
Abstract
Background
High-density Lipoprotein (HDL) attenuates endothelial cell apoptosis induced by different cell-death stimuli such as oxidation or growth factor deprivation. HDL is the main plasma carrier of the bioactive lipid sphingosine 1-phosphate (S1P), which it is a signaling molecule that promotes cell survival in response to several apoptotic stimuli. In HDL, S1P is bound to Apolipoprotein M (ApoM), a Lipocalin that is only present in around 5% of the HDL particles. The goal of this study is to characterize ApoM-bound S1P role in endothelial apoptosis protection and the signaling pathways involved.
Methods
Human umbilical vein endothelial cells (HUVEC) cultures were switched to serum/grow factor deprivation medium to induce apoptosis and the effect caused by the addition of ApoM and S1P analyzed.
Results
The addition of HDL+ApoM or recombinant ApoM-bound S1P promoted cell viability and blocked apoptosis, whereas HDL-ApoM had no protective effect. Remarkably, S1P exerted a more potent anti-apoptotic effect when carried by ApoM as compared to albumin, or when added as free molecule. Mechanistically, cooperation between S1P1 and S1P3 was required for the HDL/ApoM/S1P-mediated anti-apoptotic ability. Furthermore, AKT and ERK phosphorylation was also necessary to achieve the anti-apoptotic effect of the HDL/ApoM/S1P complex.
Conclusions
Altogether, our results indicate that ApoM and S1P are key elements of the anti-apoptotic activity of HDL and promote optimal endothelial function.
Similar content being viewed by others


Highlights
-
ApoM-bound S1P and ApoM-containing HDL are anti-apoptotic.
-
HDL/ApoM/S1P complex signals through S1P1 and S1P3.
-
ApoM-bound S1P anti-apoptotic effect is more potent than albumin-bound S1P.
Background
Apolipoprotein M (ApoM) is a member of the Lipocalin family and its structure is defined by an eight-stranded antiparallel β-barrel enclosing a hydrophobic binding pocket, where different ligands bind, e.g. retinol [1], oxidized phospholipids [2] and sphingosine 1-phosphate (S1P) [3]. Out of these, S1P is the only ApoM-ligand known to bind in vivo. An unusual property of ApoM is that its signal peptide is not cleaved off during secretion and used by the mature ApoM protein to anchor the protein to the phospholipid bilayer of high-density lipoproteins (HDL) [4, 5]. The plasma concentration of ApoM is approximately 0.9 μM and around 5% of all HDL particles in circulation carry ApoM and S1P [6, 7]. ApoM is the major carrier of S1P in circulation (~65%), the remaining S1P in plasma being bound to albumin (~35%) [7].
Sphingolipids have multiple key physiological functions that are important for the regulation of cell growth and survival. Ceramide and sphingosine are inducers of growth arrest and apoptosis and many stress stimuli increase the cellular levels of these compounds. In contrast, S1P is associated with suppression of apoptosis [8, 9].Five different membrane-bound, G-protein coupled S1P receptors (S1PR, S1P1-5) are known and binding of S1P to these receptors activates multiple receptor-specific downstream signaling pathways. In this way, S1P is able to regulate several biologic processes, such as immune cell trafficking, angiogenesis, cell migration and cell survival [10]. Indeed, S1PR represent important drug therapeutic targets. For instance, FTY720, also known as Fingolimod, is phosphorylated by endogenous kinases and works as a functional antagonist of S1P1 that has been approved for the treatment of multiple sclerosis [11].
The integrity of endothelial cells lining the vessels is crucial for vascular homeostasis and endothelial cell-death triggers vascular leakage and promotes inflammation in adjacent tissues [12]. Additionally, apoptotic endothelial cells become pro-coagulant and may provoke formation of blood clots [13]. Thus, increased endothelial cell apoptosis is associated with several cardiovascular pathologies, in particular with thrombosis and atherosclerosis [14].
HDL particles are potently anti-atherogenic and reduce endothelial cell apoptosis [15, 16]. Cholesterol efflux is one of the mechanisms underlying HDL protection of endothelium, and importantly, ApoM-containing HDL enhances cholesterol efflux [17, 18]. Likewise, it is known that free S1P attenuates apoptosis in endothelial cells [15, 19]. The goal of the present study was to further characterize the role of S1P in the regulation of human endothelial cell apoptosis and to define the signaling pathways involved. For that purpose, we took into account that HDL-associated S1P is bound to ApoM in plasma. We have used human ApoM-containing HDL (HDL+ApoM) and ApoM-lacking HDL (HDL-ApoM) to study regulation of apoptosis in human endothelial cells. Moreover, we have elucidated whether the anti-apoptotic properties of S1P are carrier dependent by comparing the anti-apoptotic effects of albumin-bound S1P, ApoM-bound S1P and S1P as a free molecule.
Methods
Cell culture and apoptosis induction
Human Umbilical Vein Endothelial Cells (HUVEC) were obtained from Gibco, grown in 1% gelatin pre-coated plates in M200 medium containing 1% penicillin and streptomycin and low serum growth supplement (LSGS) (all from Gibco) at 37 °C in a humidified 5% CO2 incubator. The culture medium was replaced every 2 days, and cells were subcultured at 90–95% confluence. Cells were studied between passages 2–8.
LSGS contains fetal bovine serum (FBS), human epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), heparin and hydrocortisone. Removal of all these components was used to induce apoptosis in HUVEC. This treatment will be referred as serum/GF deprivation. For that, cells were washed twice with M200 medium without LSGS. The absence of S1P in M200 medium without LSGS was verified by mass spectrometry as it was previously described in [7, 20].
Purifications (ApoM and HDL)
Recombinant soluble human ApoM (residues 22–188, without the signal peptide, Swiss-Prot entry O95445) was expressed in E. coli, purified from inclusion bodies and refolded as described in Ahnström et al. [1]. ApoM binding to S1P was confirmed by intrinsic fluorescence quenching and isoelectric focusing as described in Sevvana et al. [3]. ApoM loading with S1P was performed as in Ruiz et al.[21].
HDL was isolated from human plasma obtained from the Blood Bank at Växjö Hospital, Sweden, as described in Ruiz et al. [21]. Briefly, HDL were separated by ultracentrifugation followed by size exclusion chromatography. HDL+ApoM and HDL-ApoM were isolated by immunoaffinity chromatography with M23 and M58 monoclonal antibodies against ApoM.
S1P levels in HDL preparations were quantified by mass spectrometry as it was previously described [7, 20]. S1P was ~0.146 μM/mg protein in total HDL, ~0.417 μM/mg of protein in HDL+ApoM and ~0.008 μM/mg protein in HDL-ApoM.
Protein quantification, protein electrophoresis and western blot
Sample protein concentration was quantified using BCA protein assay kit (Pierce) according manufacturer’s instructions.
Electrophoresis was done in 4–15% gradient pre-casted SDS-gels (Bio-Rad) under reducing conditions. Western blotting was done after separation in a Trans-Blot Turbo transfer system (Bio-Rad). An Antibody against phospho-ERK1 (T202/Y204) / phospho-ERK2 (T185/Y187) ERK1/2 was from R&D systems; antibodies against total ERK (#9102), pSer473 AKT (D9E), total AKT (C67E7) were from Cell Signaling and an antibody against GAPDH was from Santa Cruz Biotechnology (#20357).
Annexin V staining and flow cytometry
Cells were detached with TrypLE Express (Gibco), washed and resuspended in Annexin V binding buffer (BD Bioscience). Then, cells were stained with PE Annexin V and 7-ADD according manufacturer’s instructions (BD Bioscience) and analyzed in a Cytomics FC500 (Beckman Coulter) flow cytometer. Data were analyzed with FlowJo X v.10.0 7r2. Early apoptotic cells were defined by Annexin V+ and 7-ADD−.
Measurement of caspase-3 activity
Caspase-3 activity was measured using a colorimetric assay kit according to manufacturer’s instructions (Abcam). Briefly, cell lysates (50 μg total protein) were incubated in the presence of N-acetyl-Asp-Glu-Val-Asp-p-nitroanilide (Ac-DEVD-pNA, 200 μM) and the release of pNA was measured using a plate reader (TECAN Infinite F200) at 405 nm.
Cell viability assay
Cell viability was evaluated by the MTT assay following manufacturer’s instructions (Roche). Briefly, viable cells are defined by their ability to reduce MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to formazan, which is a measure of an active metabolism. The conversion was quantified using a plate reader (TECAN Infinite F200) at 570 nm and optical density value was utilized as an indicator of cell viability.
Quantitative real-time PCR (qPCR)
Total cellular RNA was isolated using RNeasy Kit according to the manufacturer’s instructions (Qiagen) and quantified using a NanoDrop spectrophotometer (ND2000, Thermo Scientific). qPCR were performed with a CFX384 C1000 thermal cycler (Bio-Rad) using the Super Scrip III Platinum One Step qRT-PCR kit (Invitrogen) and TaqMan probes (Applied Biosystems): 4326317E (GAPDH), Hs00173499_m1 (S1P1), AJ39RQ5 (S1P2), Hs00245464_s1 (S1P3), Hs02330084_s1 (S1P4) and Hs00928195_s1 (S1P5) according manufacturer’s instructions. Samples were measured as quadruplicates. The relative expression of each gene was calculated according to the ΔΔCT method [22]. Expression of the housekeeping gene GAPDH was used to normalize for variations in RNA input.
Other reagents
Sphingosine-1-Phosphate (d18:1; Lipid Maps LMSP01050001) was purchased from Avanti Polar Lipids and Sigma; bovine fatty acid free albumin was from Sigma; W146, CAY10444 and ML-031 were from Cayman Chemical; SEW2871 and CYM5541 were from Tocris Bioscience; LY294002, U0126 and PD98059 were from R&D systems.
Statistical analysis
Statistical analyses were performed with SigmaPlot 11.0 software (Systat Software Inc.). A value of p < 0.05 was defined as threshold for significant changes. Student t-test and Mann-Whitney U-test were used for two-sample comparisons and ANOVA was used when assaying for multiple comparisons. The particular tests used for post hoc analyses depended on homoscedasticity, and are stated in the figure legends.
Results
HDL+ApoM protects endothelial cells against apoptosis and promotes cell survival
Endothelial cells undergo apoptosis when deprived of serum and growth factors (Fig. 1a) [15, 16, 23]. However, HDL addition to the cell medium mitigates serum/GF deprivation induced cell death [15, 16]. To assess the role of ApoM and S1P in HDL mediated protection we isolated HDL+ApoM and HDL-ApoM. Then, HUVEC were serum/GF deprived in the presence of HDL+ApoM or HDL-ApoM for 18 h and the amount of apoptotic cells measured by flow cytometry. HDL+ApoM reduced the percentage of apoptotic cells, whereas HDL-ApoM did not confer any protection against serum/GF deprivation (Fig. 1b and c). Consistently, total HDL also protected HUVEC against serum/GF deprivation (Fig. 1d). To confirm the anti-apoptotic effect of HDL+ApoM, we measured Caspase-3 activity in HUVEC after 24 h of serum/GF deprivation. Caspase-3 activity in cultures treated with HDL+ApoM upon serum/GF deprivation was significantly lower than in cultures treated with HDL-ApoM or without HDL (Fig. 1e). Next, we investigated whether the anti-apoptotic effect of HDL+ApoM could also be achieved after a short serum/GF deprivation time. Therefore, we quantified Caspase-3 activity 2 h after the removal of serum and growth factors and found a reduction of Caspase-3 activity in lysates from HDL+ApoM treated cells, whereas HDL-ApoM treatment did not confer protection against serum/GF deprivation induced cell-death (Fig. 1f).

HDL containing ApoM protects endothelial cells against serum/GF deprivation-induced cell death. a HUVEC were grown to confluence in full medium and then switched to serum starvation medium or serum/GF deprivation medium. The graph represents the percentage of apoptotic cells (Annexin V+ and 7-ADD−) identified by flow cytometry. Error bars correspond to SEM of n = 5. One-way ANOVA p < 0.001 followed by Holm-Sidak method multiple-comparison post hoc test. b–d Cells were serum/GF deprived and treated with ± HDL+ApoM 50 μg/ml or HDL-ApoM 50 μg/ml in b and c and ± Total HDL 500 μg/ml or HDL-ApoM 500 μg/ml in d for 18 h and then analyzed by flow cytometry. Error bars correspond to SEM. c shows dot plots from a representative experiment of B. In b, n = 4, one-way ANOVA p = 0.001 followed by Holm-Sidak method multiple-comparison post hoc test. In d, n = 10, ANOVA on ranks p = 0.016 followed by SNK method multiple-comparison post hoc test. e-f Measurements of Caspase-3 activity in HUVEC lysates. Cells were incubated in serum/GF starvation medium with ± HDL+ApoM 25 μg/ml, HDL-ApoM 25 μg/ml for 24 h in e and for 2 h in f. Data were normalized versus serum/GF starvation condition and error bars correspond to SEM. In E, n = 4, ANOVA on ranks p = 0.006 followed by SNK method multiple-comparison post hoc test. In F, n = 3, one-way ANOVA p = 0.012 followed by Holm-Sidak method multiple-comparison post hoc test. *p < 0.05
Since the HDL+ApoM treatment of HUVECs is anti-apoptotic, it is expected to have higher cell viability in those cultures. We verified this hypothesis by using the MTT assay. Serum/GF deprivation reduced HUVEC viability, but this reduction was significantly mitigated by HDL+ApoM. In contrast, HDL-ApoM did not improve cell viability either after 24 h or after 48 h of serum/GF deprivation (Fig. 2a). Next, we investigated which concentration of HDL+ApoM was required to promote cell viability upon serum/GF deprivation. Interestingly, HDL+ApoM at 50 μg/ml and 25 μg/ml significantly increased cell viability when compared to HDL-ApoM and non-HDL treatments, whereas HDL+ApoM at 10 μg/ml only significantly increased cell-viability when compared to non-HDL treatment (Fig. 2b).

HDL containing ApoM promotes endothelial cell viability upon serum/GF deprivation. a MTT assay of HUVEC after 24 h (left) or 48 h (right) of incubation in serum/GF deprivation medium with or without HDL+ApoM 50 μg/ml or HDL-ApoM 50 μg/ml. Data are expressed as mean ± SD. N = 4, one-way ANOVA p < 0.001 followed by Holm-Sidak method multiple-comparison post hoc test. b Cells were assayed as in a, but HDL+ApoM or HDL-ApoM concentrations were 50, 25 or 10 μg/ml. Data are expressed as mean ± SD. N = 4, one-way ANOVA p < 0.001 followed by Holm-Sidak method multiple-comparison post hoc test. * over the bars indicates statistical significance versus serum/GF deprivation treatment. *p < 0.05
Thus, we conclude that the HDL anti-apoptotic effect in serum/GF deprived endothelial cells is primary mediated by HDL containing ApoM and S1P.
S1P1 and S1P3 activation mediate the protective effect of ApoM-associated HDL
S1P signals through five different G-couple protein receptors known as S1P1-5. Thus, to understand the anti-apoptotic role of HDL+ApoM in the endothelium, we studied the expression of S1PR in HUVEC by qPCR. We found that HUVEC express S1P1 and S1P3, but do not express S1P2, S1P4 or S1P5 (Fig. 3a). Since S1P2 expression in HUVEC has been reported previously [24], we simultaneously run a qPCR using HEK293 cDNA as a positive control of S1P2 expression to assure the correct performance of S1P2 probe (data not shown). Then, we examined which S1P receptor/s are responsible for HDL+ApoM anti-apoptotic function. For that purpose, we followed a pharmacological approach and used receptor-specific agonists to mimic S1P stimulation. SEW2871, an S1P1 specific agonist, and CYM5541, an S1P3 specific agonist, reduced the amount of apoptotic endothelial cells upon serum/GF deprivation (Fig. 3b and c respectively). We also tested the S1P2 specific agonist ML-031. Nevertheless, ML-031 did not confer any protection against apoptosis (Fig. 3d). Next, we investigated if simultaneous pharmacological activation of S1P1 and S1P3 could confer a greater protection against serum/GF deprivation. However, the percentage of apoptotic cells treated with both, SEW2871 and CYM5541, is comparable to the cells only treated with SEW2871 or CYM5541 (Fig. 3e).

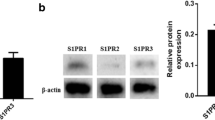
Pharmacological activation of S1P1 or S1P3 protects endothelial cells against serum/GF deprivation-induced cell death. a Relative expression of S1PR in HUVEC. Total RNA was analyzed by qPCR using Taqman probes for S1PR and normalized against GAPDH expression. S1P1 expression was chosen as reference. Error bars correspond to SD, n.d., not detected. b–e HUVEC were grown up to confluence in full medium, then switched to serum/GF deprivation medium with SEW2971 5 μM b and e, ML-031 5 μM c and e or/and CYM5541 5 μM d and e for 18 h and then analyzed by flow cytometry. The percentage of apoptotic cells (AnnexinV+ and 7-ADD−) was normalized versus serum/GF starvation condition. Error bars correspond to SEM. In b, n = 9, Mann-Whitney U-test p <0.001. In c, n = 7 Mann-Whitney U-test p = 0.026. In D, n = 4, Mann-Whitney U-test p = 0.343. In e, n = 5, one-way ANOVA p = 0.004 followed by Holm-Sidak multiple-comparison post hoc test. In e, * over the bars indicates statistical significance versus serum/GF deprivation treatment. *p < 0.05
To confirm the participation of S1P1 and S1P3 in HDL containing ApoM protection against serum/GF deprivation, we used S1P1 and S1P3 specific antagonists. Blockage of S1P1 with W146 abolished the anti-apoptotic effect of total HDL or HDL+ApoM in serum/GF deprived HUVEC (Fig. 4a and b, respectively). Additionally, W146 also abrogated the increment of viability caused by HDL+ApoM in serum/GF deprived HUVEC (Fig. 4c). Likewise, blockage of S1P3 with CAY10444 abolished the anti-apoptotic effect of total HDL in serum/GF deprived HUVEC (Fig. 4d).

S1P1 and S1P3 activation mediates the protective effect of ApoM-associated HDL in human endothelium. a-d Cells were pre-incubated with ± W146 1 μM or CAY10444 5 μM in serum/GF starvation medium for 30 min. a HUVEC were incubated in serum/GF starvation medium with total HDL 500 μg/ml and W146 1 μM for 18 h and AnnexinV+ and 7-ADD− cells were quantified by flow cytometry. As control, the experiment was replicated without W146. Error bars indicated SEM of n = 4, Mann-Whitney U-test p = 0.029 when W146 was absent and Mann-Whitney U-test p = 0.343 in W146 presence. b Cells were treated with HDL+ApoM 25 μg/ml plus W146 1 μM for for 2 h and then lysated and Caspase-3 activity measured. Data are represented as the mean ± SEM of n = 2–6. Student t-test p = 0.010 when W146 was not added; Student t-test p = 0.388 in W146 presence. c Cell viability was assayed by MTT. Cells were switched to serum/GF starvation medium with ± HDL+ApoM 25 μg/ml, HDL-ApoM 25 μg/ml and ± W146 1 μM for 24 h. Error bars represent SD of n = 3. Control condition (no W146) data were analyzed by one-way ANOVA p = 0.004 followed by Holm-Sidak method multiple-comparison post hoc test. W146 condition data were analyzed by ANOVA on ranks p = 0.059. d Cells were assayed as in A, but using CAY10444 5 μM instead of W146. Error bars indicated SEM of n = 4-5, Mann-Whitney U-test, p = 0.029 when CAY10444 was not present: Mann-Whitney U-test p = 0.690 in CAY10444 presence. *p < 0.05
In conclusion, HDL required S1P1 and S1P3 signaling to achieve their anti-apoptotic effect in serum/GF deprived HUVEC. However, pharmacological activation of S1P1 or S1P3 was sufficient to mimic HDL protection.
ApoM-bound S1P confers longer protection to endothelial cells against serum/GF deprivation
Plasma S1P is mostly carried by ApoM in HDL, but a fraction is bound to albumin [7]. Therefore, we elucidated if albumin-bound S1P could also protect endothelial cells against serum/GF deprivation induced cell-death. In order to have a direct comparison between ApoM-bound S1P and albumin-bound S1P, we produced soluble recombinant human ApoM in E. coli and loaded it with S1P. Previous work has studied S1P in apoptosis by directly adding S1P as a free molecule to the cell medium (for instance [9, 25–27]). Therefore, we also included free S1P in our study. First, as a visual approximation, we performed a DNA fragmentation assay. Endothelial cells were serum/GF deprived for 24 h in the presence of free S1P, ApoM, ApoM-bound S1P or albumin-S1P. Interestingly, ApoM-bound S1P treated cells showed a lower level of DNA fragmentation than free S1P or albumin-bound S1P treated cells (Fig. 5a). To confirm this result we carried out Caspase-3 activity assays. Importantly, free S1P, ApoM-bound S1P and albumin-bound S1P decreased Caspase-3 activity after 24 h of serum/GF deprivation (Fig. 5b). However, ApoM-bound S1P and albumin-bound S1P did it more efficiently than free S1P. Remarkably, when we looked at more prolonged protection, 48 h of serum/GF deprivation, only ApoM-bound S1P reduced Caspase-3 activity in HUVEC upon serum/GF deprivation (Fig. 5c). In consonance with this finding, free S1P, ApoM-bound S1P and albumin-bound S1P treatments improved cell viability upon serum/GF deprivation, but ApoM-bound S1P was significantly more effective than free S1P and albumin-bound S1P (Fig. 5d).

ApoM-bound S1P confers longer protection to endothelial cells against serum/GF deprivation. a–e Cells were treated with free S1P 1 μM, ApoM 1 μM, ApoM-bound S1P 1 μM or albumin-bound S1P 1 μM. a Cells were treated for 24 h and then a cell fragmentation assay performed. b Cells were treated for 2 h and then the Caspase 3 activity measured. ANOVA on ranks p < 0.001 followed by SNK multiple-comparison post hoc test. c As b, but 24 h treatment. ANOVA on ranks p = 0.007 followed by SNK multiple-comparison post hoc test. d Cell viability was determined by MTT after 24 h of treatment. Error bars correspond to SD. N = 4-5. One-way ANOVA p < 0.001 followed by Holm-Sidak method multiple-comparison post hoc test. e-f Cells were pre-incubated with ± W146 1 μM or CAY10444 10 μM in serum/GF deprivation medium for 30 min. and then treated as in c, but with the addition of W146 1 μM in e and CAY10444 10 μM in F. In E, n = 3, data were analyzed by one-way ANOVA p = 0.003 followed by Holm-Sidak method multiple-comparison post hoc test and in F, n = 3, by One-way ANOVA p < 0.001 followed by Holm-Sidak method multiple-comparison post hoc test. * over the bars indicates statistical significance versus serum/GF starvation treatment. *p < 0.05
Since anti-apoptotic and pro-survival effects of S1P were carrier dependent, we investigated if differences can be due to particular activation of S1PR. To study this, we performed Caspase-3 assays as in Fig. 5b, but in the presence of the S1P1 antagonist W146 or the S1P3 antagonist CAY10444. Interestingly, all three alternative ways to supply S1P to endothelial cells required S1P1 and S1P3 signaling to become anti-apoptotic (Fig. 5e and f).
Thus, we concluded that the anti-apoptotic effect of S1P in serum/GF deprived endothelial cells was carrier dependent, ApoM-bound S1P being the most powerful of all three carriers. Furthermore, anti-apoptotic activity of S1P was mediated by S1P1 and S1P3 with independence of which S1P carrier was used.
PI3K/AKT and ERK1/2 signaling pathways are implicated in the anti-apoptotic effect of S1P in serum/GF deprived cells
It has been shown that the anti-apoptotic activities of S1P and HDL are mediated by PI3K/AKT and ERK1/2 signaling pathways [15, 16, 28]. Moreover, a previous study demonstrated that phosphorylation of AKT and ERK is induced by HDL+ApoM and albumin-S1P, but not by HDL-ApoM [7]. To link these antecedents, we used the PI3K/AKT inhibitor LY249002 and the MEK inhibitors U0126 and PD98059. We added these inhibitors to serum/GF deprived cells in the presence of free S1P, ApoM, ApoM-bound S1P or albumin-bound S1P. Then, we lysed the cells and analyzed them by western-blot or Caspase-3 activity., The phosphorylation of AKT and ERK by free S1P, ApoM-bound S1P or albumin-S1P was abolished when cells were treated with LY249002 or U0126 (Fig. 6a). LY294002, U0126 and PD98059 also canceled the inhibitory effect of free S1P, ApoM-bound S1P or albumin-S1P on Caspase-3 activation in serum/GF deprived HUVEC (Fig. 6b).

AKT or ERK inhibition blocks S1P anti-apoptotic effect. a-b Cells were pre-incubated with ± W146 1 μM, CAY10444 10 μM, LY294002 50 μM, U0126 50 μM or PD98050 50 μM for 30 min and then treated with free S1P 1 μM, ApoM 1 μM, ApoM-bound S1P 1 μM or albumin-bound S1P 1 μM for 10 min in a or 2 h in b. In a, western blots to analyze the phosphorylation of AKT and ERK induced by the treatments mentioned above. Total AKT, ERK and GAPDH were analyzed as controls. In B, Caspase-3 activity in cell lysates after the different treatments indicated above. Data are expressed as mean ± SEM of n = 3–6. No inhibitor set: ANOVA on ranks p < 0.001 followed by SNK method multiple-comparison post hoc test. LY294002 set: one-way ANOVA p = 0.010 followed by Holm-Sidak method multiple-comparison post hoc test. U0126 set: ANOVA on ranks p = 0.018 followed by Dunn’s method multiple-comparison post hoc test. PD98050 set: one-way ANOVA p < 0.001 followed by Holm-Sidak method multiple-comparison post hoc test. *p < 0.05
Next, we determined whether S1PR activation mediated the phosphorylation of AKT and ERK by S1P. The selective S1P1 antagonist W146 dramatically reduced the phosphorylation of AKT and ERK by ApoM-bound S1P and albumin-bound S1P. However, when S1P was added as a free molecule, W146 decreased ERK phosphorylation but surprisingly not AKT phosphorylation. In contrast, CAY10444 reduced the phosphorylation of AKT and ERK mediated by free S1P, ApoM-bound S1P and albumin-S1P (Fig. 7a and b).

S1P1 or S1P3 antagonists block AKT and ERK phosphorylation. a Cells were pre-incubated with ± W146 1 μM or CAY10444 10 μM for 30 min and then treated with free S1P 1 μM, ApoM 1 μM, ApoM-bound S1P 1 μM or albumin-bound S1P 1 μM for 10 min. Then, cells were lysed and analyzed by western blot for pAKT and pERK. Total AKT, ERK and GAPDH were analyzed as controls. b Quantification of relevant pairs of A. Student t-test of n = 3 independent experiments. Error bars correspond to SD. *p < 0.05
In conclusion, S1P anti-apoptotic effect on serum/GF deprived endothelial cells went via S1P1 and S1P3 and required the phosphorylation of AKT and ERK1/2 (Additional file 1: Fig. S1).
Discussion
Previous studies have pointed out the protective role of HDL on endothelial cells upon different cell-death stimuli, including oxidized LDL [29, 30] and serum/GF deprivation [15, 16]. Likewise, anti-apoptotic properties of free S1P have been demonstrated [9, 25–27, 30, 31]. Here we connect previous findings and show that ApoM-containing HDL, and therefore S1P, have anti-apoptotic and pro-survival properties in serum/GF deprived endothelial cells (Figs. 1 and 2). Importantly, S1P also promotes survival in cardiomyocytes [32], macrophages [31] and other cell types [33–36]. Now, it would be relevant to study S1P protection in other human cell types taking in account ApoM. It is important to highlight that HDL particles are highly heterogenic in protein and lipid composition and additional cytoprotective mechanisms are possible [37]. Which ones are relevant may depend on the cell-death stimulus, time and concentration used.
de Souza et al. [29] isolated HDL subpopulations and found that small and dense HDL3, which are enriched in S1P [and ApoM, [38]], have cytoprotective activity superior to that of large and light HDL2. Interestingly, reconstituted HDL (rHDL) with added S1P did not enhance the anti-apoptotic effect achieved by rHDL without S1P [29]. Similarly, S1P-fortified HDL subfractions did not to significantly improve the anti-apoptotic effect of non-S1P-fortified HDL. Both scenarios could be explained by the fact that the exogenous S1P was not bound to ApoM and therefore may not properly interact with S1PR. This explanation concurs with Fig. 5a-d, where ApoM-S1P displayed significantly elevated anti-apoptotic activity as compared to free S1P or albumin-S1P. In agreement, apoptosis was not inhibited when albumin-S1P was used at 1–100 nM [29]. Likewise, rHDL anti-apoptotic ability is enhanced when plasmalogens are incorporated to rHDL [39], but the molecular mechanism behind has not been described yet. Several endothelial cell types express ApoM [40] and the S1P transporter Spns2 [41]. Possibly, rHDL including plasmalogens are better acceptors for ApoM and S1P than plasmalogen-free rHDL.
Riwanto et al. [42] demonstrated that ApoJ enhances HDL anti-apoptotic effect on endothelial cells. However, ApoJ is absent in our HDL+ApoM preparations [17] and, therefore, ApoM-S1P anti-apoptotic effect cannot be ascribed to ApoJ. In contrast, HDL anti-apoptotic activity is impaired in HDL enriched in ApoC-III [42], which it is less abundant in HDL+ApoM than in HDL-ApoM [17]. Thus, the poor anti-apoptotic capacity of ApoC-III containing HDL can be explained by the low content in ApoM-S1P.
Endothelial-cell survival is enhanced by free S1P via S1P1 and S1P3 [19]. We corroborated this finding and demonstrated that parallel activation of both S1P1 and S1P3 by HDL+ApoM is required to achieve S1P anti-apoptotic and pro-survival effects (Figs. 4 and 5). Furthermore, we show that S1P1 and S1P3 activation requirement is independent of the S1P carrier (Fig. 5e-f). However, activation by ApoM-S1P renders a longer protection than albumin-S1P. These apparently conflicting data can be explained by S1P carrier specific degradation of S1P1 [43, 44]. Following activation of S1P1 by albumin-S1P, S1P1 is internalized and degraded by the proteasome, whereas S1P1 is internalized and recycled to the plasma membrane after ApoM-S1P activation. Unfortunately, no data on S1P-carrier dependent biology of S1P3 are available. However, an analogous situation to S1P1 may be plausible for S1P3.
Beyond S1PR, other plasma membrane receptors connect apoptosis, HDL and its major component, ApoA1. First, HDL3 acts via Scavenger Receptor Class B Type I (SR-BI) to inhibit apoptosis on endothelial cells [45]. Indeed, Li et al. [46] over-expressed SR-BI in CHO cells and elaborated an attractive model in which SR-BI is a pro-apoptotic receptor in absence of HDL. This model needs to be validated in endothelial cells, but the fact that HDL+ApoM are more efficient than HDL-ApoM in stimulating cholesterol efflux suggests that HDL+ApoM may have higher affinity for SR-BI than in HDL-ApoM. Additionally, stimulation of F1-ATPase by lipid-free ApoA1 inhibits endothelial cell apoptosis [45], but interactions between HDL and F1-ATPase have not been reported.
AKT and ERK1/2 phosphorylation mediate HDL and S1P cytoprotective actions [15, 16, 27, 28]. Moreover, HDL+ApoM and albumin-S1P, but not HDL-ApoM, phosphorylate AKT and ERK [7]. Here, we confirmed S1P-dependent phosphorylation of ERK and AKT and demonstrated that blockage of AKT and ERK signaling abolishes S1P anti-apoptotic effects (Fig. 6). Importantly, activation of S1P1 and S1P3 by ApoM-S1P or albumin-S1P phosphorylate AKT and ERK (Fig. 7). Interestingly, S1P induces AKT activation and protects against ischemia/reperfusion in mouse cardiomyocytes via S1P2 and S1P3 [47, 48]. This suggests that the pattern of S1PR activated by S1P to achieved cytoprotection may be tissue/organism dependent.
Retinol binding protein (RBP) is another member of the Lipocalin family and transports retinol in plasma. Interestingly, apo-RBP is pro-apoptotic, whereas holo-RBP is anti-apoptotic [49]. We did not identify any pro-apoptotic activity of apo-ApoM per se in endothelial cells. However, ApoM over-expression promotes apoptosis in the human hepatoma derived cell line HepG2 [50]. Moreover, two other plasma Lipocalins: Lipocalin-type prostaglandin D2 synthase (L-PGDS) and Apolipoprotein D (ApoD) mitigate cell-death and promote viability [51, 52]. Interestingly, ApoM and ApoD are HDL enriched in HDL3 [38]. Thus, abundance of Lipocalins in HDL3 could explain the high cytoprotective ability of HDL3.
Conclusions
Taken together, our results demonstrate that the HDL/ApoM/S1P-complex plays an essential role in vascular biology and protects endothelial cells from apoptosis. This is especially relevant in pathologies where endothelial cell apoptosis is altered such as in thrombosis and atherosclerosis.
Abbreviations
- ApoM:
-
Apolipoprotein M
- HDL:
-
High-density lipoproteins
- HDL+ApoM :
-
ApoM-containing HDL
- HDL-ApoM :
-
ApoM-lacking HDL
- HUVEC:
-
Human umbilical vein endothelial cells
- S1P:
-
Sphingosine 1-phosphate
- S1PR:
-
S1P receptor
References
Ahnstrom J, Faber K, Axler O, Dahlback B. Hydrophobic ligand binding properties of the human lipocalin apolipoprotein M. J Lipid Res. 2007;48(8):1754–62.
Elsoe S, Ahnstrom J, Christoffersen C, Hoofnagle AN, Plomgaard P, Heinecke JW, Binder CJ, Bjorkbacka H, Dahlback B, Nielsen LB. Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis. 2012;221(1):91–7.
Sevvana M, Ahnstrom J, Egerer-Sieber C, Lange HA, Dahlback B, Muller YA. Serendipitous fatty acid binding reveals the structural determinants for ligand recognition in apolipoprotein M. J Mol Biol. 2009;393(4):920–36.
Christoffersen C, Ahnstrom J, Axler O, Christensen EI, Dahlback B, Nielsen LB. The signal peptide anchors apolipoprotein M in plasma lipoproteins and prevents rapid clearance of apolipoprotein M from plasma. J Biol Chem. 2008;283(27):18765–72.
Liu M, Allegood J, Zhu X, Seo J, Gebre AK, Boudyguina E, Cheng D, Chuang CC, Shelness GS, Spiegel S, et al. Uncleaved ApoM signal peptide is required for formation of large ApoM/sphingosine 1-phosphate (S1P)-enriched HDL particles. J Biol Chem. 2015;290(12):7861–70.
Axler O, Ahnstrom J, Dahlback B. An ELISA for apolipoprotein M reveals a strong correlation to total cholesterol in human plasma. J Lipid Res. 2007;48(8):1772–80.
Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A. 2011;108(23):9613–8.
Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta. 2006;1758(12):2016–26.
Zhang Y, Pan Y, Bian Z, Chen P, Zhu S, Gu H, Guo L, Hu C. Ceramide Production Mediates Aldosterone-Induced Human Umbilical Vein Endothelial Cell (HUVEC) Damages. Plos One. 2016;11(1):e0146944.
Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res. 2014;55(8):1596–608.
Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9(11):883–97.
Winn RK, Harlan JM. The role of endothelial cell apoptosis in inflammatory and immune diseases. J Thromb Haemost. 2005;3(8):1815–24.
Bombeli T, Karsan A, Tait JF, Harlan JM. Apoptotic vascular endothelial cells become procoagulant. Blood. 1997;89(7):2429–42.
Affara M, Dunmore B, Savoie C, Imoto S, Tamada Y, Araki H, Charnock-Jones DS, Miyano S, Print C. Understanding endothelial cell apoptosis: what can the transcriptome, glycome and proteome reveal? Philos Trans R Soc Lond B Biol Sci. 2007;362(1484):1469–87.
Kimura T, Sato K, Kuwabara A, Tomura H, Ishiwara M, Kobayashi I, Ui M, Okajima F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J Biol Chem. 2001;276(34):31780–5.
Nofer JR, Levkau B, Wolinska I, Junker R, Fobker M, von Eckardstein A, Seedorf U, Assmann G. Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J Biol Chem. 2001;276(37):34480–5.
Christoffersen C, Nielsen LB, Axler O, Andersson A, Johnsen AH, Dahlback B. Isolation and characterization of human apolipoprotein M-containing lipoproteins. J Lipid Res. 2006;47(8):1833–43.
Plomgaard P, Dullaart RP, de Vries R, Groen AK, Dahlback B, Nielsen LB. Apolipoprotein M predicts pre-beta-HDL formation: studies in type 2 diabetic and nondiabetic subjects. J Intern Med. 2009;266(3):258–67.
Kimura T, Sato K, Malchinkhuu E, Tomura H, Tamama K, Kuwabara A, Murakami M, Okajima F. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler Thromb Vasc Biol. 2003;23(7):1283–8.
Frej C, Andersson A, Larsson B, Guo LJ, Norstrom E, Happonen KE, Dahlback B. Quantification of sphingosine 1-phosphate by validated LC-MS/MS method revealing strong correlation with apolipoprotein M in plasma but not in serum due to platelet activation during blood coagulation. Anal Bioanal Chem. 2015;407(28):8533–42.
Ruiz M, Frej C, Holmer A, Guo LJ, Tran S, Dahlback B: High-density lipoprotein-associated apolipoprotein M Limits endothelial inflammation by delivering sphingosine-1-phosphate to the Sphingosine-1-phosphate receptor 1. Arterioscler Thromb Vasc Biol. 2017;37(1):118–129. doi:10.1161/ATVBAHA.116.308435.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (–Delta Delta C (T)) Method. Methods. 2001;25(4):402–8.
Levkau B, Scatena M, Giachelli CM, Ross R, Raines EW. Apoptosis overrides survival signals through a caspase-mediated dominant-negative NF-kappa B loop. Nat Cell Biol. 1999;1(4):227–33.
Fernandez-Pisonero I, Duenas AI, Barreiro O, Montero O, Sanchez-Madrid F, Garcia-Rodriguez C. Lipopolysaccharide and sphingosine-1-phosphate cooperate to induce inflammatory molecules and leukocyte adhesion in endothelial cells. J Immunol. 2012;189(11):5402–10.
Lee YJ, Jeong JK, Lee JH, Park YG, Moon JH, Seol JW, Jackson CJ, Park SY. Sphingosine-1-phosphate protects against bisphosphonateinduced HUVEC cell death via regulation of c-JunNterminal kinase signaling. Int J Mol Med. 2013;31(4):811–6.
Moriue T, Igarashi J, Yoneda K, Nakai K, Kosaka H, Kubota Y. Sphingosine 1-phosphate attenuates H2O2-induced apoptosis in endothelial cells. Biochem Biophys Res Commun. 2008;368(4):852–7.
Nowatari T, Murata S, Nakayama K, Sano N, Maruyama T, Nozaki R, Ikeda N, Fukunaga K, Ohkohchi N. Sphingosine 1-phosphate has anti-apoptotic effect on liver sinusoidal endothelial cells and proliferative effect on hepatocytes in a paracrine manner in human. Hepatol Res. 2015;45(11):1136–45.
Kimura T, Watanabe T, Sato K, Kon J, Tomura H, Tamama K, Kuwabara A, Kanda T, Kobayashi I, Ohta H, et al. Sphingosine 1-phosphate stimulates proliferation and migration of human endothelial cells possibly through the lipid receptors, Edg-1 and Edg-3. Biochem J. 2000;348(Pt 1):71–6.
de Souza JA, Vindis C, Negre-Salvayre A, Rye KA, Couturier M, Therond P, Chantepie S, Salvayre R, Chapman MJ, Kontush A. Small, dense HDL 3 particles attenuate apoptosis in endothelial cells: pivotal role of apolipoprotein A-I. J Cell Mol Med. 2010;14(3):608–20.
Kontush A, Therond P, Zerrad A, Couturier M, Negre-Salvayre A, de Souza JA, Chantepie S, Chapman MJ. Preferential sphingosine-1-phosphate enrichment and sphingomyelin depletion are key features of small dense HDL3 particles: relevance to antiapoptotic and antioxidative activities. Arterioscler Thromb Vasc Biol. 2007;27(8):1843–9.
Feuerborn R, Becker S, Poti F, Nagel P, Brodde M, Schmidt H, Christoffersen C, Ceglarek U, Burkhardt R, Nofer JR. High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis. 2016;257:29–37.
Theilmeier G, Schmidt C, Herrmann J, Keul P, Schafers M, Herrgott I, Mersmann J, Larmann J, Hermann S, Stypmann J, et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation. 2006;114(13):1403–9.
Castillo SS, Teegarden D. Sphingosine-1-phosphate inhibition of apoptosis requires mitogen-activated protein kinase phosphatase-1 in mouse fibroblast C3H10T 1/2 cells. J Nutr. 2003;133(11):3343–9.
Guo L, Zheng Z, Ai J, Howatt DA, Mittelstadt PR, Thacker S, Daugherty A, Ashwell JD, Remaley AT, Li XA. Scavenger receptor BI and high-density lipoprotein regulate thymocyte apoptosis in sepsis. Arterioscler Thromb Vasc Biol. 2014;34(5):966–75.
Rutherford C, Childs S, Ohotski J, Mcglynn L, Riddick M, Macfarlane S, Tasker D, Pyne S, Pyne NJ, Edwards J, et al. Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis. 2013;4:e927.
Schmitz EI, Potteck H, Schuppel M, Manggau M, Wahydin E, Kleuser B. Sphingosine 1-phosphate protects primary human keratinocytes from apoptosis via nitric oxide formation through the receptor subtype S1P (3). Mol Cell Biochem. 2012;371(1–2):165–76.
Tran-Dinh A, Diallo D, Delbosc S, Varela-Perez LM, Dang QB, Lapergue B, Burillo E, Michel JB, Levoye A, Martin-Ventura JL, et al. HDL and endothelial protection. Br J Pharmacol. 2013;169(3):493–511.
Davidson WS, Silva RA, Chantepie S, Lagor WR, Chapman MJ, Kontush A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol. 2009;29(6):870–6.
Sutter I, Velagapudi S, Othman A, Riwanto M, Manz J, Rohrer L, Rentsch K, Hornemann T, Landmesser U, von Eckardstein A. Plasmalogens of high-density lipoproteins (HDL) are associated with coronary artery disease and anti-apoptotic activity of HDL. Atherosclerosis. 2015;241(2):539–46.
Ruiz M, Frej C, Holmer A, Guo LJ, Tran S, Dahlback B. High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arterioscler Thromb Vasc Biol. 2016;37(1):118–29.
Fukuhara S, Simmons S, Kawamura S, Inoue A, Orba Y, Tokudome T, Sunden Y, Arai Y, Moriwaki K, Ishida J, et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest. 2012;122(4):1416–26.
Riwanto M, Landmesser U. High density lipoproteins and endothelial functions: mechanistic insights and alterations in cardiovascular disease. J Lipid Res. 2013;54(12):3227–43.
Galvani S, Sanson M, Blaho VA, Swendeman SL, Obinata H, Conger H, Dahlback B, Kono M, Proia RL, Smith JD, et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal. 2015;8(389):ra79.
Wilkerson BA, Grass GD, Wing SB, Argraves WS, Argraves KM. Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: high density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J Biol Chem. 2012;287(53):44645–53.
Radojkovic C, Genoux A, Pons V, Combes G, de Jonge H, Champagne E, Rolland C, Perret B, Collet X, Terce F, et al. Stimulation of cell surface F1-ATPase activity by apolipoprotein A-I inhibits endothelial cell apoptosis and promotes proliferation. Arterioscler Thromb Vasc Biol. 2009;29(7):1125–30.
Li XA, Guo L, Dressman JL, Asmis R, Smart EJ. A novel ligand-independent apoptotic pathway induced by scavenger receptor class B, type I and suppressed by endothelial nitric-oxide synthase and high density lipoprotein. J Biol Chem. 2005;280(19):19087–96.
Means CK, Xiao CY, Li Z, Zhang T, Omens JH, Ishii I, Chun J, Brown JH. Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;292(6):H2944–51.
Morel S, Christoffersen C, Axelsen LN, Montecucco F, Rochemont V, Frias MA, Mach F, James RW, Naus CC, Chanson M, et al. Sphingosine-1-phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovasc Res. 2016;109(3):385–96.
Chen YR, Feng F, Wang L, Qu SY, Zhang ZQ, Liu L, Qin HY, Liang YM, Han H. Deletion of RBP-J in dendritic cells compromises TLR-mediated DC activation accompanied by abnormal cytoskeleton reorganization. Mol Biol Rep. 2012;40(2):1531–9.
Hu YW, Chen ZP, Hu XM, Zhao JY, Huang JL, Ma X, Li SF, Qiu YR, Wu XJ, Sha YH, et al. The miR-573/apoM/Bcl2A1-dependent signal transduction pathway is essential for hepatocyte apoptosis and hepatocarcinogenesis. Apoptosis. 2015;20(10):1321–37.
Bajo-Graneras R, Ganfornina MD, Martin-Tejedor E, Sanchez D. Apolipoprotein D mediates autocrine protection of astrocytes and controls their reactivity level, contributing to the functional maintenance of paraquat-challenged dopaminergic systems. Glia. 2011;59(10):1551–66.
Fujimori K, Fukuhara A, Inui T, Allhorn M. Prevention of paraquat-induced apoptosis in human neuronal SH-SY5Y cells by lipocalin-type prostaglandin D synthase. J Neurochem. 2012;120(2):279–91.
Acknowledgements
We thank Li J. Guo for technical assistance throughout the project.
Funding
This work was supported by the Swedish Research Council (#07143), the Swedish Heart and Lung Foundation, Söderberg’s Foundation, and Österlund’s Foundation. Funding agencies did not participate in the experimental design of the study, collection, analysis, interpretation of data, decision to publish or manuscript preparation.
Availability of data and materials
Data available on request from the authors.
Authors’ contributions
BD conceived the original project. MR, HO and BD designed the experiments. MR and HO performed the experiments. MR, HO and BD wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not-applicable.
Ethics approval and consent to participate
Not-applicable.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
Schematic representation summarizing the anti-apoptotic effect of ApoM-bound S1P in endothelial cells. (PPTX 631 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ruiz, M., Okada, H. & Dahlbäck, B. HDL-associated ApoM is anti-apoptotic by delivering sphingosine 1-phosphate to S1P1 & S1P3 receptors on vascular endothelium. Lipids Health Dis 16, 36 (2017). https://doi.org/10.1186/s12944-017-0429-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-017-0429-2