
Abstract
Undeniably, cancer immunotherapies have expanded the spectrum of cancer treatment, however, some patients do not respond to immunotherapies. This scenario is no different for lung cancer, whose two main types, non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), still pose a serious clinical challenge. Adoptive T-cell therapies (ATC), which primarily include cytokine-induced killer (CIK) cell therapy, chimeric antigen receptor T-cell (CAR T-cell) therapy and γδ-T-cell therapy, strengthen the patient’s immune system in combating cancer. Combining ATC with immune checkpoint inhibitors (ICI) further enhances the effectiveness of this approach to eradicate cancer. With a particular emphasis on CIK cell therapy, which recently completed 30 years, we highlight the role of the PD-1/PD-L1 axis in NSCLC and SCLC. Besides, we provide insights into the potential synergies of PD-1/PD-L1 inhibitors with adoptive T-cell immunotherapy in reshaping the treatment paradigm for lung cancer.
Similar content being viewed by others

Introduction
Lung cancer remains the second most commonly diagnosed cancer worldwide [1], being categorized into two main types: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). Approximately 85% of patients exhibit a group of histological subtypes referred as NSCLC, of which lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) represent the most common subtypes [2]. In the United States, 5-year survival between 2008 and 2014 was 24% for all patients with NSCLC and 5.5% for those with distant metastases [3]. Likewise, the median overall survival of German advanced NSCLC patients was equally as low as that of other countries as shown in a German retrospective data analysis [4]. Unlike NSCLC, the SCLC accounts for approximately 13%-15% of all lung cancers with a 5-year survival rate of less than 7%, a rapid doubling time and a high propensity to metastasize. SCLC is considered a “recalcitrant” cancer as no significant improvements in survival and therapeutic approaches have been achieved for more than 30 years [5, 6].
Since cancer cells have several mechanisms to evade immune surveillance, including the PD-1/PD-L1 axis, this axis has also been focused on for LC. Thus, the programmed cell death protein 1 (PD-1 [also known as CD279]) and its ligand, programmed death ligand 1 (PD-L1 [also known as CD274]), have been utilized in several standard first-line LC treatments. PD-L1, also known as Cluster of Differentiation (CD274) or B7 homolog 1 (B7-H1), belongs to the growing B7 family of immune molecules and is involved in the regulation of cellular and humoral immune responses. B7-H1 belongs to the cell surface immunoglobulin superfamily with two Ig-like domains in the extracellular region and a short cytoplasmic domain within the extracellular region and a short cytoplasmic domain. Subsequent to nivolumab [7, 8], there have been additional PD-1 (pembrolizumab and cemiplimab) and PD-L1 (atezolizumab and durvalumab) inhibitors approved by the FDA. Unlike NSCLC cells, the efficacy of a combination of nivolumab and ipilimumab was enhanced in patients with high tumor mutation burden (TMB) in the nonrandomized or randomized cohorts of CheckMate 032 [9], possibly due to the nearly universal association of SCLC with smoking [10, 11]. However, in addition to improving survival, PD-1/PD-L1 inhibitors were associated with a wide range of unfavourable effects, for instance, immune-related adverse events (irAEs), including rash, colitis, hepatitis, endocrinopathies, and pneumonitis [12]. Thus, it is imperative to explore new therapeutic strategies to alleviate irAEs and improve the efficacy of immune effector cells.
Undeniably, the availability of the PD-1/PD-L1 axis also opens up the avenue for its combination with various cancer immunotherapies. One among them is cytokine-induced killer (CIK) cell therapy, pioneered by Ingo Schmidt-Wolf, et al. in 1991 [13]. CIK cells were described to have dual cytotoxic functions of innate immunity via NK-specific activating receptors and adaptive immunity via polyclonal TCR repertoire [14], and more than 80 clinical trials involving CIK cells ranging from solid tumors to hematologic malignancies (clinicaltrial.gov). Concerning CIK treatment for lung cancer, 12 clinical trials have been conducted. Of these, 10 studies reported that CIK treatment improved median progression-free survival, and 7 studies improved overall survival [15]. Like CIK cells, another alternatively novel immunotherapy chimeric antigen receptor T-cell (CAR-T) also showed great promise for NSCLC. However, unlike hematologic malignancies, the clinical application of CAR-T cells has remained limited success due to on-target/off-tumor as well as neurological toxicity [16]. Meanwhile, γδ T cell immune therapy also presented promising outcomes in a recent clinical trial on lung cancer patients [17]. Nevertheless, it is reasonable to optimize CIK therapy, for instance, a combination of PD-1/PD-L1 inhibitor based on T cell immune therapy. Particularly, the response of cancer cells to immunotherapy will be determined by both intrinsic properties of the cancer cells and specific interactions with the microenvironment [18]. In this review, we will discuss the PD-1/PD-L1 axis in the lung cancer microenvironment, current T cell adoptive immunotherapy combined with PD-1/PD-L1 blockade combination, as well as future directions.
Landscape of PD-1/PD-L1 axis in NSCLC and SCLC
The role of PD-1/PD-L1 in the tumor immune microenvironment of NSCLC
Cancer stem cell (CSC)
Human lungs are composed of two distinct areas: the conducting airway, including the trachea, bronchi, and bronchioles, and the gas exchange regions, alveolar spaces. The division of these stem cells is thought to be sufficient to renew the lung’s structure of the lung during normal adult life. In the trachea and main bronchi, the tracheal epithelium consists mainly of columnar and mucus secreting goblet cells. In addition, airway basal cells are considered as a stem cell population, which can maintain the balance between their proliferation and differentiation. The imbalance can contribute to squamous cell metaplasia or dysplasia, which are precursors of squamous cell lung carcinoma [19, 20]. Meanwhile, in bronchioles and alveoli, non-ciliated club cells (Clara cells) are located in the bronchiolar and alveolar epithelium and could differentiate into ciliated cells after exposure to oxidant induced damage [21, 22]. Club cells are shown to survive KRAS mutations and to form lung tumors after tobacco carcinogen exposure [23]. The high frequency of club cell-like cells in papillary adenocarcinoma could be a useful histological marker for ALK+ lung cancers [24]. In addition, Club and Alveolar type 2 (AT2) cells give rise to EML4-ALK lung adenocarcinoma in mice model [25]. Pulmonary neuroendocrine (PNE) cells are thought to serve as a precursor for the progress of small cell lung carcinoma. Clara cells are not necessary for PNE cell hyperplasia since proliferation and hyperplasia of PNE cells occurred in the conditional Clara cell ablation [25]. It is also believed that adenocarcinomas can originate from broncho alveolar stem cells or pneumocytes of type I and type II [26]. (Fig. 1).

The putative origin of lung cancer stem cells in lung anatomical sites. In the trachea and main bronchi, airway basal cells are considered as a stem cell population, precursors of squamous cell lung carcinoma. Pulmonary neuroendocrine (PNE) cells are thought to serve as a precursor for the progress of small cell lung carcinoma. It is also believed that adenocarcinomas can originate from broncho alveolar stem cells or pneumocytes of type I and type II
CSCs are thought to be responsible for cancer initiation, progression, metastasis, recurrence, and drug resistance. The presence of CSCs with PD-L1 expression in the metastatic lymph nodes (LNs) in lung cancer patients might correlate with immunotherapy [27]. Additional data showed that PD-L1+ lung cancer stem cells modified the metastatic lymph-node immune microenvironment in NSCLC patients, positively correlated with the percentage of Tregs, PD-1+ CD4+ T cells and Tim3+ CD4+ T cells, while negatively correlated with that of CD4+ T cells and CD28+ CD4+ T cells [28]. Indeed, this suppressive immunophenotype correlated with tumor PD-L1 can be evaluated by endobronchial ultrasound-guided transbronchial needle aspiration before immune therapy [29] (Fig. 2A).

Schematic representation of the negative regulation of anti-tumor immune responses of PD-1/PD-L1 in NSCLC and SCLC. (A) PD-L1 expression in lung cancer cell lines was significantly upregulated by co-culture with M2-differentiated tumor-associated macrophages (TAMs). PD-L1 overexpression on macrophages might be induced via STAT3 activation by cancer cell-derived GM-CSF. The interaction of CD47 on tumor cells and signal‐regulated protein (SIRPα) expressed on the surface of macrophages can protect cells from being “eaten” by macrophages in NSCLC patients. TGF-β secreted from cancer-associated fibroblasts (CAF cells) also reduced the proliferation and activation of CD8+ T cells. In addition, PD-L1+ lung cancer stem (CSC) cells as well as FoxP3+ Treg T cells may modify the metastatic lymph-node immune microenvironment in NSCLC patients. Besides, soluble PD-L1 (sPD-L1) might interrupt PD-1 and Anti-PD-1 mAb whereas interaction of sPD-1/sPD-L1 or sPD-1/PD-L1 may reduce inhibition of sPD-L1 or enhance Anti-PD-1 mAb. The interaction of PD-1/PD-L1 between TCRVγ9Vδ2+γδ tumor-infiltrating lymphocytes (TILs) and αβ T cells could restrain the activation of T cells. Intratumoral Vδ1 T cells demonstrated natural killer and CD8+ T cell function. (B) The role of PD-L1 in SCLC. In the SCLC-Y subtype, YAP1 not only affects PD-L1, but also upregulates CXCL5 to recruit myeloid-derived suppressor cells (MDSCs). YAP1 and Notch1 are complementary and each suppresses neuroendocrine (NE) differentiation
Tumor-associated macrophages (TAMs)
Tumor-associated macrophages (TAMs) play an important role on the tumorigenesis of lung cancer. TAMs conduct pro-angiogenic effects and polarization of TAMs can affect the proliferation, migration, invasion. TGF-β and IL-10 secreted from TAMs are important factors that form the microenvironment of immunosuppressive tumors [30]. PD-L1 on cancer cells engages with PD-1 on immune cells, contributing to cancer immune escape [31]. Additionally, TAMs are known to be instrumental in the immunosuppressive effects of the PD-1/PD-L1 pathway in the cancer microenvironment. PD-L1 was significantly higher in macrophages in both the tumor and stromal compartment compared with other immune cells in NSCLC patients, correlating with better overall survival [32]. This self-protective immune escape could aid cells to evade being eliminated by neighboring cells, which might also benefit from PD-1/PD-L1 inhibitors. PD-L1 overexpression on macrophages induced via STAT3 activation by cancer cell-derived GM-CSF was suggested to promote cancer progression in lung adenocarcinoma in vitro and vivo animal models [33]. Furthermore, data from lung adenocarcinoma patients has confirmed that high PD-L1 expression on macrophages was correlated with the presence of EGFR mutation, a lower cancer grade, and a shorter cancer-specific overall survival [33]. Watanabe H. et al. reported a case of a 72-year-old man with PD-L1-nagative lung adenocarcinoma harboring an EGFR mutation who responded to nivolumab for more than 2 years. The pathological evidence demonstrated infiltration of PD-L1+ TAM and CD8+ lymphocytes in the tumor environment, revealing that PD-L1 high expression in TAM might be an indicator of a positive response to anti-PD-1 antibodies [34]. Another study revealed that in patients with early-stage lung adenocarcinoma, expression of PD-L1 on the cell surface of tumor cells was observed, which was accompanied by an increase in TAMs, cytotoxic CD8+ T cells, and regulatory FoxP3+ T cells [35]. Additional in vitro investigations revealed that PD-L1 expression in lung cancer cell lines was significantly upregulated by co-culture with M2-differentiated macrophages, whereas it was downregulated by a transforming growth factor‐β inhibitor. It is known that the interaction of CD47 on tumor cells and signal‐regulated protein (SIRPα) expressed on the surface of macrophages can protect cells from being “eaten” by macrophages, as has been reported in NSCLC patients. And that dual targeting of CD47/PD-L1 innate and adaptive checkpoints may serve as new combined dual-targeting immunotherapy to enhances macrophage phagocytosis [36]. Taken together, tumor‐infiltrating TAM are extrinsic regulators of tumor PD‐L1 expression, indicating that combination therapy targeting both tumor PD‐L1 and stromal TAM molecular might be a possible strategy for effective treatment of lung cancer (Fig. 2A). Recently, Carfilzomib modulates tumor microenvironment via driving M2 macrophages to express M1 cytokines to potentiate PD-1 antibody immune therapy for lung cancer [37]. Liu M. et al. suggested that the transcription factor c-Maf critically regulates human M2 macrophages/monocytes infiltrating the tumor and circulating monocytes from patients with NSCLC, as inhibition of c-Maf partially overcomes resistance to anti-PD-1 therapy in a subcutaneous LLC tumor model [38]. How c-Maf promotes immunoregulation of PD-1-expressed TAMs or CD8+ T cells in NSCLC patients is still unknown, while it has been already been demonstrated in multiple sclerosis and relapsed/refractory classic Hodgkin lymphoma [39, 40].
Cancer-associated fibroblasts (CAF)
Some clinical studies on immunotherapy via immune checkpoints emphasized the role of TGF-β. In metastatic urothelial cancer (mUC), TGF-β attenuates tumor response to PD-L1 blockade (atezolizumab) by contributing to exclusion of T cells [41]. The lack of response was associated with a transforming growth factor β (TGF-β) signature in fibroblasts, particularly in patients with CD8+ T cells excluded from the tumor parenchyma and instead found in the fibroblast and collagen-rich peritumoral stroma. In an animal model, these immune exclusion properties were also recapitulated, and a TGF-β-blocking antibody together with anti-PD-L1 reduced TGF-β signaling in stromal cells, facilitated T cell entry into the center of the tumor, and elicited potent anti-tumor immunity [42]. TGF-β signaling primarily mediates extracellular matrix (ECM) remodeling in the human NSCLC cell line A549. Ln-γ2, a member of the laminin family of ECM, was transcriptionally activated by TGF-β1 secreted from cancer-associated fibroblasts via JNK/AP1 signaling, and the mediated cell exclusion attenuates the response to anti-PD-1 therapy [42] (Fig. 2A).
TCR-αβ T cells exhaustion
T cell repertoire analysis has revealed that in early-stage NSCLC patients, there is greater homology of the T-cell repertoire between the tumor and the uninvolved tumor-adjacent lung, suggesting a less tumor-focused T-cell response as well as an association with inferior survival. Furthermore, the accumulation of regulatory CD4+ T cells in the tumor center may impair the ability of CD8+ T cells to proliferate in response to antigens [43], although the increased proliferation of PD-1+ CD8+ T cells in peripheral blood after PD-1-targeting therapy in lung cancer patients may overcome it [44]. Conversely, PD-L1+ CD8+ T cells exerted regulatory functions that inhibit CD8+ T cell proliferation and cytotoxic capabilities via the PD-L1/PD-1 axis. Moreover, tumor-derived IL-27 promotes PD-L1+ CD8+ T cell development through STAT1/STAT3 signaling [45]. Therefore, it is plausible that adoptive T-cell therapy combined with PD-1/PD-L1 inhibitors might increase clinical benefits because of cytotoxic T-cell infusion (Fig. 2A).
Soluble PD-1/PD-L1
Soluble PD-1/PD-L1 forms are generated by proteolytic shedding or alternative splicing of pre-mRNA, presenting an active circulating protein, with immune-modulatory functions [46]. In the recent study, PD-L1–vInt4, a splicing variant of PD-L1, was reported to be detectable in clinical samples of lung squamous cell carcinoma (LUSC) and its secretion resisted anti–PD-L1 antibody treatment via alternative polyadenylation. It is noteworthy that several LUSC samples presented a lack of capability of antigen presentation due to the loss of HLA expression overexpression in vitro experiment of PD-L1–vInt4 in vitro experiment [47]. Besides, sPD-L1expression in plasma of small cell lung cancer is associated with disease progression [48]. In contrast, some animal outcomes have been documented the anti-tumor effect of sPD-1 [49, 50]. An increased or stable sPD-1 level independently correlated with longer PFS in two cycles of nivolumab-treated metastatic NSCLC patients, suggesting sPD-1 as a predictive biomarker of response to ICI treatment in patients with lung cancer [51, 52]. Considering the analysis of sPD-L1 with immune-assays (ELISA) method may not distinguish between vesicular and soluble forms in circulation of cancer patients in some studies, dynamic blood PD-L1 expression for immune checkpoint inhibitors in advanced NSCLC patients might be more reliable as a biomarker [53]. Nevertheless, further work is required to better understand the functions of soluble PD-1/PD-L1 variants in lung cancer (Fig. 2A).
TCR-γδ T cells tumor-infiltrating lymphocytes (TILs)
γ/δ T lymphocytes localize in different epithelial tissues and are phenotypically distinct from peripheral γ/δ T cell-population. In about one-fourth of human lung cancers, γ/δ T cells represented a significant proportion of freshly isolated tumor-infiltrating lymphocytes [54]. Ferrarini M. et al. found that half of the Vδ1+ (as well as Vδ1−Vδ2−) γ/δ-lymphocytes that could be selectively expanded from human lung cancers also co-expressed the CD8α/α homodimer [55]. Furthermore, it has been reported that TCS1+ γ/δ+ tumor-infiltrating lymphocytes from human lung carcinomas lysed only autologous tumor cells and K-562 [56]. Human normal lung tissues and NSCLCs harbor resident populations of γδ T cells, particularly enriched in the Vδ1 subtype. Intratumoral Vδ1 T cells exhibited stemness characteristics and were skewed toward cytolysis and helper T cell type 1 function, similar to intratumoral natural killer and CD8+ T cells, which are considered beneficial to the patients [57]. Based on deconvolution of human cancers microarrays, TCRVγ9Vδ2+γδ TIL abundance in lung carcinoma was also reassessed by a study in ∼10,000 cancer biopsies. However, TCRVγ9Vδ2+ γδ TIL was not positively correlated with cytolytic activity and favorable outcome like αβ TIL [58]. In contrast, Cazzetta V. et al. revealed that NKG2A expression identifies a subset of human Vδ2 T cells exerting the highest antitumor effector functions [59]. This apparent discrepancy illustrates the complexity of the relationship between TCRVγ9Vδ2+ γδ TIL and lung cancer and also depends on differences in measurement methodologies (whole-tissue transcriptomic analysis, phenotypic analysis or immunohistochemistry analysis).
The role of PD-1/PD-L1 in small-cell lung carcinoma (SCLC)
SCLC is broadly classed as limited stage-small cell lung cancer (LS-SCLC) and extensive stage- small cell lung cancer (ES-SCLC). Although SCLC harbors a high mutation rate (tumor mutational burden/TMB, a biomarker of sensitivity to immunotherapy in SCLC) [60], the pooled prevalence of PD-L1 expression stands at 26.0%, and 22.0% respectively, after excluding the potential outlier studies, which remains lower compared to NSCLC [61]. In addition, low expression of MHC-I, which leads to a decrease of cytotoxic T-lymphocytes (TILs) infiltrating the SCLC tumor, and excess of regulatory T cells (Tregs), which can inhibit activation, expansion and effector functions of other T cells [62], may contribute to low response to immune checkpoint inhibitors (ICIs) (approximately 10% with anti-PD-1 monotherapy) [63, 64].
In SCLC, small cell lung cancer stem cells display mesenchymal properties and exploit immune checkpoint pathways in activated cytotoxic T lymphocytes [65]. A subpopulation of pulmonary neuroendocrine cells are reserve stem cells regulated by the tumor suppressors Rb, p53, and Notch, and are considered tumor-initiating cells for SCLC [66].
A new classification of SCLC subtypes has been defined by differential expression of four key transcription regulators: achaete-scute homologue 1 (ASCL1; also known as ASH1), neurogenic differentiation factor 1 (NeuroD1), yes-associated protein 1 (YAP1) and POU class 2 homeobox 3 (POU2F3) [67]. YAP1 has been shown to contribute to inducing immunosuppressive TME by upregulating PD-L1 [68] or stimulating cytokines such as CXCL5 from tumor cells to recruit tumor-infiltrating macrophages, myeloid-derived suppressor cells (MDSCs) [69] and Tregs cells. In fact, it has been associated with mutations in the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR signaling pathway [70, 71]. Additionally, Notch signaling has been documented to play a critical role in the response to ICB in SCLC [72]. YAP1 and Notch1 are complementary and may be involved in cell proliferation, EMT, drug resistance, and neuroendocrine (NE) differentiation [73]. The mechanism of SCLC has not been fully understood (Fig. 2B).
The possible mechanism of PD-1/PD-L1 in NSCLC cells
Several extrinsic factors (e.g., release of interferon-γ by immune cells that upregulate PD-L1 expression) and intrinsic factors regulate PD-L1 expression in NSCLC. As one of extrinsic factors, environmental tobacco smoke (ETS), contributes to a distinct PI3K (phosphatidylinositol 3-kinase)–Akt pathway that leads to cell survival in adenocarcinoma [74]. Genomic alterations that activate KRAS, EGFR, and ALK, as well as the loss of PTEN, have been associated with increased PD-L1 expression. Several tumorigenic intrinsic factors such as activation of the mechanistic target of rapamycin (mTOR), mitogen-activated protein kinase (MAPK) and Myc pathways can increase PD-L1. Alternatively, methylation, allelic loss and gene silencing expression might be considered [75]. Also, the molecular involvement of PD-L1 single nucleotide polymorphisms and non-synonymous single nucleotide polymorphisms (nsSNPs) with oncogenic potential in NSCLC, cannot be excluded [76, 77].
PD-1, is a type I transmembrane protein encoded by the PDCD1 gene of the CD28 immunoglobulin superfamily, comprised of a single Ig variable-type (IgV) extracellular domain, a transmembrane domain and a cytoplasmic domain. N-terminal and C-terminal tyrosine residues in the cytoplasmic domain are involved in the formation of immunoreceptor tyrosine-based inhibitory motifs (ITIMs) and immunoreceptor tyrosine-based switch motifs (ITSMs) [78]. The engagement of PD-1 in T cells and PD-1 ligands leads to the recruitment of SHP-1/2 (Src homology 2-containing tyrosine phosphatase 1/2) to the C-terminal of the ITSM. SHP-2 then dephosphorylates TCR-associated CD-3ζ and ZAP70, resulting in the inhibition of downstream signaling [79]. The advent of human T cell non-Hodgkin lymphomas and relative animal models has revealed that the activity of PD-1 enhances levels of the tumor suppressor PTEN and attenuates signaling by the kinases PI3K/AKT and PKCθ/NF-κB pathways in oncogenic T cells [80]. PD-1 on CD4+ T regulates cell cycle and inhibits T cell proliferation via Akt and Ras pathways [81]. Moreover, Lck signaling has been shown to contribute to PD-1+ T cell exhaustion via CD28 co-stimulation in a humanized mouse model [82] (Fig. 3A and B).

Interaction of PD-1/PD-L1 between T cells and NSCLC cells. (A-B) The engagement of PD-1 in T cells and PD-1 ligands leads to the recruitment of SHP-1/2 (Src homology 2-containing tyrosine phosphatase 1/2) to the C-terminal of the ITSM. SHP-2 then dephosphorylates TCR-associated CD-3ζ and ZAP70, resulting in the inhibition of downstream signaling and T cell inactivation (A). In the presence of Anti-PD-1 mAb, T cells might be reactivated via PD-1/PD-L1 axis (B). (C-D) The effect of tumor cell-intrinsic PD-1 on NSCLC cells. PD-L1 expressed by NSCLC cells or other cells acts on PD-1+ tumor cells to mediate PD-1 signaling in tumor cells via ITIM and ITSM. The AKT and ERK1/2 pathways can suppress tumor growth by dampening AKT and ERK signaling (C). Anti-PD 1 mAb blocks PD-1/PD-L1-mediated tumor suppression, leading to hyperprogression in NSCLC cells (D)
In recent years, the role of intrinsic PD-1 in NSCLC cells raises consideration. Wang X. et al. proposed that PD-1 and PD-L1 reduces tumor growth by suppressing AKT and ERK1/2. In the absence of an adaptive immune system, tumor cell-intrinsic PD-1/PD-L1 mediates the resistance to anti-PD-1/PD-L1 antibodies by activating AKT and ERK1/2, which induces tumor growth [83] (Fig. 3C and D).
Current adaptive T cells clinical trials combined with PD-1/PD-L1 blockades in lung cancer
Blockade of PD-1 immunosuppression boosts CIK/DC-CIK therapy
Shortly after injection of CIK cells, a bioluminescent signal was detected in the lung followed by the liver and spleen in the animal model [84]. CIK cells are heterogenous T cells, composed of CD3+CD8+, CD3+CD4+, CD3+CD56+ subpopulations and the main dominant of CIK cells is TCR αβ T cells [14]. Mechanistically, cell signaling not only through TCR/CD3 but also through NKG2D, DNAM-1, and NKp30 leads to CIK cell activation resulting in granule exocytosis, cytokine secretion, and cytotoxicity [14, 85]. FasL is another major effector mechanism used by CIK cells to induce the apoptosis of tumor cells [86]. Alternatively, it has been reported that TCR αβ CD3+CD56+CIK cells can be retargeted to exert antibody-dependent cellular cytotoxicity (ADCC) function by antigen-specific mAbs [87]. Although CIK/DC-CIK therapy has improved anti-tumor responses in a total of 12 clinical trials targeting lung cancer in recent years [15], the dynamic phenotype profiles of checkpoint molecules on CIK cells derived from patients with NSCLC patients has revealed that CIK cells may partly be exhausted before the clinical transfusion. This has been also characterized by the elevated expression of PD-L1, LAG-3, TIM-3 and the reduced expression of PD-1 and CTLA-4. Based on these findings, blocking PD-1/PD-L1 improve the efficiency of CIK therapy for NSCLC patients [88]. Furthermore, a clinical report on the enhancement of autologous CIK cells after treatment with PD-1 blocking antibodies in patients with advanced NSCLC provided additional evidence for this combination strategy [89].
Alternatively, in a phase I clinical trial in patients with advanced solid tumors including NSCLC, pembrolizumab-activated DC-CIK cells demonstrated superior antitumor properties with increased IFN-γ secretion in ex vivo and an overall disease control rate of 64.5% [90]. A case report of a 63-year-old man with squamous cell carcinoma has provided further clinical evidence. The patient received a diagnosis of squamous cell carcinoma after biopsy of the right lower lobe lung mass and had developed multiple metastases on CT scans. After first-line and second-line chemotherapy, the disease was processed. After receiving pembrolizumab in combination with seven cycles of CIK cell transfer, the patient continued to be in remission 185 days posttreatment and had no adverse events. The tumor cells from pretreatment biopsies showed strongly expression of PD-L1 instead of PD-1, and large number of tumor-infiltrating CD3+ T cells were observed [91]. Taken together, this study suggested that a combination of CIK cells and pembrolizumab could synergistically enhance therapeutic efficacy in metastatic NSCLC patients, especially when PD-L1 expression in tumor biopsies was high. Han Y. et al. also documented that autologous CIK cells improved the clinical response to PD-1 blocking antibodies in patients with advanced NSCLC [89]. Recently, a Phase IB Trial of autologous CIK cells in combination with Sintilimab, (mAb PD-1), plus chemotherapy in patients with advanced NSCLC also showed encouraging efficacy (NCT03987867) [92]. Additionally, Mankor JM. et al. observed that the efficacy of nivolumab and ipilimumab in patients with malignant pleural mesothelioma was related to a subtype of cytotoxic T cells effector memory [93]. Therefore, it is important to stratify patients using accessible markers of T-cell status or tumor genomic detection to design therapeutic trials.
PD-1/PD-L1 blockades in CAR T-cell therapy in lung cancer
The concept of adaptive immunotherapy resurfaced with the advent of CAR T-cell therapy. The first report of a combined approach of CAR T cells with PD-1 blockade in Her-2 transgenic mice was reported in 2013 [94, 95]. It was demonstrated that anti-PD-1 antibody can potently enhance CAR T-cell therapy by the eradication of established tumors without severe toxicity. Beyond PD-1 blockade, constructed CAR T cells that target PD-L1 were found to exert cytotoxic activity against PD-L1high NSCLC cells and xenograft tumors. An additional subtherapeutic dose of local radiotherapy improved the efficacy of PD-L1-CAR T cells against PD-L1low NSCLC cells and tumors [96]. Novel CAR.αPD1-T cells were generated based on the anti-CD19 CAR, and constitutive anti-PD-1 secretion was more efficient in tumor eradication than parental anti-CD19 CAR T cells in human lung carcinoma xenograft tumors [97]. Due to dual expression of mesothelin (MSLN) in both lung cancer and normal mesothelium, the development of mesothelin-specific CAR-T cell therapy that incorporates an HLA-gated safety mechanism can selectively kill MSLN(+)A*02(-) malignant cells [98]. Furthermore, image-guided intrapleural delivery of CAR T cells using intracavitary or intratumoral routes and pembrolizumab treatment after CAR-T infusion is feasible, repeatable and safe across anatomically variable pleural cancers (NCT 02414269) [99].
Currently, there are 17 clinical trials worldwide investigating the safety and efficacy of CAR-T cell therapy in the treatment of lung cancer. They are specifically targeting epidermal growth factor receptor (EGFR); human epidermal growth factor receptor 2 (HER2); mesothelin (MSLN); prostate stem cell antigen (PSCA); mucin 1 (MUC1); carcinoembryonic antigen (CEA); tyrosine kinase-like orphan receptor 1 (ROR1); programmed death ligand 1 (PD-L1) and CD80/CD86. There are three clinical trials involving PD-1/PD-L1 (NCT03525782, NCT02862028 and NCT03198052). In NCT03525782, 8 patients with NSCLC (IIIb to IV stage) were infused with anti-MUC1 CAR-T cells combined with PD-1 knockout engineered T cells. All patients experienced significant symptoms in the first 2 weeks after infusion. Serum CYFRA 21, a tumor marker of squamous cell carcinomas, declined following infusion and subsequently increased 4 weeks after treatment. In 2/6 patients, lung tumor size shrunk significantly within 4 weeks after treatment, while the effect on metastasis was limited. No cytokine release syndrome (CRS) or adverse effects were observed in patients [100]. This study suggests that combined MUC1-CAR+/PD-1-KO therapy is feasible but effective individually. Additional studies of CAR-T in LC are ongoing (NCT02862028, NCT03198052).
To date, one phase 1 clinical trial (NCT03392064) has been conducted using AMG 119, a chimeric antigen receptor (CAR) T cell therapy targeting DLL3 for the treatment of relapsed/refractory SCLC patients [101]. The target gene in this study is delta-like ligand 3 (DLL3), an inhibitory ligand of Notch receptors that regulate neuroendocrine differentiation in SCLC. Given the serious side effects of CAR-T cells targeting healthy tissue [102, 103], DLL3 is highly restricted to SCLC with a neglectable expression in the normal adult tissues, making it an ideal target for cancer immunotherapy [104]. A new clinical trial using DLL3-directed chimeric antigen receptor T-cells in patients with extensive stage small cell lung cancer (NCT05680922) has also been listed but not yet recruiting. Another study in in vitro and xenograft mouse models demonstrated that the PD-1 inhibitory antibody dramatically improved the anti-tumor efficacy of the DLL3 bispecific antibody [105], suggesting the feasibility of DLL3 CAR-T combined with PD-1 inhibitors. In addition, regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab showed promising outcomes in a Phase I Trial [106]. Alternatively, a phase I clinical trial to evaluate the safety and tolerability of autologous mesothelin (MSLN)-targeted chimeric antigen receptor (MSLN-CAR) T cells secreting PD-1 nanobodies (αPD1-MSLN-CAR T cells) in patients with solid tumors achieved good outcomes (NCT04503980). All patients showed expansion of CAR-T cells and increased PD-1 nanobodies in circulation. The CAR T-cell therapy is a relatively safe therapeutic option safe and the total objective response rate was 63.64% [107].
Recently, a Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors establish the preliminary feasibility and safety of CRISPR-engineered CAR-T cells with PD-1 disruption (NCT03747965). Lung squamous cell carcinoma cell line NCI-H226 was used in vitro experiment. PD-1/PD-L1 pathway inhibits CAR-T cell function in vitro. knocking out the PDCD1 gene would enhance the antitumor effect of CAR-T cells. PDCD1 knockout P4 (MPK-CAR-T) cells were generated via electroporation of Cas9-sgRNA ribonucleoprotein (RNP), had substantial cytotoxic effects against the cell line PD-L1+ cell line and released substantial amounts of IFN-γ in vitro. A total of 15 patients received one or more infusions of MPTK-CAR-T cells without prior lymphodepletion. No dose-limiting toxicity or unexpected adverse events were observed in any of the 15 patients. The best overall response was stable disease (2/15 patients) [108]. Therefore, we conclude that CAR T-cells are uniquely equipped with specific molecules and a combination with PD-1/PD-L1 blockade is feasible. Continued safety monitoring in future trials is essential.
Blockade of PD-1 immunosuppression enhances γδ T cells therapy in lung cancer
δ T cells represent a unique T cell subpopulation capable of recognizing cancer cells not only through a TCR-dependent pathway, but also through natural killer receptors (NKRs) that are not restricted to the major histocompatibility complex (MHC). As aforementioned, human Vδ1 T cells exerted anti-tumoral function in the tissues of NSCLC patients [57]. Human Vδ1 T cells can be isolated from the peripheral blood mononuclear cells (PBMC) of healthy donors and proliferated in vitro induced by mitogen concanavalin A (Con A) [109]. The cytotoxic Vδ1 T cells against B-cell chronic lymphocytic leukemia cells were correlated with the expression of Vδ1 TCR, CD56, and CD95. In contrast, using the same expansion method, Vδ2 T cells can be expanded by targeting chronic myeloid leukemia-derived cells, which correlate with Vδ2 TCR and NKG2D [109]. Although peripheral blood Vδ1 T cells typically represent a minority population compared to the more dominant Vγ9Vδ2 [110], allogeneic CAR Vδ1 T cells expanded from PBMCs and genetically modified to express a 4-1BB/CD3z CAR against GPC-3 display robust antitumor efficacy against hepatocellular carcinoma [111].
Human Vγ2Vδ2 T cells constitute important circulating γδ T cells that bridge adaptive and innate immunity. γδ T cells can be effectively expanded using synthetic antigens such as pyrophosphomonoesters and nitrogen-containing bisphosphonates (N-BPs). Adoptive transfer of autologous or allogeneic Vγ9Vδ2-T cells expanded with zoledronate in patients with refractory NSCLC [17, 112, 113] resulted in good antitumor responses. High PD-1 levels on the activated γδ T cells also suggest the potential of combination therapy involving γδ T cells and PD-1 ICIs. Of particular interest, HDAC inhibitors attenuate the antitumor cytotoxic potential of γδ T cells, whereas PD-1 blockade enhances the antitumor effector functions of HDAC inhibitor-treated γδ T cells [114]. However, Vδ T cells are double-faced immunocytes in cancer treatment. The interaction between PD-1 on αβ T cells and its ligand PD-L1 on γδ T cells restrains αβ T cell activation [115]. It is reasonable to conduct γδ T cell functional identification and elimination of suppressive subgroup before transferring the γδ-T cells into the patients. In addition, CD16 has been shown to mediate ADCC in γδ T cells by PD-1 blockade in follicular lymphoma [116]. Considering the percentage of γδ CD16+CD3+CD56+ CIK cells accounts for approximately 31% of total CD16+ CIK cells [87], isolation and expansion of this subset of CIK cells may expand the scope of immune therapy in the future.
Three clinical trials using γδ-T cells against lung cancer have been reported. NCT03183232 was originally designed to evaluate the safety and efficiency of autologous PBMC-derived γδ-T cells against lung cancer. However, PBMCs from the majority of cancer patients cannot be effectively expanded in terms of cell number, purity and function. Moreover, cancer patients cannot afford to donate 100 ml of blood for culture every 2–3 weeks. Therefore, they optimized the protocol of ex vivo expansion of Vγ9Vδ2-T cells derived from allogeneic PBMCs. The clinical trial has been completed but the results are not yet available. Another clinical trial NCT02459067, designed to determine the safety, tolerability, maximum tolerated dose (MTD) and efficacy of Autologous γδ T Lymphocytes (ImmuniCell®) in patients with melanoma, renal cell cancer (RCC) or non-small cell lung cancer (NSCLC) has been terminated. Taken together, γδ T-cell therapy for lung cancer is still in its early stages, and clinical data is inadequate. An overview of three types of adaptive immunotherapies in clinical trials involving the PD-1/PD-L1 axis is shown in Table 1.
Conclusion and future perspectives
A few clinical trials of adaptive T-cell therapy with PD-1/PD-L1 blockade have been conducted in recent years. However, understanding the impact of the PD-1/PD-L1 axis on lung cancer is still in the nascent stages. More importantly, we have paid little attention to PD-L2 (a second ligand of PD-1), despite the fact that its expression has been upregulated in patients with lung adenocarcinoma [117, 118] and has the potential to serve as a clinicopathological and prognostic marker [119]. Undeniably, PD-L2 has previously been considered an insignificant ligand, although it binds to PD-1 with a 2-6-fold higher affinity [120]. It can inhibit T cell activation via recruiting SHP-2, yet it does not inhibit T cells due to cell cycle arrest in G0/G1. This characteristic is similar to that of CTLA-4. Notably, cemiplimab, a human PD-1 monoclonal antibody that binds to PD-1 and blocks its interaction with PD-L1 and PD-L2 has been approved by FDA in 2018 [121]. In fact, cemiplimab monotherapy significantly improved overall survival and progression-free survival compared to chemotherapy in patients with advanced NSCLC with PD-L1 of at least 50% [122, 123], providing another option for immune therapy.
Secondly, the moderate understanding of the tumor microenvironment is still a major hurdle to the complete success of lung cancer immunotherapy. For instance, lower airway dysbiosis led to the activation of checkpoint inhibitor [124]. Of importance, this lower airway dysbiotic signature was found to be more prevalent in the stage IIIB-IV tumor-node-metastasis lung cancer group, associated with poor prognosis and accompanied by the upregulation of the IL-17, PI3K, MAPK, and ERK pathways in airway transcriptome. It is also important to mention about microbiome, it has been suggested that normal human oral flora, Veillonella parvula, may act as a key driver force in lung cancer [125]. Moreover, use of antibiotics during ICIs treatment regimen has been shown to significantly decrease their efficiency against lung cancer [126, 127]. In addition, the relationship of lung microbiota to PD-1/PD-L1 inhibitors responses has been investigated [128]. CXCL9 levels in bronchoalveolar lavage fluid (BALF) were significantly elevated in responders compared with non-responders, along with a greater diversity of the lung microbiome profile in BALF and a greater frequency of the CD56+ subset in blood T cells before initiating nivolumab. Antibiotic treatment in a preclinical lung cancer model significantly decreased CXCL9 in the lung TME, resulting in reduced sensitivity to anti-PD-1 antibody. Unfortunately, authors could not identify the specific bacterial species involved in the therapeutic effect of ICIs in this study [128]. Further research is needed to fully understand how lung microbiota can more specifically influence the outcome of cancer immunotherapy. Interestingly, Ochi N. et al. suggested that antibiotics treatment was significantly associated with a shorter median overall survival (OS), but not progression-free survival (PFS) in NSCLC patients who received antibiotics before or after ICI treatment in a retrospective study. The detrimental effect of impact of antibiotics uses on the efficacy of ICIs differed based on PD-L1 expression in patients with advanced NSCLC [129]. However, this effect was not observed at the multivariate analysis. The influence factor on overall survival needs to be explored further.
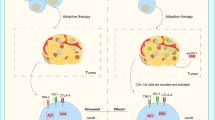
Thirdly, PD-1/PD-L1 inhibitors might promote the efficacy of CAR-T/CAR-CIK, bispecific antibody or δ T cells, however, this assumption requires experimental validation. Notably, a recent clinical trial NCT02425748 was designed to evaluate the safety and efficiency of γδ T cells in combination with DC-CIK against NSCLC (without EGFR mutation) compared to either monotherapy of DC-CIK or γδ T cells. Although the results have not been published, this study offers another promising immunotherapy approach.
Finally, a very limited number of reports are available on clinical trials of adaptive immunotherapy in SCLC. For instance, there was only one ongoing clinical trial of SCLC registered as NCT02688673. This study aims to evaluate the safety and efficacy of recombinant adenovirus-code MUC1 and Survivin-transfected dendritic cells (DC) combined with CIK cell treatment in patients with extensive-stage SCLC. Furthermore, a combination of bispecific DLL3-targeted antibody and PD-1 inhibition retained the growth of SCLC more efficiently [105]. Thus, it may also be potentially envisioned as a novel strategy for SCLC.
Similarly, recent observation suggested that PD-L1/PD-1 blockage enhanced the cytotoxicity of natural killer (NK) cells on NSCLC by granzyme B secretion. This finding was also demonstrated in NK cells isolated from NSCLC patients. Their outcomes highlight that PD-L1/PD-1 blockade enhances cellular adaptive immune therapy [130].
In this review, we emphasize the role of three T cell adoptive therapeutic approaches and discuss whether PD-1/PD-L1 blockade can effectively ‘unleash’ T cell response. In our opinion, PD-1/PD-L1 blockade could give a boost to T-cell immunotherapy if patient-centered trials can be considered.
Data availability
Not applicable.
Abbreviations
- CIK cells:
-
cytokine-induced killer cells
- CAR-T cells:
-
chimeric antigen receptor T-cells
- PD-1:
-
programmed cell death protein 1
- PD-L1:
-
programmed death ligand 1
- NSCLC:
-
non-small cell lung cancer
- SCLC:
-
small cell lung cancer
- ICI:
-
immune checkpoint inhibitors
- CSC:
-
cancer stem cell
- TAMs:
-
tumor-associated macrophages
- CAF:
-
cancer-associated fibroblasts
References
Sung H, Ferlay J, Siegel RL, et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. https://doi.org/10.3322/caac.21660.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell Lung cancer. Nature. 2018;553:446–54.
Noone AM, Howlader N, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2015. Bethesda, MD: National Cancer Institute; 2018. http://seer.cancer.gov/archive/csr/1975_2012/.
Hardtstock F, Myers D, Li T, et al. Real-world treatment and survival of patients with advanced non-small cell Lung Cancer: a German retrospective data analysis. BMC Cancer. 2020;20:260. https://doi.org/10.1186/s12885-020-06738-z.
Kahnert K, Kauffmann-Guerrero D, Huber RM. SCLC-state of the art and what does the future have in store? Clin Lung Cancer. 2016;17:325–33. https://doi.org/10.1016/j.cllc.2016.05.014.
Karachaliou N, Pilotto S, Lazzari C, et al. Cellular and molecular biology of small cell Lung cancer: an overview. Transl Lung Cancer Res. 2016;5:2–15. https://doi.org/10.3978/j.issn.2218-6751.2016.01.02.
Horn L, Spigel DR, Vokes EE, et al. Nivolumab versus Docetaxel in previously treated patients with advanced non-small-cell Lung cancer: two-year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057). J Clin Oncol. 2017;35:3924–33. https://doi.org/10.1200/JCO.2017.74.3062.
Lu S, Wang J, Cheng Y, et al. Nivolumab versus Docetaxel in a predominantly Chinese patient population with previously treated advanced non-small cell Lung cancer: 2-year follow-up from a randomized, open-label, phase 3 study (CheckMate 078). Lung Cancer. 2021;152:7–14. https://doi.org/10.1016/j.lungcan.2020.11.013.
Hellmann MD, Callahan MK, Awad MM, et al. Tumor mutation burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small cell Lung cancer. Cancer Cell. 2018;33:853–861e4. https://doi.org/10.1016/j.ccell.2018.04.001.
Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. https://doi.org/10.1038/nature12477.
Byers LA, Rudin CM. Small cell Lung cancer: where do we go from here? Cancer. 2015;121:664–72. https://doi.org/10.1002/cncr.29098.
Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the Immune-related adverse effects of Immune Checkpoint inhibitors. Rev JAMA Oncol. 2016;2:1346–53. https://doi.org/10.1001/jamaoncol.2016.1051.
Schmidt-Wolf IG, Negrin RS, Kiem HP, et al. Use of a SCID mouse/human Lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. 1991;174:139–49. https://doi.org/10.1084/jem.174.1.139.
Pievani A, Borleri G, Pende D, et al. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. 2011;118:3301–10. https://doi.org/10.1182/blood-2011-02-336321.
Zhang Y, Schmidt-Wolf IGH. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. J Cell Physiol. 2020;235:9291–303. https://doi.org/10.1002/jcp.29827.
Qu J, Mei Q, Chen L, et al. Chimeric antigen receptor (CAR)-T-cell therapy in non-small-cell Lung cancer (NSCLC): current status and future perspectives. Cancer Immunol Immunother. 2021;70:619–31. https://doi.org/10.1007/s00262-020-02735-0.
Xu Y, Xiang Z, Alnaggar M, et al. Allogeneic Vγ9Vδ2 T-cell immunotherapy exhibits promising clinical safety and prolongs the survival of patients with late-stage lung or Liver cancer. Cell Mol Immunol. 2021;18:427–39. https://doi.org/10.1038/s41423-020-0515-7.
Li HY, McSharry M, Bullock B, et al. The Tumor Microenvironment regulates sensitivity of Murine Lung Tumors to PD-1/PD-L1 antibody blockade. Cancer Immunol Res. 2017;5:767–77. https://doi.org/10.1158/2326-6066.CIR-16-0365.
Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech. 2010;3:545–56. https://doi.org/10.1242/dmm.006031.
George PJ, Banerjee AK, Read CA, et al. Surveillance for the detection of early Lung cancer in patients with bronchial dysplasia. Thorax. 2007;62:43–50. https://doi.org/10.1136/thx.2005.052191.
Stevens TP, McBride JT, Peake JL, et al. Cell proliferation contributes to PNEC hyperplasia after acute airway injury. Am J Physiol. 1997;272:L486–93. https://doi.org/10.1152/ajplung.1997.272.3.L486.
Reynolds SD, Hong KU, Giangreco A, et al. Conditional clara cell ablation reveals a self-renewing progenitor function of pulmonary neuroendocrine cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1256–63. https://doi.org/10.1152/ajplung.2000.278.6.L1256.
Spella M, Lilis I, Pepe MA, et al. Club cells form lung adenocarcinomas and maintain the alveoli of adult mice. Elife. 2019;8:e45571. https://doi.org/10.7554/eLife.45571.
Miyata-Morita K, Morita S, Matsutani N, et al. Frequent appearance of club cell (Clara cell)-like cells as a histological marker for ALK-positive lung adenocarcinoma. Pathol Int. 2019;69:688–96. https://doi.org/10.1111/pin.12864.
Chen Y, Toth R, Chocarro S, et al. Club cells employ regeneration mechanisms during lung tumorigenesis. Nat Commun. 2022;13:4557. https://doi.org/10.1038/s41467-022-32052-2.
Hardavella G, George R, Sethi T. Lung Cancer Stem Cells-Characteristics, phenotype. Transl Lung Cancer Res. 2016;5:272–9. https://doi.org/10.21037/tlcr.2016.02.01.
Raniszewska A, Polubiec-Kownacka M, Rutkowska E, et al. PD-L1 expression on Lung Cancer Stem cells in metastatic lymph nodes Aspirates. Stem Cell Rev Rep. 2019;15:324–30. https://doi.org/10.1007/s12015-018-9860-7.
Raniszewska A, Vroman H, Dumoulin D, et al. PD-L1+ Lung cancer stem cells modify the metastatic lymph-node immunomicroenvironment in nsclc patients. Cancer Immunol Immunother. 2021;70:453–61. https://doi.org/10.1007/s00262-020-02648-y.
Murthy V, Katzman DP, Tsay JJ, et al. Tumor-draining lymph nodes demonstrate a suppressive immunophenotype in patients with non-small cell Lung cancer assessed by endobronchial ultrasound-guided transbronchial needle aspiration: a pilot study. Lung Cancer. 2019;137:94–9. https://doi.org/10.1016/j.lungcan.2019.08.008.
Zhu S, Luo Z, Li X, Han X, Shi S, Zhang T. Tumor-associated macrophages: role in tumorigenesis and immunotherapy implications. J Cancer. 2021;12:54–64. https://doi.org/10.7150/jca.49692.
Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21:28. https://doi.org/10.1186/s12943-021-01489-2.
Liu Y, Zugazagoitia J, Ahmed FS, et al. Immune Cell PD-L1 colocalizes with macrophages and is Associated with Outcome in PD-1 pathway blockade therapy. Clin Cancer Res. 2020;26:970–7. https://doi.org/10.1158/1078-0432.CCR-19-1040.
Shinchi Y, Ishizuka S, Komohara Y, et al. The expression of PD-1 ligand 1 on macrophages and its clinical impacts and mechanisms in lung adenocarcinoma. Cancer Immunol Immunother. 2022;1–17. https://doi.org/10.1007/s00262-022-03187-4.
Watanabe H, Ohashi K, Nishii K, et al. A long-term response to Nivolumab in a case of PD-L1-negative lung adenocarcinoma with an EGFR mutation and surrounding PD-L1-positive tumor-associated macrophages. Intern Med. 2019;58(20):3033–7. https://doi.org/10.2169/internalmedicine.2875-19.
Shima T, Shimoda M, Shigenobu T, et al. Infiltration of tumor-associated macrophages is involved in Tumor programmed death-ligand 1 expression in early lung adenocarcinoma. Cancer Sci. 2020;111:727–38. https://doi.org/10.1111/cas.14272.
Yang Z, Peng Y, Guo W, et al. PD-L1 and CD47 co‐expression predicts survival and enlightens future dual‐targeting immunotherapy in non‐small cell Lung cancer. Thorac Cancer. 2021;12:1743–51. https://doi.org/10.1111/1759-7714.13989.
Zhou Q, Liang J, Yang T, et al. Carfilzomib modulates Tumor microenvironment to potentiate immune checkpoint therapy for cancer. EMBO Mol Med. 2022;14:e14502. https://doi.org/10.15252/emmm.202114502.
Liu M, Tong Z, Ding C, et al. Transcription factor c-Maf is a checkpoint that programs macrophages in Lung cancer. J Clin Invest. 2020;130:2081–96. https://doi.org/10.1172/JCI131335.
Koto S, Chihara N, Akatani R, et al. Transcription factor c-Maf promotes immunoregulation of programmed cell death 1-Expressed CD8+ T cells in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2022;9(4):e1166. https://doi.org/10.1212/NXI.0000000000001166.
Gusak A, Fedorova L, Lepik K, et al. Immunosuppressive microenvironment and efficacy of PD-1 inhibitors in Relapsed/Refractory classic Hodgkin Lymphoma: Checkpoint molecules Landscape and Macrophage populations. Cancers (Basel). 2021;13(22):5676. https://doi.org/10.3390/cancers13225676.
Mariathasan S, Turley SJ, Nickles D, et al. TGF-β attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–8. https://doi.org/10.1038/nature25501.
Li L, Wei JR, Dong J, et al. Laminin γ2–mediating T cell exclusion attenuates response to anti–PD-1 therapy. Sci Adv. 2021;7:eabc8346. https://doi.org/10.1126/sciadv.abc8346.
Reuben A, Zhang J, Chiou SH, et al. Comprehensive T cell repertoire characterization of non-small cell Lung cancer. Nat Commun. 2020;11:603. https://doi.org/10.1038/s41467-019-14273-0.
Kamphorst AO, Pillai RN, Yang S. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in Lung cancer patients. PNAS. 2017;114:4993–8. https://doi.org/10.1073/pnas.1705327114.
Zheng Y, Han L, Chen Z, et al. PD-L1+CD8+ T cells enrichment in Lung cancer exerted regulatory function and tumor-promoting tolerance. iScinece. 2022;25:103785. https://doi.org/10.1016/j.isci.2022.103785.
Bailly C, Thuru X, Quesnel B. Soluble programmed death Ligand-1 (sPD-L1): A Pool of circulating proteins implicated in Health and Diseases. Cancers. 2021;13(12):3034. https://doi.org/10.3390/cancers13123034.
Sagawa R, Sakata S, Gong B, et al. Soluble PD-L1 works as a decoy in Lung cancer immunotherapy via alternative polyadenylation. JCI Insight. 2022;7:e153323. https://doi.org/10.1172/jci.insight.153323.
Lu F, Dong Y, Li Q, Wang M. The Change of Soluble Programmed Death Ligand 1 (sPD-L1) in Plasma of Small Cell Lung Cancer and Its Clinical Significance. Comput Math Methods Med. 2022; 2022: 8375349. https://doi.org/10.1155/2022/8375349.
Yuan Y, He Y, Wang X, et al. Investigation on the effects of soluble programmed death-1 (sPD-1) enhancing anti-tumor immune response. J Huazhong Univ Sci Technolog Med Sci. 2004;24(6):531–4. https://doi.org/10.1007/bf02911345.
He L, Zhang G, He Y, et al. Blockade of B7-H1 with sPD-1 improves immunity against Murine Hepatocarcinoma. Anticancer Res. 2005;25(5):3309–13.
Meyo MT, Jouinot A, Giroux-Leprieur E, et al. Predictive value of Soluble PD-1, PD-L1, VEGFA, CD40 Ligand and CD44 for Nivolumab Therapy in Advanced Non-small Cell Lung Cancer: a case-control study. Cancers. 2020;12:473. https://doi.org/10.3390/cancers12020473.
Ohkuma R, Ieguchi K, Watanabe M, et al. Increased plasma soluble PD-1 concentration correlates with Disease Progression in patients with Cancer treated with Anti-PD-1 antibodies. Biomedicines. 2021;9(12):1929. https://doi.org/10.3390/biomedicines9121929.
Yang Q, Chen M, Gu J, et al. Novel biomarkers of dynamic blood PD-L1 expression for Immune checkpoint inhibitors in Advanced Non-small-cell Lung Cancer patients. Front Immunol. 2021;12:665133. https://doi.org/10.3389/fimmu.2021.665133.
Ferrarini M, Pupa SM, Zocchi MR, et al. Distinct pattern of HSP72 and monomeric laminin receptor expression in human Lung Cancers infiltrated by gamma/delta T lymphocytes. Int J Cancer. 1994;57(4):486–90. https://doi.org/10.1002/ijc.2910570408.
Ferrarini M, Heltal S, Chiesa G, et al. Vδ1+ Gamma/Delta T Lymphocytes Infiltrating Human Lung Cancer Express the CD8α/α Homodimer. Scand J Immunol. 1994;40(3):363–7. https://doi.org/10.1111/j.1365-3083.1994.tb03475.x.
Zocchi MR, Ferrarini M, Rugarli C. Selective lysis of the autologous Tumor by delta TCS1+ gamma/delta+ tumor-infiltrating lymphocytes from human lung carcinomas. Eur J Immunol. 1990;20(12):2685–9. https://doi.org/10.1002/eji.1830201224.
Wu Y, Biswas D, Usaite I, et al. A local human Vδ1 T cell population is associated with survival in nonsmall-cell Lung cancer. Nat Cancer. 2022;3(6):696–709. https://doi.org/10.1038/s43018-022-00376-z.
Tosolini M, Pont F, Poupot M, et al. Assessment of tumor-infiltrating TCRV γ 9V δ 2 γδ lymphocyte abundance by deconvolution of human cancers microarrays. Oncoimmunology. 2017;6(3):e1284723. https://doi.org/10.1080/2162402X.2017.1284723.
Cazzetta V, Bruni E, Terzoli S, et al. NKG2A expression identifies a subset of human Vδ2 T cells exerting the highest antitumor effector functions. Cell Rep. 2021;37(3):109871. https://doi.org/10.1016/j.celrep.2021.109871.
George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell Lung cancer. Nature. 2015;524:47–53. https://doi.org/10.1038/nature14664.
Acheampong E, Abed A, Morici M, et al. Tumour PD-L1 expression in small-cell Lung Cancer: a systematic review and Meta-analysis. Cells. 2020;9:2393. https://doi.org/10.3390/cells9112393.
Hamilton G, Rath B. Immunotherapy for small cell Lung cancer: mechanisms of resistance. Expert Opin Biol Ther. 2019;19:423–32. https://doi.org/10.1080/14712598.2019.1592155.
Antonia SJ, López-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell Lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17:883–95. https://doi.org/10.1016/S1470-2045(16)30098-5.
Ott PA, Elez E, Hiret S, et al. Pembrolizumab in patients with extensive-stage small-cell Lung cancer: results from the phase ib KEYNOTE-028 study. J Clin Oncol. 2017;35:3823–9. https://doi.org/10.1200/JCO.2017.72.5069.
Kursunel MA, Taskiran EZ, Tavukcuoglu E, et al. Small cell Lung cancer stem cells display mesenchymal properties and exploit immune checkpoint pathways in activated cytotoxic T lymphocytes. Cancer Immunol Immunother. 2022;71:445–59. https://doi.org/10.1007/s00262-021-02998-1.
Ouadah Y, Rojas ER, Riordan DP, et al. A subpopulation of pulmonary neuroendocrine cells are reserve stem cells regulated by the Tumor suppressors rb, p53, and Notch. Cell. 2019;179:403–416e23. https://doi.org/10.1016/j.cell.2019.09.010.
Rudin CM, Poirier JT, Byers LA, et al. Molecular subtypes of small cell Lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer. 2019;19:289–97. https://doi.org/10.1038/s41568-019-0133-9.
Lee BS, Park DI, Lee DH, et al. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem Biophys Res Commun. 2017;491:493–9. https://doi.org/10.1016/j.bbrc.2017.07.007.
Wang G, Lu X, Dey P, et al. Targeting YAP-dependent MDSC infiltration impairs Tumor progression. Cancer Discov. 2016;6:80–95. https://doi.org/10.1158/2159-8290.CD-15-0224.
Shibata M, Ham K, Hoque MO. A time for YAP1: tumorigenesis, immunosuppression and targeted therapy. Int J Cancer. 2018;143:2133–44. https://doi.org/10.1002/ijc.31561.
Horie M, Saito A, Ohshima M, et al. YAP and TAZ modulate cell phenotype in a subset of small cell Lung cancer. Cancer Sci. 2016;107:1755–66. https://doi.org/10.1111/cas.13078.
Roper N, Velez MJ, Chiappori A, et al. Notch signaling and efficacy of PD-1/PD-L1 blockade in relapsed small cell Lung cancer. Nat Commun. 2021;12:3880. https://doi.org/10.1038/s41467-021-24164-y.
Saito H, Tenjin Y, Yamada T, et al. The role of YAP1 in small cell Lung cancer. Hum Cell. 2022;35:628–38. https://doi.org/10.1007/s13577-022-00669-6.
Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers-a different Disease. Nat Rev Cancer. 2007;7:778–90. https://doi.org/10.1038/nrc2190.
Lamberti G, Sisi M, Andrini E, et al. The mechanisms of PD-L1 Regulation in Non-small-cell Lung Cancer (NSCLC): which are the involved players? Cancers. 2020;12:3129. https://doi.org/10.3390/cancers12113129.
Re MD, Cucchiara F, Rofi E, et al. A multiparametric approach to improve the prediction of response to immunotherapy in patients with metastatic NSCLC. Cancer Immunol Immunother. 2021;70(6):1667–78. https://doi.org/10.1007/s00262-020-02810-6.
Dhakar R, Dakal TC, Sharma A. Genetic determinants of Lung cancer: understanding the oncogenic potential of somatic missense mutations. Genomics. 2022;114(4):110401. https://doi.org/10.1016/j.ygeno.2022.110401.
Shinohara T, Taniwaki M, Ishida Y, et al. Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics. 1994;23:704–6. https://doi.org/10.1006/geno.1994.1562.
Jiang X, Wang J, Deng X, et al. Role of the Tumor microenvironment in PD-L1/PD-1-mediated Tumor immune Escape. Mol Cancer. 2019;18:10. https://doi.org/10.1186/s12943-018-0928-4.
Wartewig T, Kurgyis Z, Keppler S, et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature. 2017;552:121–5. https://doi.org/10.1038/nature24649.
Patsoukis N, Brown J, Petkova V, et al. Selective effects of PD-1 on akt and ras pathways regulate Molecular Components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5:ra46. https://doi.org/10.1126/scisignal.2002796.
Gulati P, Rühl J, Kannan A, et al. Aberrant lck Signal via CD28 Costimulation augments Antigen-Specific functionality and Tumor Control by Redirected T Cells with PD-1 blockade in Humanized mice. Clin Trial Clin Cancer Res. 2018;24:3981–93. https://doi.org/10.1158/1078-0432.CCR-17-1788.
Wang X, Yang X, Zhang C, et al. Tumor cell-intrinsic PD-1 receptor is a Tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc Natl Acad Sci U S A. 2020;117:6640–50. https://doi.org/10.1073/pnas.192144511.
Edinger M, Cao Y, Verneris MR, et al. Revealing Lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood. 2003;101:640–8. https://doi.org/10.1182/blood-2002-06-1751.
Wu X, Sharma A, Oldenburg J, et al. NKG2D Engagement alone is sufficient to Activate Cytokine-Induced Killer cells while 2B4 only provides limited coactivation. Front Immunol. 2021;12:731767. https://doi.org/10.3389/fimmu.2021.731767. Published online 2021 Oct 7.
Verneis MR, Kornacker M, Mailänder V, Negrin RS. Resistance of ex vivo expanded CD3+CD56+ T cells to Fas-mediated apoptosis. Cancer Immunol Immunother. 2000;49(6):335–45. https://doi.org/10.1007/s002620000111.
Cappuzzelloa E, Tosia A, Zanovelloa P, et al. Retargeting cytokine-induced killer cell activity by CD16 engagement with clinical-grade antibodies. Oncoimmunology. 2016;5:e1199311. https://doi.org/10.1080/2162402X.2016.1199311.
Zhang L, Wang J, Wei F, et al. Profiling the dynamic expression of checkpoint molecules on cytokine-induced killer cells from non-small-cell Lung cancer patients. Oncotarget. 2016;7:43604–15. https://doi.org/10.18632/oncotarget.9871.
Han Y, Mu D, Liu T, et al. Autologous cytokine-induced killer (CIK) cells enhance the clinical response to PD‐1 blocking antibodies in patients with advanced non‐small cell Lung cancer: a preliminary study. Thorac Cancer. 2021;12:145–52. https://doi.org/10.1111/1759-7714.13731.
Chen CL, Pan QZ, Weng DS, et al. Safety and activity of PD-1 blockade-activated DC-CIK cells in patients with advanced solid tumors. Oncoimmunology. 2018;7:e1417721. https://doi.org/10.1080/2162402X.2017.1417721.
Wang Z, Liu X, Till B, Sun M, Li X, Gao Q. Combination of Cytokine-Induced Killer cells and programmed cell Death-1 Blockade Works synergistically to enhance therapeutic efficacy in metastatic renal cell carcinoma and Non-small Cell Lung Cancer. Front Immunol. 2018;9:1513. https://doi.org/10.3389/fimmu.2018.01513.
Zhou L, Xiong Y, Wang Y, et al. A phase IB Trial of Autologous Cytokine-Induced Killer cells in combination with sintilimab, monoclonal antibody against programmed cell Death-1, plus Chemotherapy in patients with Advanced Non-small-cell Lung Cancer. Clin Lung Cancer. 2022;23(8):709–19. https://doi.org/10.1016/j.cllc.2022.07.009.
Mankor JM, Disselhorst MJ, Poncin M, et al. Efficacy of nivolumab and ipilimumab in patients with malignant pleural Mesothelioma is related to a subtype of effector memory cytotoxic T cells: translational evidence from two clinical trials. EBioMedicine. 2020;62:103040. https://doi.org/10.1016/j.ebiom.2020.103040.
John LB, Devaud C, Duong CP, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19:5636–46. https://doi.org/10.1158/1078-0432.CCR-13-0458.
John LB, Kershaw MH, Darcy PK. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology. 2013;2:e26286. https://doi.org/10.4161/onci.26286.
Liu M, Wang X, Li W, et al. Targeting PD-L1 in non-small cell Lung cancer using CAR T cells. Oncogenesis. 2020;9:72. https://doi.org/10.1038/s41389-020-00257-z.
Li S, Siriwon N, Zhang X, et al. Enhanced Cancer Immunotherapy by chimeric Antigen receptor–modified T cells Engineered to secrete checkpoint inhibitors. Clin Cancer Res. 2017;23:6982–92. https://doi.org/10.1158/1078-0432.
Tokatlian T, Asuelime GE, Mock JY, et al. Mesothelin-specific CAR-T cell therapy that incorporates an HLA-gated safety mechanism selectively kills Tumor cells. J Immunother Cancer. 2022;10:e003826. https://doi.org/10.1136/jitc-2021-003826.
Ghosn M, Cheema W, Zhu A, et al. Image-guided interventional radiological delivery of chimeric antigen receptor (CAR) T cells for pleural malignancies in a phase I/II clinical trial. Lung Cancer. 2022;165:1–9. https://doi.org/10.1016/j.lungcan.2022.01.003.
Chen S, Lin Y, Zhong S, et al. Anti-MUC1 CAR-T cells combined with PD-1 knockout engineered T cells for patients with non-small cell Lung cancer (NSCLC): a pilot study. Ann Oncol. 2018;29(Supplement 10):x11–6. https://doi.org/10.1093/annonc/mdy485.
Byers LA, Chiappori A, Smit MAD. Phase 1 study of AMG 119, a chimeric antigen receptor (CAR) T cell therapy targeting DLL3, in patients with relapsed/refractory small cell Lung cancer (SCLC). J Clin Oncol. 2019;37:TPS8576. https://doi.org/10.1200/JCO.2019.37.15_suppl.TPS8576.
Zhong S, Cui Y, Liu Q, Chen S. CAR-T cell therapy for Lung Cancer: a Promising but Challenging Future. J Thorac Dis. 2020;12:4516–21. https://doi.org/10.21037/jtd.2020.03.118.
Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-Cell therapy. Mol Ther Oncolytics. 2016;3:16011. https://doi.org/10.1038/mto.2016.11.
Owen DH, Giffin MJ, Bailis JM, et al. DLL3: an emerging target in small cell Lung cancer. J Hematol Oncol. 2019;12:61. https://doi.org/10.1186/s13045-019-0745-2.
Chen X, Amar N, Zhu Y, et al. Combined DLL3-targeted bispecific antibody with PD-1 inhibition is efficient to suppress small cell Lung cancer growth. J Immunother Cancer. 2020;8:e000785. https://doi.org/10.1136/jitc-2020-000785.
Adusumilli PS, Zauderer MG, Rivière I, et al. A phase I Trial of Regional Mesothelin-targeted CAR T-cell therapy in patients with malignant Pleural Disease, in combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021;11:2748–63. https://doi.org/10.1158/2159-8290.CD-21-0407.
Liu Z, Xia Y, Li L, Sun Y, Lin Z, Rong L, et al. Abstract CT134: non-viral mesothelin-targeted CAR-T cells armored with IFNg-induced secretion of PD-1 nanobody in treatment of malignant Mesothelioma in phase I clinical trial. Cancer Res. 2023;83(8Supplement):CT134. https://doi.org/10.1158/1538-7445.AM2023-CT134.
Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. 2021;18:2188–98.
Siegers GM, Dhamko H, Wang XH, et al. Human Vδ1 γδ T cells expanded from peripheral blood exhibit specific cytotoxicity against B-cell chronic lymphocytic leukemia-derived cells. Cytotherapy. 2011;13:753–64. https://doi.org/10.3109/14653249.2011.553595.
Davey MS, Willcox CR, Baker AT, et al. Recasting human Vδ1 lymphocytes in an adaptive role. Trends Immunol. 2018;39:446–59. https://doi.org/10.1016/j.it.2018.03.003.
Makkouk A, Yang XC, Barca T, et al. Off-the-shelf Vδ1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J Immunother Cancer. 2021;9(12):e003441. https://doi.org/10.1136/jitc-2021-003441.
Sakamoto M, Nakajima J, Murakawa T, et al. Adoptive immunotherapy for advanced non-small cell Lung cancer using zoledronate-expanded γδ T cells: a phase I clinical study. J Immunother. 2011;34:202–11. https://doi.org/10.1097/CJI.0b013e318207ecfb.
Kakimi K, Matsushita H, Masuzawa K, et al. Adoptive transfer of zoledronate-expanded autologous Vγ9Vδ2 T-cells in patients with treatment-refractory non-small-cell Lung cancer: a multicenter, open-label, single-arm, phase 2 study. J Immunother Cancer. 2020;8:e001185. https://doi.org/10.1136/jitc-2020-001185.
Bhat SA, Vedpathak DM, Chiplunkar SV. Checkpoint blockade rescues the repressive effect of histone deacetylases inhibitors on γδ T cell function. Front Immunol. 2018;9:1615. https://doi.org/10.3389/fimmu.2018.01615.
Peters C, Oberg HH, Kabelitz D, Wesch D. Phenotype and regulation of immunosuppressive Vδ2-expressing γδ T cells. Cell Mol Life Sci. 2014;71(10):1943–60. https://doi.org/10.1007/s00018-013-1467-1.
Bezombes C, Rossi C, Gravelle P, et al. Boosting Gamma Delta T cells-mediated ADCC by PD-1 blockade in Follicular Lymphoma. Blood. 2018;132(Supplement 1):5381. https://doi.org/10.1182/blood-2018-99-112404.
Shinchi Y, Komohara Y, Yonemitsu K, et al. Accurate expression of PD-L1/L2 in lung adenocarcinoma cells: a retrospective study by double immunohistochemistry. Cancer Sci. 2019;110:2711–21. https://doi.org/10.1111/cas.14128.
Takamori S, Takada K, Azuma K, et al. Prognostic impact of programmed death-ligand 2 expression in primary lung adenocarcinoma patients. Ann Surg Oncol. 2019;26:1916–24. https://doi.org/10.1245/s10434-019-07231-z.
Matsubara T, Takada K, Azuma K, et al. A clinicopathological and prognostic analysis of PD-L2 expression in surgically resected primary lung squamous cell carcinoma. Ann Surg Oncol. 2019;26:1925–33. https://doi.org/10.1245/s10434-019-07257-3.
Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. https://doi.org/10.1038/85330.
Markham A, Duggan S. Cemiplimab: First Global approval. Drugs. 2018;78:1841–6. https://doi.org/10.1007/s40265-018-1012-5.
Sezer A, Kilickap S, Gümüş M, et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell Lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet. 2021;397:592–604. https://doi.org/10.1016/S0140-6736(21)00228-2.
Moreno V, Garrido P, Papadopoulos KP, et al. Tolerability and antitumor activity of cemiplimab, a human monoclonal anti-PD-1, as monotherapy in patients with pretreated non-small cell Lung cancer (NSCLC): data from the phase 1 NSCLC expansion cohort. Lung Cancer. 2021;155:151–5. https://doi.org/10.1016/j.lungcan.2021.02.034.
Tsay JJ, Wu BG, Sulaiman I, et al. Lower airway dysbiosis affects Lung cancer progression. Cancer Discov. 2021;11:293–307. https://doi.org/10.1158/2159-8290.CD-20-0263.
Zitvogel L, Kroemer G. Lower Airway dysbiosis exacerbates Lung cancer. Cancer Discov. 2021;11:224–6. https://doi.org/10.1158/2159-8290.CD-20-1641.
Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7. https://doi.org/10.1126/science.aan3706.
Derosa L, Hellmann MD, Spaziano M, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell Lung cancer. Ann Oncol. 2018;29:1437–44. https://doi.org/10.1093/annonc/mdy103.
Masuhiro K, Tamiya M, Fujimoto K, et al. Bronchoalveolar lavage fluid reveals factors contributing to the efficacy of PD-1 blockade in Lung cancer. JCI Insight. 2022;7:e157915. https://doi.org/10.1172/jci.insight.157915.
Ochi N, Ichihara E, Takigawa N, et al. The effects of antibiotics on the efficacy of immune checkpoint inhibitors in patients with non-small-cell Lung cancer differ based on PD-L1 expression. Eur J Cancer. 2021;149:73–81. https://doi.org/10.1016/j.ejca.2021.02.040.
Qiao DR, Cheng JY, Yan WQ, et al. PD-L1/PD-1 blockage enhanced the cytotoxicity of natural killer cell on the non-small cell Lung cancer (NSCLC) by granzyme B secretion. Clin Transl Oncol. 2023;25:2373–83. https://doi.org/10.1007/s12094-023-03120-w.
Acknowledgements
Not applicable.
Funding
The CIO Aachen Bonn Köln Düsseldorf is kindly supported by the Deutsche Krebshilfe, Bonn, Germany (grant No. 70113470).
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Author Contributions: Conceptualization, Y.L., A.S. and I.G.H.S-W.; Writing - original draft, Y.L.; Visualization: Y.L. and A.S.; Supervision, I.G.H.S-W. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Informed consent statement
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Sharma, A. & Schmidt-Wolf, I.G. Evolving insights into the improvement of adoptive T-cell immunotherapy through PD-1/PD-L1 blockade in the clinical spectrum of lung cancer. Mol Cancer 23, 80 (2024). https://doi.org/10.1186/s12943-023-01926-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01926-4