayavalasa, and Henrik Palmgren. Thanks also to our lipidomics collaborators, Jan Borén and Marcus Henricsson, as well as my wife, Xiao-Rong Peng, who got us started on this adventure.
Funding
I thank the funding agencies that have made our research possible, and in particular the Swedish Research Council, Cancerfonden, SSF, Diabetesfonden and Carl Tryggers Stiftelsen. Open Access funding provided by University of Gothenburg.
Author information
Authors and Affiliations
Contributions
MP wrote the article and prepared the figure. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declared that he has no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pilon, M. Paradigm shift: the primary function of the “Adiponectin Receptors” is to regulate cell membrane composition. Lipids Health Dis 20, 43 (2021). https://doi.org/10.1186/s12944-021-01468-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-021-01468-y