
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an incurable cancer resistant to traditional treatments, although a limited number of early-stage patients can undergo radical resection. Immunotherapies for the treatment of haematological malignancies as well as solid tumours have been substantially improved over the past decades, and impressive results have been obtained in recent preclinical and clinical trials. However, PDAC is likely the exception because of its unique tumour microenvironment (TME). In this review, we summarize the characteristics of the PDAC TME and focus on the network of various tumour-infiltrating immune cells, outlining the current advances in PDAC immunotherapy and addressing the effect of the PDAC TME on immunotherapy. This review further explores the combinations of different therapies used to enhance antitumour efficacy or reverse immunodeficiencies and describes optimizable immunotherapeutic strategies for PDAC. The concordant combination of various treatments, such as targeting cancer cells and the stroma, reversing suppressive immune reactions and enhancing antitumour reactivity, may be the most promising approach for the treatment of PDAC. Traditional treatments, especially chemotherapy, may also be optimized for individual patients to remodel the immunosuppressive microenvironment for enhanced therapy.
Similar content being viewed by others


Introduction
PDAC remains one of the deadliest malignancies with a poor outcome, and very few regimens have been successfully used to treat this lethal cancer. The 5-year overall survival (OS) rate of PDAC patients is abysmal at less than 5% [1]. PDAC was the fourth leading cause of cancer-related death in 2012 [2] and is projected to become the third most common cancer in the United States by 2030. Although PDAC-associated morbidity does not rank highly in cancer epidemiology [3], the mortality rate is nearly the highest among of all cancers. Surgical resection is the sole curable approach for localized PDAC, but no more than 20% of tumours are resectable at the time of diagnosis due to the lack of early symptoms and the aggressive biological nature of this carcinoma [4]. Most patients relapse after surgery even after routine adjuvant therapies have been used systematically [5]. Neoadjuvant treatment increases the resectable rate and benefits OS, but the results are unclear [6]. Even for patients with localized and resectable tumours, the 5-year OS rate is only approximately 27% [7]. Chemotherapy based on gemcitabine (Gem) is currently the standard treatment for metastatic PDAC, and the combination of Gem with oxaliplatin, irinotecan, leucovorin and 5-fluorouracil (FOLFIRINOX) can reduce the mortality rate but has been shown to increase toxicity and to have a poor survival benefit and high cost burden [8, 9]. Therefore, the exploration of new therapies for PDAC is urgently needed. Immunotherapy, including strategies such as monoclonal antibody (mAb) therapy, immune checkpoint inhibitor (ICI) therapy, adoptive cell therapy/adoptive cell transfer (ACT), vaccines and other agents that enhance the antitumour response and/or reverse the immunosuppressive functions of regulatory immune cells in the TME, has made great progress in cancer treatment in recent decades. However, no immunotherapeutic approaches have produced promising results thus far despite similar strategies making notable progress in other cancers. For reasons unknown, the TME plays a critical role in the development, progression, and metastasis of PDAC as well as to its sensitivity to immunotherapy.
TME of PDAC
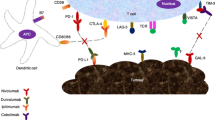
The TME of PDAC consists of the cancer cell nest and stroma. The stroma contains various components, primarily the stromal matrix and various cells. Here, we concisely summarize the existing knowledge about the TME of PDAC (Fig. 1) and emphasize the immune cell network established around cancer cells (Fig. 2).

The graphic abstract of PDAC TME.• From the right upper side to the left low side, we summarize the progression of PDAC from PanIN and the distribution of different cells in TME. The yellow area represent the area mainly comprising different advanced stage of epithelial tissue from normal acinar to PanIN and invasive cancer nest, as well as monocyte-like cells; the reddish area present the area comprising mainly matrix including fibrotic matrix, pancreatic stellate cells, cancer associated fibroblasts, TLS, as well as accumulated effector lymphocytes. The cancer nests look like islands in the stroma desert; Treg cells surround the PanIN and establish a TSA specific suppressive condition to support PDAC progression; MDSCs appear at very early stage of the PDAC progression and disperse the whole lesion of tumor; TAMs locate majorly at the invasive front of the tumor and promote angiogenesis, lymphogenesis and metastasis; DCs are scarce and restricted in PanIN and TLS; CAFs and PSCs are the major source of tumor stromal matrix, they can also adhere infiltrating T lymphocytes, keep them outside of cancer nest and result effector T cell anergy; TLS localize in the tumor stroma and consist of proliferating effector cells as well as Treg cells, tumor specific anti-tumor and pro-tumor reactivity present concordantly

The molecular interaction of different cells in TME. The cancer cells of PDAC exploit several mechanisms including cell surface molecule and soluble factors to establish immunosuppressive TME through accumulating and activating immune suppressive cells, and inhibiting antitumor effector cells directly and indirectly; suppressive cells can inhibit the function of effector cells through nutrition depletion, phenotype alternation, apoptosis and anergy; Treg cells may play a central role in the establishment of immunosuppressive TME of PDAC since they are in favor of establishing tumor specific immunotolerance and have extensive interaction with other cells
PDAC epithelial cells
Tumour-associated antigens (TAAs) have been identified in PDAC but are limited, and the absence of TAAs hinders naturally occurring antitumour reactivity. Deficiencies in antigen processing and epitope presentation are another critical mechanism of immune evasion. PDAC cells generally downregulate the expression of major histocompatibility (MHC) class I molecules [10,11,12], and MHC class I/II molecules may also develop genetic mutations that result in impaired antigen presentation. Aberrant expression of the receptor Fas and Fas ligand extensively occurs in most PDAC patients and results in immune tolerance. Normal pancreatic ductal cells express the Fas receptor but not the Fas ligand, while PDAC cells express a non-functional form of the Fas receptor, which results in resistance to Fas-mediated apoptosis; furthermore, PDAC cells express the Fas ligand to induce apoptosis in immune effector cells [13]. PDAC cells recruit immunosuppressive tumour-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) from the peripheral circulation via the CCL2/CCR2 axis [14]. PDAC cells express high levels of CCL5 to recruit regulatory T cells (Treg cells) through CCR5 [15], and this process may partially explain the recruitment of Treg cells to PDAC lesions [16]. Approximately 12.5% of PDAC patients are reported to positively express programmed cell death protein ligand-1 (PD-L1) [17], which induces T cell anergy and apoptosis through programmed cell death protein-1 (PD-1) expressed on T cells, resulting in immune system evasion [18]. PDAC cells can also programme the TME by directly secreting soluble cytokines, such as transforming growth factor (TGF-β) and interleukin (IL)-10, to inhibit dendritic cell (DC) differentiation and maturation in favour of Treg cell accumulation [19, 20]. PDAC cells produce indoleamine 2,3-dioxygenase (IDO) to catalyse the degradation of tryptophan, which is necessary for T cell survival and activation, thereby inducing T cell apoptosis and anergy [21, 22].
PDAC stroma
A high-density fibrotic stromal reaction, termed “desmoplasia”, may be one of the most prominent characteristics of the PDAC stroma, as almost 90% of the tumour mass is composed of the stroma, which facilitates immunosuppression and fibrosis progression [23, 24]. The carcinogenic nests appear as islands surrounded by the stromal desert, as depicted in Fig. 1. The PDAC stroma has been demonstrated to not only promote tumour progression but to also dampen the delivery of antitumour regimens [24,25,26], even increasing the number of immunosuppressive cells and inactivating cytotoxic CD8+ T cells [27, 28]. Controversial results have been reported recently, including those of Wang and Knudsen et al., who divided PDAC into three classes according to the stromal density and demonstrated that stromal density and volume had a positive association with patient OS [29, 30]. Özdemir et al. interpreted the mechanisms in a preclinical study in which cancer-associated fibroblasts (CAFs) were depleted, which had extensive effects on the TME, such as reducing collagen and matrix reorganization, decreasing angiogenesis, enhancing hypoxia, increasing cancer stem cell numbers, and increasing Treg cell frequency, all of which contributed to a poor outcome [31]. The numbers of pancreatic stellate cells (PSCs), special CAFs unique to PDAC, increase abundantly during the progression of the disease [32]. Activated PSCs can restrain tumour-infiltrating CD8+ T cells in the stroma but not cancer nests through the production of CXCL12 since activated CD8+ T cells express high levels of CXCR4 [33]. The chemokine ligand/receptor has been demonstrated to be a strong chemoattractant for lymphocytes [34]. PSCs also induce T cell apoptosis and anergy by expressing galectin-1 [35]. PSCs may crosstalk with TAMs in PanIN, and these cell populations activate each other by secreting various soluble factors. This process may be the major mechanism of desmoplasia; interestingly, the deposition of collagen preferentially excludes TAMs [32].
Infiltrating immune cells
The results of research on PDAC-infiltrating immune cells are often vague and controversial. Here, we summarize them concisely with a distinctive view.
Antitumour effector cells and immunodeficiency
Immune cells comprise nearly 50% of the PDAC cellular component [36], but only a few are antitumour effector cells. The low number of antitumour effector cells could possibly be attributed to the cells being disabled by several mechanisms (Fig. 2). Some studies have evaluated the function of tumour-associated neutrophils (TANs) in PDAC progression, which has been reviewed extensively [37]. In a recent clinical study, neutrophils were found to have an unexpected positive correlation with CD8+ T cells [38]; the correlation was surprising since these cells might play a role in excluding infiltrating T cells from PDAC tissue in mouse models [39, 40]. These controversial results may be interpreted as a function of the different neutrophil frequencies in humans and mice. The characteristics of natural killer (NK) cells within PDAC tumours have been investigated, but few reports describe the role of NK cells in normal and PDAC tissues [36, 41]. A study demonstrated that CD3+ T cells were the major immune cell type in PDAC, and the majority of resectable PDAC samples displayed intermediate to high levels of CD3+ T cell infiltration, which predominantly occurred in the stroma instead of the cancer cell nest centre [42]. CD3+ conventional T (Tconv) cells localize in tertiary lymphoid structures (TLSs) (Fig. 1) and co-localize with DCs, Treg cells, B cells, and high endothelial venules (HEVs). Localized proliferation, not merely migration, was shown to be a major source of activated T cells. Clonal T cell expansion was observed within the TLSs throughout the tumour lesions, indicating a tumour antigen-specific reaction within the TLSs [42]. In a subsequent study, heavy lymphocyte infiltration was observed in TLSs, but in situ proliferation was not observed [38]. Both of the above studies demonstrated a positive relationship between TLSs and OS in PDAC patients, suggesting that the potential antitumour response in PDAC is suppressed. Most of the tumour-infiltrating lymphocytes (TILs) displayed an antigen-experienced and memory-related phenotype [38, 42,43,44], which further supported this conclusion. The frequencies of CD4+ and CD8+ lymphocytes were variable among specimens; CD4+ T cells, especially CD4+ Tconv cells, were predominant, but CD8+ T cells were not [38, 42], suggesting a deficiency in the cytotoxic activity of CD8+ T cells. The accumulation of CD8+ T cells in PDAC is extremely variable; the frequency of CD8+ T cells among CD45+ leucocytes may be as high as 15–30% or less than 7%. These effector cells are functionally deficient, as they express various co-inhibitory molecules [38, 42].
CD4+ and CD8+ T cells are subtly synchronized with each other within PDAC tumours; only patients with both CD4- and CD8-positive T cells have a significantly increased OS rate, and the CD4/CD8 double-positive T cell status is an independent prognostic factor [45, 46]. Among CD4+ Tconv cells, only the Th1 subset can facilitate the antitumour response, and the function of Th17 cells is controversial. Th2 cells are generally considered factors promoting tumour progression. Notably, Th2 cells are the major population of CD4+ T cells within PDAC tumours, and the Th2 CD4+ T cell number is higher than not only the Th1 CD4+ T cell number but also the FoxP3+ Treg cell number [47]. CD4+ T cells are inclined to polarize towards the Th2 phenotype, and this skewing is specific for carcinoembryonic Ag (CEA) [47]. These findings indicate that PDAC can induce TAA-specific immune impairment through CD4+ T cells. DCs in PDAC are generally functionally impaired. In a recent preclinical study, DCs were observed to abundantly infiltrate the tumour lesion, and DC accumulation increased as the disease progressed from PanIN to PDAC. However, the expression of the maturation marker MHC class II and the costimulatory molecules CD86 and CD40 was downregulated by Treg cells in a cell contact-dependent manner (Fig. 3) [48]. All of these molecules were indispensable for CD8+ T cell activation, and Treg cells could even suppress the in vivo expansion of tumour-infiltrating DCs [48]. PDAC epithelial cells can also exploit variable strategies to decrease the function of DCs, such as downregulating the expression of HLA-DR and CD40 to produce immature DCs and secreting DC-suppressing cytokines and chemokines [12, 49, 50]. Immature DCs can directly suppress the effector T cell response by expressing IDO [48]. DCs may execute antigen-specific suppressive functions by presenting tissue-specific antigens (TSAs) and even neoantigens to Treg cells to induce tumour-specific immunosuppression. Both DCs and Treg cells accumulate in TLSs with a high density of endothelial venules [38, 42], which are generally found in lymph nodes and responsible for antigen presentation. These facts highlight the possibility that tumour-specific immune tolerance exists in these structures through DC–Treg interactions.

The mechanisms of Treg inhibit Tconv through APC. Treg and Tconv contact directly with the same APC and establish tumor specific suppressive TME. a: Treg capture and degradate CD86 on DC with CTLA4, the process occurs in LN/TLS and PanIN, activating Treg migrate to established tumor and transform to resting Treg and execute suppression; b: Treg (also Tconv) contact with APC through various pairs of ligand-receptor including of TCR/MHC, CD28/CD86, CD28/CD80, CTLA4/CD86, CTLA4/CD80, mature DC dominantly express high level of CD86 and combine with CD28 and CTLA4, MDSC preferentially express CD80 and combine with CTLA4, immature/inducible DC express both CD86 and CD80. Notably, MDSC express low level of MHC and enhance suppressive function of Treg with weak TCR signal, whereas DC express high level of MHC and promote Treg activation and proliferation; c: APC could transform each other with the effect of Treg and Tconv concordantly; d: APC inhibit Tconv through several soluble factors and induce Tconv anergy through weak/downregulating TCR signal; e: APC inhibit CD4+ Tconv directly and CD8+ Tconv indirectly mainly by downregulating IL-2 and IFN-γ et al., Treg cells could inhibit Tconv by depriving IL-2. Biophysical stability of CTLA4/CD28-CD80/CD86 polymer: CTLA4-CD80 > CTLA4-CD86 > CD28-CD86 > CD28- CD80
Protumour regulatory cells and immunosuppression
Nearly all TAMs exhibit an M2 phenotype, identified by the surface markers CD163 and CD206 and cytokines, such as IL-10 and TGF-β, but they also display M1 characteristics [51]. TAM infiltration begins at a very early disease stage and persists in PDAC [36]. TAMs are generally located at the invasive front of the tumour (Fig. 1) [36, 52]. This process occurs in both murine and human PDAC and is accompanied by perineural invasion [53], lymphatic angiogenesis, lymph node metastasis [52, 54], cancer cell epithelial-mesenchymal transition (EMT) and extravasation [51]. Several factors can recruit monocytes to PDAC lesions and differentiate these cells into TAMs, including the hypoxic TME [55], vascular endothelial growth factor (VEGF)/epidermal growth factor receptor (EGFR) 2 axis [56], CCL2/CCR2 axis [14] and CSF1/CSF1R axis [57]. In an extensive study, Kaneda et al. [58] demonstrated that TAMs exploited numerous mechanisms to drive PDAC progression, including secreting immunosuppressive factors such as arginase-1 (Arg1) and TGF-β to inhibit antitumour CD8+ T cells and promoting PDAC desmoplasia and cancer cell metastasis via the chemotactic factor PDGF-BB. Therefore, the major role of TAMs in PDAC seems to be tightly regulating invasion and metastasis rather than inhibiting the immune response.
MDSCs are Gr1 and CD11b double-positive in mice and CD14-negative and CD11b-positive in humans. A subset of MDSCs express the granulocyte marker Ly6G at a high level and the monocyte marker Ly6C at an intermediate level; the other MDSC pool expresses high levels of Ly6C not Ly6G [59]. Therefore, MDSCs are categorized into two major subsets: granulocytic MDSCs (Gr-MDSCs) and monocytic MDSCs (Mo-MDSCs). MDSCs, especially Gr-MDSCs, are rare in the normal pancreas, and their accumulation increases progressively as the disease becomes invasive. MDSCs are widely dispersed throughout the tumour in invasive PDAC [36, 59]. PDAC cells highly express granulocyte macrophage colony-stimulating factor (GM-CSF), which was demonstrated to be a necessary and sufficient factor for functional and suppressive MDSC generation [39]. The function of MDSCs in PDAC was reviewed extensively in a previous publication [60]. Most investigators focus on the function of MDSCs in immunosuppression through the secretion of modulatory factors and direct contact with effector cells via checkpoint molecules. One important property of MDSCs worthy of emphasis is that although they are antigen presenting, they express low levels of the MHC II complex [59] and high levels of CD80 to induce antigen-specific immunosuppression via Treg cells (Fig. 3) [61]. Treg cells have T cell receptors (TCRs) with relatively high affinities for TSAs and constitutively express cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), which preferentially binds with CD80 and outcompetes CD86 binding [62]. Gabrilovich et al. suggested that MDSCs might be involved in Treg cell differentiation [63]. These results indicate that TSA-specific and/or even neoantigen-specific immunosuppressive mechanisms mediated through the MDSC-Treg axis and antibodies against CD80 or CTLA-4 may have similar effects.
Treg cells have extensive interactions with various cells (Fig. 2), and the tight relationship between Treg and antigen-presenting-like cells has been repeatedly highlighted in numerous studies. However, overall conclusions are still obscure; Treg cells and antigen-presenting cells (APCs) cannot be defined restrictedly, and the molecular biophysical interactions between these two subsets of cells (particularly MHC/TCR, CTLA-4-CD28 and CD80-CD86 interactions) are controversial despite numerous researchers focusing on this field. We present an overview of the mechanism by which Treg cells inhibit Tconv cells via concordant contact with APCs (Fig. 3). Treg cells exert suppressive effects by recognizing self-TSAs presented by APCs but can inhibit effector cells in an antigen-independent fashion [64, 65]. Moreover, because the TCRs of Treg cells have higher affinities for epitopes than the corresponding TCRs of Tconv cells, Treg cells can recognize antigens at concentrations lower than those required for Tconv cell activation [65], suggesting that Treg cells may be activated by immature APCs with weak antigen presentation. Treg cells accumulate within tumours and tumour-draining lymph nodes at a very early stage in PanIN, and their numbers increase upon progression to PDAC [20, 48]. Upon the establishment of invasive tumours, Treg cells are generally localized within the TLSs with follicular DCs and HEVs (Fig. 1) [38, 42]. The prevalence of Treg cells is tightly correlated with the prognosis of PDAC [38, 48, 66,67,68] and generally has a negative relationship with patient OS. There are two major types of Treg cells: naturally occurring Treg cells (nTreg cells) derived from the thymus and resident in tissues and inducible Treg cells (iTreg cells) derived from naïve CD4+ T cells in the peripheral blood. PDAC cells produce CCL5 and VEGF to attract Treg cells through CCR5 [15, 16] and neuropilin-1 [48, 69]. Stromal cells recruit Treg cells by CXCL10 on PSCs [70] and CCL5 on MDSCs [71] through CXCR3 and CCR5, respectively (Fig. 2). These interactions may be the mechanism of iTreg cell accumulation since nTreg cells are generally resident cells. However, researchers have demonstrated that Treg cells accumulate in PDAC through proliferation and conversion in situ rather than via the infiltration of peripheral nTreg cells and naïve T cells [67]. Peripheral blood Treg cell depletion with an anti-CD25 antibody and functional inhibition do not affect the Treg cell frequency within tumours [72]. Localized proliferation is exploited by nTreg cells to drive accumulation within PDAC tissue at an early stage and is mediated by activation of tissue-resident nTreg cells by resident DCs through the presentation of self-antigens. Localized proliferation might also be the mechanism of iTreg cell accumulation within TLSs in which follicular DCs and HEVs are present. The function of Treg cells in PDAC immune editing also remains controversial, although most studies have demonstrated that Treg cells regulate CD4+ and CD8+ lymphocytes through monocyte-type cells. However, the pathway and target cells are not yet clear. In a pilot study, Qureshi et al. demonstrated that CTLA-4 molecules could capture and endocytose CD86 expressed on the cell surface, which resulted in CD86 degradation, and the subsequent activation of Treg cells prevented DCs from priming naïve T cells (Fig. 3) [73]. This may be the mechanism by which nTreg cells inhibit tissue-resident DCs in early PanIN lesions since tissue-resident DCs rarely express CD86 rather than CD80. On the other hand, Treg cells may regulate infiltrating CD4+ cells rather than CD8+ T cells in PDAC through the CTLA-4/CD80 pathway by contacting MDSCs because blockade of CTLA-4 on Treg cells or blockade of CD80 on MDSCs was shown to produce the same results [72]. Based on these observations, MDSCs appear to have a high probability of being monocyte-type cells targeted by iTreg cells in invasive PDAC. Treg cells express TCRs that recognize self-TSAs and may be activated by self-TSAs in the presence of APCs [74, 75]. This property of Treg cells may be exploited by cancer cells and immature APCs to produce immune tolerance. It has been previously demonstrated that immature APCs can preferentially induce Treg cells [76, 77]. Immature APCs may have a better potential to facilitate the suppressive function of Treg cells than mature APCs because of their higher expression levels of CD80 [62], which generally forms a dimer and preferentially binds with CTLA-4 molecules, which are constitutively expressed on Treg cells (Fig. 3). Targeting tissue-specific Treg cells and/or blocking the interaction between Treg cells and monocyte-like cells may be an interesting direction of research for PDAC immunotherapy.
mAb therapy for PDAC
mAb-based therapy has been used as an established treatment strategy for multiple solid tumours for decades. The functional mechanisms of mAbs in cancer therapy are limited to not only the direct killing of cells through antibody-dependent cellular cytotoxicity (ADCC) and similar pathways but also to the regulation of the immune microenvironment by blocking the corresponding signalling pathway, reversing immunosuppression and enhancing the activity of antitumour effector cells. mAbs could even be used for the delivery of various therapeutic reagents (Table 1).
In this chapter, we focus on mAb therapy directed against cancer and stromal cells. Mesothelin (MSLN) is extensively expressed in several solid tumours and in almost 100% of PDAC cells [87]. MSLN plays a critical role in the development of pancreatic cancer, especially at an early stage, and in peritoneal metastasis by binding with its single ligand MUC16; however, the intracellular mechanism remains unclear [88]. Furthermore, overexpression of MSLN is associated with poor outcomes for PDAC patients [89]. Several preclinical and clinical trials of MSLN-targeted mAb-based therapy have been summarized by several reviews [90,91,92]. In brief, the functional mechanisms of anti-MSLN mAb include not only ADCC but also alteration of intracellular signalling in cancer cells through endocytosis. This phenomenon has been exploited to deliver cytotoxins to kill cancer cells [93]. Anti-MSLN antibodies can also block the binding of MSLN with MUC16 and inhibit the expansion and metastasis of cancer cells [88]. MORAb-009 is a humanized antibody known as amatuximab. Baldo demonstrated that amatuximab exerts therapeutic efficacy by inducing ADCC and inhibiting the binding of MSLN with MUC16 [94]. Hassan, Fujisaka and their colleagues successively reported two phase I clinical studies including PDAC and other solid tumours expressing MSLN. They demonstrated the safety of amatuximab but observed no apparent objective responses despite stable disease occurring in some patients [95, 96].
MUC1 is restricted to apical surface expression on normal epithelial cells [97] and is overexpressed in approximately 90% of PDAC cells [98] on the basolateral membrane [97]. Biochem and colleagues demonstrated that an antibody similar to the anti-MUC1 antibody GP1.4 could trigger the internalization of EGFR on PDAC cells. This process could inhibit ERK signalling and result in the inhibition of cancer cell proliferation and migration [78], but the mechanism was unclear. Wu et al. [79] recently reported that MUC1-C, an isoform of MUC1, was highly expressed in 60.6% of human PDAC tissue samples compared to normal tissue samples. They used the same anti-MUC1 antibody on human pancreatic cell lines and a xenograft mouse model and demonstrated that the anti-hMUC1 antibody could pass through the membrane, inactivate MUC1 signalling and then suppress tumour growth in vivo. Since GP1.4 can be internalized by cancer cells, whether it can be exploited as a carrier of a cytotoxin would be an interesting investigation.
VEGF can promote vascularization in cancer lesions, and although PDAC does not have high vessel density, the cancer cells aberrantly express VEGF. This conclusion is supported by an early preclinical study that used the murine-derived anti-VEGF antibody A.4.6.1 to supress tumour growth [80]. Another anti-VEGF antibody, bevacizumab, has been the subject of multicentre-based investigations in combination with chemotherapy, but the results have not yet been published. Treatment combining the anti-EGFR antibody erlotinib with Gem was recently carefully assessed, and mild efficacy and tolerable adverse effects were concluded (Table 1) [99, 100].
AnxA6 is expressed in almost all PDACs by CAFs and localizes at the invasive front of the tumours, where it forms a complex structure with LDL receptor-related protein 1 and thrombospondin and participates in crosstalk between cancer cells and the stroma. The structure has shown strong correlations with cancer cell survival and perineural invasion [101]. O’Sullivan et al. isolated a novel antibody against AnxA6, 9E1, and demonstrated in an ex vivo experiment that the antibody could reduce the invasive capacity of pancreatic cancer cells by reducing MMP-9 expression and modulating ERK and MEK signalling [81].
Delta-like ligand 4 (DLL4) may be another possible mAb target for PDAC treatment since the DLL4 signalling pathway is important for PDAC cancer stem cell (CSC) survival. Demcizumab is a humanized anti-DLL4 antibody that has the potential to reverse chemotherapy resistance, and a study showed that demcizumab combined with paclitaxel and Gem was safe but not efficacious [82]. Two clinical trials on the use of demcizumab for PDAC treatment were completed recently, but the results have not yet been published (Table 1).
Antibodies or antibody fragments can also be conjugated with radioisotopes to deliver localized radiotherapy, known as radioimmunotherapy, and is emerging as an important selection for PDAC patients [83]. Recently, CD147 [84] and B7-H3 [85] were explored as targets of radioimmunotherapy for cancer cells and CSCs, respectively, with a 90Y-labelled antibody (059–053) and a 212Pb-labelled antibody (376.96) and investigated in preclinical experiments; both achieved promising results and demonstrated potential therapeutic efficacy for PDAC (Table 1).
Mutation of the Kras gene may be a promising target for mAbs in PDAC since more than 90% of PDAC cases bear a mutation at position G12 [102]. In a pilot study, Meng et al. demonstrated that tumour-infiltrating B cell (TIB)-derived IgGs could recognize most G12 mutations occurring in PDAC and noted that TIBs might be a source of antitumour antibodies targeting neoantigens [86]. This study established a novel way to produce neoantigen-targeting antibodies for personalized mAb immunotherapy.
Strategies reversing immunosuppressive mechanisms
ICI therapy
Only approximately 4% of all PDAC cells, including cancer cells (5.5% ± 1.1), CD163+ TAMs (9.3% ± 3.6) and CAFs, express PD-L1 [38]. Although the majority of PDAC cases show intermediate to high numbers of infiltrating T cells, CD4+ T cells, rather than CD8+ T cells, are the main component [38, 42]. The objective response of malignancy to ICI therapy is positively associated with the mutational burden, which is relatively low in PDAC [103, 104]. All of these factors indicate a dismal response to ICI therapy by PDAC compared to other solid tumours [104,105,106,107]. Investigators are trying to improve the effect of ICI therapy through different approaches. GM-CSF-secreting tumour cells (GVAX) can significantly upregulate PD-L1 expression and improve the effect of anti-CTLA-4 and anti-PD-1/PD-L1 antibodies [17, 108]. Oncolytic virotherapy [109], chemotherapy and radiotherapy [110, 111], a CSF1 blockade [57], an anti-IL-6 antibody [112], a CXCL12/CXCR4 axis inhibitor and stromal cell depletion [113] have also been tested to enhance the efficacy of ICI therapy on PDAC. Among these efforts, the combination of ICI therapy and chimeric antigen receptor (CAR) T cell infusion may hold the most promise [114, 115], as this strategy can simultaneously increase the number of tumour-targeting effector cells and prevent infused cell anergy.
Strategies targeting immunosuppressive cells
Treg cells
Chemotherapy reverses immunological tolerance for a prolonged period [116], and the mechanism was demonstrated by selectively depleting Treg cells [117]. Cyclophosphamide (Cy) is the most commonly used agent to deplete Treg cells to enhance cytotoxic and helper T cell responses [118]. Treg cells lack the ATP-binding cassette (ABC) transporter, which can extrude Cy out of cells, causing Treg cells to be more susceptible to Cy than other T cells [119]. Gem is another chemotherapeutic drug selectively capable of depleting Treg cells. Shevchenko et al. observed that in a mouse model, the depletion of local Treg cells with a low dose of Gem significantly improved the modest survival rate without affecting tumour growth or metastasis [67]. While Beatty et al. demonstrated that the depletion of Treg cells in the peripheral blood did not affect the Treg cell frequency in the tumour lesion and had no effect on tumour progression, a CD40 agonist used in combination with Gem decreased the Treg cell numbers and the accumulation of CD4+ and/or CD8+ cells in xenograft and/or orthotopic tumours [110], indicating that Gem, which can deplete tumour-infiltrating Treg cells, may restore the antitumour effects of CD40 agonists and ICIs. These results suggested that tumour-infiltrating Treg cells rather than circulating Treg cells accounted for the overall Treg function; targeting local proliferating/accumulating Treg cells but not peripheral Treg cells might be more advantageous and have fewer adverse effects on the immune system. Treg cell depletion can also enhance the effect of a PDAC vaccine. Lei Zheng and colleagues treated PDAC patients with a low dose of Cy in combination with GVAX and observed Cy-dependent Treg cell depletion and lymphoid aggregate formation in the PDAC TME. In addition, decreased Treg cell numbers in the lymphoid aggregates not only enhanced existing effector T cell activation but also facilitated more effector T cell trafficking into PDAC tumours [120]. Even premalignant PanIN lesions could benefit from Treg cell depletion; Treg cell depletion combined with the LM-Kras vaccine (attenuated Listeria monocytogenes strain expressing KrasG12D) could recruit CD4+ and CD8+ effector T cells to the premalignant lesion and inhibit PanIN progression. This strategy could also enhance the recruitment of Gr-1+ cells but repolarize them into an antitumour phenotype to enable cytokine production and the induction of an inflammatory response [121]. This study further verified the tight correlation between Treg cells and MDSCs.
MDSCs and TAMs
The subtle distinction between Gr-MDSCs and Mo-MDSCs should be noted. In a preclinical study to test the potential of targeting MDSCs, Stromnes et al. demonstrated an extensive effect of depleting Gr-MDSCs on the prognosis of PDAC patients and determined the rational mechanism. They selectively depleted Gr-MDSCs with the anti-Ly6G mAb 1A8. Compared with untreated mice, treated mice showed a 4- to 5-fold increase in Mo-MDSC numbers in the spleen and PDAC lesions, and the gross number of tumour-infiltrating CD45+ cells increased approximately 2-fold in 1A8-treated mice [59]. Further study indicated that the numbers of proliferating and activated CD8+ T cells with high granzyme B levels increased absolutely, and these cells were found in not only the stroma but also in the proximity of tumour cells. Decreased stromal matrix deposition and integrity, increased caspase-3-positive tumour cell numbers and blood vessels were observed in 1A8-treated tumours [59]. There was no observed reduction in tumour size due to an influx of tumour-reactive effector cells, a phenomenon known as tumour pseudoprogression [122]. The compensatory increase in Mo-MDSCs synchronized with the depletion of Gr-MDSCs was remarkable, and a similar result was reported in another study in which the decrease in TAMs/Mo-MDSCs was accompanied by an increase in Gr-MDSCs. The checks and balances between Gr-MDSCs and Mo-MDSCs may indicate some therapeutic value; although these cells share some similar phenotypic molecules and show similar suppressive functions, these two myeloid cell subsets might have very distinct final fates and should be handled separately. TAMs are a pool of cells with heterogeneous functions and phenotypes, and their versatile plasticity allows their transformation into each other according to the local conditions. Both the CSF1/CSF1R and CCL2/CCR2 axes are critical for the accumulation and differentiation of TAMs from their progenitors in the blood. A CSF1/CSF1R blockade can not only decrease the number of TAMs in PDAC lesions but also reprogram TAMs to enhance their antigen-presenting ability, resulting in enhanced antitumour T cell responses [57]. In a contemporary preclinical study [123], Mitchem et al. investigated an axis-targeting treatment combined with chemotherapy and demonstrated that CCR2 and/or CSF1R inhibitors displayed only modest effects. Gem alone could increase the number of TAMs in PDAC lesions, and CCR2 and/or CSF1R inhibitors could reverse this increase and dramatically reduce tumour masses. In addition, the researchers observed significant CD4+ and CD8+ T cell infiltration and decreased Treg cell infiltration after treatment. Remarkably, they found that a CCR2 and/or CSF1R blockade could decrease the numbers of both TAM and Mo-MDSC, which was potentially the result of a phenotypic overlap between these two monocyte subsets. However, a modest increase in Gr-MDSC numbers was observed, which was potentially due to a compensatory relationship between the two types of MDSCs. Specifically, blocking either CCR2 or CSF1R could disrupt this interaction and reverse chemotherapy resistance [123]. TAMs generally localize at the invasive front of PDAC lesions and are involved in angiogenesis and EMT, which are important for cancer cell invasion and metastasis. Investigations of methods to reverse or inhibit this function of TAMs would be interesting.
Strategies enhancing the antitumour response
Costimulatory molecule agonists
In a pilot study, Beatty et al. demonstrated an unexpected function of a CD40 agonist, as treated F4/80+ macrophages in the peripheral blood were activated and infiltrated tumour lesions. However, although the expected T lymphocyte infiltration was not observed, the PDAC stroma was destroyed, and cancer cells were killed by the infiltrating macrophages [124]. The researchers further demonstrated that this agonist of CD40 upregulated the expression of MHC class II and CD86, suggesting an enhanced antigen-presenting ability of the macrophages. Nevertheless, T cells did not infiltrate tumours and remained in the peripancreatic lymph nodes adjacent to the tumours, suggesting that an additional mechanism excluded these antitumour effector cells. In a subsequent study [125], the same team found that the agonist of CD40 induced heavy T cell infiltration into tumours upon combination with Gem and resulted in CD4+ and/or CD8+ T cell-dependent tumour regression. They explained the controversial results by concluding that circulating macrophages may have dual roles in regulating immunoreactivity in PDAC but did not interpret the role of Gem in the treatment. Gem combined with the CD40 agonist could induce tumour regression even after circulating macrophages were depleted [125]. This result suggested that the chemotherapeutic agent in the experiment targeted some unknown immunosuppressive cells that could exclude effector T cells. Rationally, these cells were probably Treg cells since Gem has been demonstrated to be a potent Treg cell-depleting agent in PDAC [67]. In a multicentre phase I clinical study by Beatty and his collaborators, an agonistic anti-CD40 antibody was applied in combination with Gem for PDAC treatment; while only a mild effect was observed, the safety of the combination was established [126]. In addition, the CD40 agonist and Gem combination could also reverse resistance to ICI therapy via promoting the accumulation of robust antitumour CD8+ T cells in PDAC tumours [110]. These results potentially demonstrate that the combination of reprogramming macrophages to enhance their antigen-presenting ability with Treg cell depletion and ICI administration is a promising approach. The stromal destruction observed with both Gr-MDSC depletion (increase in tumour-infiltrating Mo-MDSC numbers) [59] and TAM reprogramming [124] indicates that Mo-MDSCs and TAMs share an overlapping role.
ACT
ACT is a very active field of investigation in PDAC immunotherapy and is performed using lymphocytes with or without gene editing and TILs (Table 2). Substantial progress has been made over the last three years regarding PDAC.
ACT with genetically engineered cells
CAR-engineered T cell (CAR-T) ACT for PDAC was very recently thoroughly reviewed [127,128,129,130,131]. Various artificial gene-design strategies targeting the cancer stroma and overcoming immunosuppressive factors have been explored to improve the effect of CAR-T ACT on PDAC. Rataj et al. genetically engineered ovalbumin (OVA)-specific CD4+ and CD8+ T cells with a PD-1-CD28 fusion protein. They observed significant synergy between the two cell populations correlating with the number of CD4+ T cells, indicating that the PD-1/PD-L1 suppressive signal was reversed and that the helper function of CD4+ T cells and antitumour effect of CD8+ T cells were enhanced [132]. Mohammed et al. performed a similar experiment [133] wherein they engineered the T cell population with two genes simultaneously, a first-generation PSCA-specific CAR and an inverted cytokine receptor (ICR) with an IL-4 extracellular domain and an IL-7 intracellular domain to yield CAR/ICR T cells. CAR/ICR T cells could reverse the IL-4-derived inhibitory signal to the T cell proliferation signal and showed enhanced antitumour activity. Genetically engineered TCR T cell (TCR-T) infusion is another ACT strategy. Stromnes et al. conducted ground-breaking research in this field, wherein a series of pilot and extensive experiments generated valuable data [134]. They screened a TCR for an endogenous unmutated MSLN epitope, which worked in an MHC class I-independent manner. TCR-Ts accumulated preferentially in orthotopic PDAC lesions and induced cancer cell death as well as stromal remodelling. Serial TCR-T infusion was performed, and improved survival was observed without increased toxicity [134].
TILs and neoantigens
CD3+ T cells were shown to comprise up to 90% of all tumour-infiltrating cells [41] and for almost all CD45RO+ memory cells [38, 42,43,44]. Recently, Hall and Meng reported the successful extraction of TILs from PDAC specimens and the expansion of these cells in vitro [135, 136]. However, they used different protocols to isolate and expand the TILs from tumour fragments. Hall et al. used medium containing a high dose of IL-2 and obtained TILs composed primarily of CD4+ T cells, while Meng et al. cultured fragments with medium containing the cytokines IL-2, IL-15, and IL-21 and expanded TILs composed primarily of CD8+ T cells. Both research teams demonstrated autologous tumour cell killing activity in an HLA-dependent manner. In a pilot study [42], Poschke et al. observed clonal tumour-reactive T cell expansion in PDAC, and they isolated and expanded TILs with a success rate similar to that achieved in melanoma. The authors reported that ex vivo culture appeared to reverse the exhausted phenotype of the freshly isolated TILs, but the proportion of tumour-reactive T cells was very low in the final pools, and these cells displayed no effect against an autologous PDAC xenograft. The researchers further interpreted the phenomenon of TCR repertoire alteration during ex vivo expansion. The regulatory cells within TIL populations should be carefully considered because they may exist in the fragment culture for a long time and bias the nonspecific expansion of TILs. Since TCR repertoire alteration might be the major hurdle for TIL treatment in PDAC, the identification of tumour-specific TCRs and/or TIL clones may be an alternative approach. In a very recent study, Meng et al. reported the production of three TIL cell lines and two autologous tumour cell lines; they screened, sequenced and synthesized mutation-derived neopeptides and observed neoantigen-specific tumour killing in an HLA-dependent fashion. They demonstrated the presence of neoantigen-specific TIL clones in both CD8+ and CD4+ T cell pools, which functioned in HLA class I- and HLA class II-dependent manners, respectively. Importantly, they reported that peripheral blood mononuclear cells (PBMCs) as well as TILs could be used to screen neoantigens. These results pave the way for highly specific and personalized ACT [137] since targeting personalized mutations has been demonstrated to be a durable approach for the treatment of metastatic solid tumours with a relatively low mutation burden [138].
Vaccines
The vaccines used for PDAC therapy are diverse and employ very different mechanisms (Table 3). Briefly, there are three major vaccine platforms for PDAC: DC-based vaccines, tumour cell-based vaccines and bacterium-based vaccines. DCs are the most common platform, and DC-based vaccines have been tested in numerous clinical trials and thoroughly reviewed [139, 140]. Another PDAC vaccine platform is the whole-tumour cell vaccine platform using autologous and/or allogeneic cancer cells with or without genetic editing. GVAX is a whole-cell vaccine system used extensively for treating various cancers, including PDAC. GVAX vaccines for PDAC are derived from two pancreatic cancer cell lines engineered with the GM-CSF gene; these vaccines can be injected intradermally and secrete high levels of GM-CSF to attract APCs and promote their maturation. The vaccines have been demonstrated to be safe but to have modest effects [141, 142]. It should be noted that GM-CSF alone is not sufficient for APC maturation, and the simultaneous presence of IL-4 is indispensable. Algenpantucel-L is another whole-cell vaccine consisting of two pancreatic cancer cell lines genetically engineered to express α-galactosyl (α-gal) epitopes on membrane glycoproteins and glycolipids [143]; these epitopes are not expressed in human cells [144] and induce complement- and antibody-dependent cytotoxicity since there are large amounts of anti-α-gal antibodies in human serum [145]. Algenpantucel-L combined with chemotherapy moderately improved the 1-year OS rate of patients with resectable PDAC without severe adverse effects [143]. Tanemura and Doki et al. subsequently produced whole-cell vaccines expressing α-gal epitopes based on cancer cell lines and tumour lysates separately and demonstrated therapeutic potency in preclinical studies; notably, both vaccines could target both cancer cells and CSCs [146, 147]. Recently, a bacterium-based vaccine, CRS-207, was developed that comprises a recombinant live-attenuated Listeria monocytogenes strain engineered to secrete MSLN into the cytoplasm of infected APCs. This strategy could not only enhance the ability of APCs but also target an antigen universally expressed by PDAC. It has been demonstrated to be safe, and the combination of GVAX and CRS-207 has shown a survival benefit [148, 149]. The fact that the epitopes used to enhance effector cell antitumour reactivity can also be presented to Treg cells and result in tumour-specific immune tolerance is an important phenomenon that should be emphasized and can be used to interpret the mild effect of whole-cell and DC vaccines [150, 151]. How to overcome suppressive cells, especially tumour antigen-specific Treg cells, is a critical issue that needs to be resolved [152].
Conclusion
In this review, we summarized the characteristics of the PDAC TME, including the cancer epithelial cell properties, the role of stromal cells and matrix in the immunosuppressive TME, the complex network among tumour-infiltrating immune cells and how these cells orchestrate the shape and programme of the PDAC TME. We have also covered the current and future aspects of immunotherapy for PDAC from various perspectives in this review. mAb-based immunotherapy still has the potential to enhance the treatment of PDAC. However, the absence of TAAs restricts its progression, and the strategy for improving the suboptimal selection of mAb-based therapy involves combinations with other approaches or exploration of TSAs, especially neoantigen-targeting mAbs, from TIBs [86], as the latter is emerging as a promising field. Vaccines may have dual roles in the treatment of PDAC. On the one hand, they can theoretically induce or enhance the naturally occurring antitumour response and improve the functions of transferred antitumour effector cells. However, they may have the adverse effect of inducing tumour-specific immune tolerance through Treg cells, which at least in part underlies the modest effect observed with vaccine treatment. For GVAX vaccines, GM-CSF alone might not be sufficient to induce APC maturation. Recent advances in isolating neoantigen-targeting antibodies from TIBs have given rise to a promising approach for both vaccine and mAb therapies as well as for selecting scFvs for CAR-T therapy. ACT with genetically engineered cells has achieved promising results in some solid tumours in preclinical studies but not in any clinical trials. ACT-based therapy must be immensely improved to exploit PDAC-targeting cells because PDAC has relatively few TAAs. Furthermore, the high stromal density and absence of angiogenesis dampen the infiltration of infused cells, and the suppressive TME also inactivates infiltrating cells. Promisingly, substantial progress has been made regarding PDAC TILs in recent years [135,136,137]. These results exploited potential tools to obtain multiple tumour-specific colonies and even a single TIL colony specific for endogenous tumour cells. The strategy of identifying and sequencing neoantigen-specific TCRs to engineer lymphocytes for ACT is expected, as Rosenberg and his team have made significant progress in this field [153,154,155].
Availability of data and materials
Not applicable.
Abbreviations
- ABC:
-
ATP-binding cassette
- ACT:
-
Adoptive cell therapy
- ACT:
-
Adoptive cell therapy/adoptive cell transferring
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- APCs:
-
Antigen-specific cells
- Arg1:
-
Arginase-1
- CAFs:
-
Cancer-associated fibroblasts
- CAR:
-
Chimeric antigen receptor
- CEA:
-
Carcinoembryonic antigen
- CSCs:
-
Cancer stem cells
- CTLA-4:
-
Cytotoxic T lymphocyte-associated antigen 4
- Cy:
-
Cyclophosphamide
- DCs:
-
Dendritic cells
- DLL4:
-
Delta-like ligand 4
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-to-mesenchymal transition
- Gem:
-
Gemcitabine
- GM-CSF/CSF2:
-
Granulocyte macrophage colony-stimulating factor
- GVAX:
-
GM-CSF-secreting tumour cells
- HEV:
-
High endothelial venules
- ICI:
-
Immune checkpoint inhibitor
- ICI:
-
Immune checkpoint inhibitor
- IDO:
-
Indoleamine 2,3-dioxygenase
- iNOS:
-
Inducible nitric oxide synthase
- mAb:
-
Monoclonal antibody
- M-CSF/CSF1:
-
Macrophage colony-stimulating factor
- MDSCs:
-
Myeloid-derived suppressive cells
- MSLN:
-
Mesothelin
- OS:
-
Overall survival
- OVA:
-
Ovalbumin
- PanIN:
-
Pancreatic intraepithelial neoplasia
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PD-L1:
-
Programmed cell death protein ligand-1
- PSCs:
-
Pancreatic stellate cells
- ROS:
-
Reactive oxygen species
- TAAs:
-
Tumours associated antigens
- TAMs:
-
Tumour-associated macrophages
- TANs:
-
Tumour-associated neutrophils
- Tconv:
-
Conventional T cells
- TGF-β:
-
Transforming growth factor-β
- TIBs:
-
Tumour-infiltrating B cells
- TILs:
-
Tumour-infiltrating lymphocytes
- TLS:
-
Tertiary lymphoid structures
- TME:
-
Tumour microenvironment
- Treg cells:
-
Regulatory T cells
- TSAs:
-
Tissue-specific antigens
- VEGF:
-
Vascular endothelial growth factor
References
Paulson AS, Cao HST, Tempero MA, Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013;144(6):1316–26.
Siegel R, Naishadham D, Jemal A. Cancer statistics. 2012. CA Cancer J Clin. 2012;62:10–29.
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21.
Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318–48.
Neoptolemos JP. Adjuvant treatment of pancreatic cancer. Eur J Cancer. 2011;47:S378–80.
Dhir M, Malhotra GK, Sohal DP, Hein NA, Smith LM, O’Reilly EM, Bahary N, Are C. Neoadjuvant treatment of pancreatic adenocarcinoma: a systematic review and meta-analysis of 5520 patients. World J Surg Oncol. 2017;15(1):183.
Howlader N, Noone A, Krapcho M, Garshell J, Neyman N, Altekruse S, Kosary C, Yu M, Ruhl J, Tatalovich Z. SEER Cancer Statistics Review, 1975–2012, National Cancer Institute; 2015; 2016.
McBride A, Bonafede M, Cai Q, Princic N, Tran O, Pelletier C, Parisi M, Patel M. Comparison of treatment patterns and economic outcomes among metastatic pancreatic cancer patients initiated on nab-paclitaxel plus gemcitabine versus FOLFIRINOX. Expert Rev Clin Pharmacol. 2017;10(10):1153–60.
Kang J, Hwang I, Yoo C, Kim K-p, Jeong JH, Chang H-M, Lee SS, Park DH, Song TJ, Seo DW, et al. Nab-paclitaxel plus gemcitabine versus FOLFIRINOX as the first-line chemotherapy for patients with metastatic pancreatic cancer: retrospective analysis. Investig New Drugs. 2018;36(4):732–41.
Ryschich E, Nötzel T, Hinz U, Autschbach F, Ferguson J, Simon I, Weitz J, Fröhlich B, Klar E, Büchler MW, et al. Control of T-cell–mediated immune response by HLA class I in human pancreatic carcinoma. Clin Cancer Res. 2005;11(2):498–504.
Pandha H, Rigg A, John J, Lemoine N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin Exp Immunol. 2007;148(1):127–35.
Birnbaum DJ, Finetti P, Lopresti A, Gilabert M, Poizat F, Turrini O, Raoul JL, Delpero JR, Moutardier V, Birnbaum D, et al. Prognostic value of PDL1 expression in pancreatic cancer. Oncotarget. 2016;7(44):71198–210.
Wv B, Spanjaard RA, Chan AK, Lockhart DC, Sadanaga N, Wood I, Peiper M, Goedegebuure PS, Eberlein TJ. Pancreatic cancer cells can evade immune surveillance via nonfunctional Fas (APO-1/CD95) receptors and aberrant expression of functional Fas ligand. Surgery. 1999;125(1):73–84.
Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19(13):3404–15.
Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh C-S, Linehan DC. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182(3):1746–55.
Wang X, Lang M, Zhao T, Feng X, Zheng C, Huang C, Hao J, Dong J, Luo L, Li X, et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3(+)Treg cells in pancreatic ductal adenocarcinoma. Oncogene. 2017;36(21):3048–58.
Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, Wamwea A, Bigelow E, Lutz E, Liu L, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38(1):1–11.
Basso D, Fogar P, Falconi M, Fadi E, Sperti C, Frasson C, Greco E, Tamburrino D, Teolato S, Moz S. Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: role of inhibitory co-stimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study. PloS one. 2013;8(1):e54824.
Bellone G, Turletti A, Artusio E, Mareschi K, Carbone A, Tibaudi D, Robecchi A, Emanuelli G, Rodeck U. Tumor-associated transforming growth factor-β and interleukin-10 contribute to a systemic Th2 immune phenotype in pancreatic carcinoma patients. Am J Pathol. 1999;155(2):537–47.
Linehan DC, Goedegebuure PS. CD25+ CD4+ regulatory T-cells in cancer. Immunol Res. 2005;32(1–3):155–68.
Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2, 3-dioxygenase. Nat Med. 2003;9(10):1269–74.
Peng Y-P, Zhang J-J, Liang W-b, Tu M, Lu Z-P, Wei J-S, Jiang K-R, Gao W-T, Wu J-L, Xu Z-K, et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer. 2014;14(1):738.
Neesse A, Algül H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut. 2015;64(9):1476–84.
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–76.
Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68(3):918–26.
Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012;11(7):1282–90.
Wörmann S, Diakopoulos K, Lesina M, Algül H. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33(23):2956–67.
Zheng L, Xue J, Jaffee EM, Habtezion A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology. 2013;144(6):1230–40.
Knudsen ES, Vail P, Balaji U, Ngo H, Botros IW, Makarov V, Riaz N, Balachandran V, Leach S, Thompson DM, et al. Stratification of pancreatic ductal adenocarcinoma: combinatorial genetic, stromal, and immunologic markers. Clin Cancer Res. 2017;23(15):4429–40.
Wang LM, Silva MA, D'Costa Z, Bockelmann R, Soonawalla Z, Liu S, O'Neill E, Mukherjee S, McKenna WG, Muschel R, et al. The prognostic role of desmoplastic stroma in pancreatic ductal adenocarcinoma. Oncotarget. 2016;7(4):4183–94.
Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–34.
Shi C, Washington MK, Chaturvedi R, Drosos Y, Revetta FL, Weaver CJ, Buzhardt E, Yull FE, Blackwell TS, Sosa-Pineda B, et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab Investig. 2014;94(4):409–21.
Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145(5):1121–32.
Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J Exp Med. 1996;184(3):1101–9.
Tang D, Gao J, Wang S, Yuan Z, Ye N, Chong Y, Xu C, Jiang X, Li B, Yin W, et al. Apoptosis and anergy of T cell induced by pancreatic stellate cells-derived galectin-1 in pancreatic cancer. Tumour Biol. 2015;36(7):5617–26.
Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–27.
Felix K, Gaida MM. Neutrophil-derived proteases in the microenvironment of pancreatic cancer-active players in tumor progression. Int J Biol Sci. 2016;12(3):302–13.
Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol Res. 2017;5(11):978–91.
Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–35.
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–47.
Ademmer K, Ebert M, Muller-Ostermeyer F, Friess H, Buchler MW, Schubert W, Malfertheiner P. Effector T lymphocyte subsets in human pancreatic cancer: detection of CD8+CD18+ cells and CD8+CD103+ cells by multi-epitope imaging. Clin Exp Immunol. 1998;112(1):21–6.
Poschke I, Faryna M, Bergmann F, Flossdorf M, Lauenstein C, Hermes J, Hinz U, Hank T, Ehrenberg R, Volkmar M, et al. Identification of a tumor-reactive T-cell repertoire in the immune infiltrate of patients with resectable pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5(12):e1240859.
Emmrich J, Weber I, Nausch M, Sparmann G, Koch K, Seyfarth M, Löhr M, Liebe S. Immunohistochemical characterization of the pancreatic cellular infiltrate in normal pancreas, chronic pancreatitis and pancreatic carcinoma. Digestion. 1998;59(3):192–8.
Shibuya KC, Goel VK, Xiong W, Sham JG, Pollack SM, Leahy AM, Whiting SH, Yeh MM, Yee C, Riddell SR, et al. Pancreatic ductal adenocarcinoma contains an effector and regulatory immune cell infiltrate that is altered by multimodal neoadjuvant treatment. PLoS One. 2014;9(5):e96565.
Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, Kato K, Kurokawa T, Suzuoki M, Nakakubo Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28(1):e26–31.
Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, Hiraoka N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Brit J Cancer. 2013;108(4):914–23.
Tassi E, Gavazzi F, Albarello L, Senyukov V, Longhi R, Dellabona P, Doglioni C, Braga M, Di Carlo V, Protti MP. Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J Immunol. 2008;181(9):6595–603.
Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. 2017;20(3):558–71.
Bellone G, Carbone A, Smirne C, Scirelli T, Buffolino A, Novarino A, Stacchini A, Bertetto O, Palestro G, Sorio C, et al. Cooperative induction of a tolerogenic dendritic cell phenotype by cytokines secreted by pancreatic carcinoma cells. J Immunol. 2006;177(5):3448–60.
Bharadwaj U, Li M, Zhang R, Chen C, Yao Q. Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res. 2007;67(11):5479–88.
Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B, Lee B, Lee T, Mak SY, Ho YS, et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5(8):e1191731.
Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, Takao S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011;167(2):e211–9.
Zeng L, Guo Y, Liang J, Chen S, Peng P, Zhang Q, Su H, Chen Y, Huang K. Perineural invasion and TAMs in pancreatic ductal adenocarcinomas: review of the original pathology reports using immunohistochemical enhancement and relationships with clinicopathological features. J Cancer. 2014;5(9):754–60.
Kurahara H, Takao S, Maemura K, Mataki Y, Kuwahata T, Maeda K, Sakoda M, Iino S, Ishigami S, Ueno S, et al. M2-polarized tumor-associated macrophage infiltration of regional lymph nodes is associated with nodal lymphangiogenesis and occult nodal involvement in pN0 pancreatic cancer. Pancreas. 2013;42(1):155–9.
Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70(19):7465–75.
Dineen SP, Lynn KD, Holloway SE, Miller AF, Sullivan JP, Shames DS, Beck AW, Barnett CC, Fleming JB, Brekken RA. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008;68(11):4340–6.
Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69.
Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, Schmid MC, Sun P, Mose E, Bouvet M, et al. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016;6(8):870–85.
Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, Hingorani SR. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769–81.
Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017;24(3):100–5.
Yang R, Cai Z, Zhang Y, Yutzy WH, Roby KF, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma-associated gr-1+CD11b+ myeloid cells. Cancer Res. 2006;66(13):6807–15.
Zheng Y, Manzotti CN, Liu M, Burke F, Mead KI, Sansom DM. CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J Immunol. 2004;172(5):2778–84.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74.
Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164(1):183–90.
Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10(12):1969–80.
Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y, Ni B, Lu B, Wang H. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS One. 2014;9(3):e91551.
Shevchenko I, Karakhanova S, Soltek S, Link J, Bayry J, Werner J, Umansky V, Bazhin AV. Low-dose gemcitabine depletes regulatory T cells and improves survival in the orthotopic Panc02 model of pancreatic cancer. Int J Cancer. 2013;133(1):98–107.
Liyanage UK, Moore TT, Joo H-G, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169(5):2756–61.
Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, Albert J, Sparwasser T, Sakaguchi S, Westendorf AM, et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med. 2012;209(11):2001–16.
Lunardi S, Lim SY, Muschel RJ, Brunner TB. IP-10/CXCL10 attracts regulatory T cells: implication for pancreatic cancer. Oncoimmunology. 2015;4(9):e1027473.
Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, Cerwenka A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol. 2012;189(12):5602–11.
Bengsch F, Knoblock DM, Liu A, McAllister F, Beatty GL. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol Immun. 2017;66(12):1609–17.
Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–3.
Walker LS, Chodos A, Eggena M, Dooms H, Abbas AK. Antigen-dependent proliferation of CD4+ CD25+ regulatory T cells in vivo. J Exp Med. 2003;198(2):249–58.
Yamazaki S, Iyoda T, Tarbell K, Olson K, Velinzon K, Inaba K, Steinman RM. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J Exp Med. 2003;198(2):235–47.
Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000;192(9):1213–22.
Jonuleit H, Schmitt E, Steinbrink K, Enk AH. Dendritic cells as a tool to induce anergic and regulatory T cells. Trends Immunol. 2001;22(7):394–400.
Hisatsune A, Nakayama H, Kawasaki M, Horie I, Miyata T, Isohama Y, Kim KC, Katsuki H. Anti-MUC1 antibody inhibits EGF receptor signaling in cancer cells. Biochem Bioph Res. 2011;405(3):377–81.
Wu G, Maharjan S, Kim D, Kim JN, Park BK, Koh H, Moon K, Lee Y, Kwon HJ. A novel monoclonal antibody targets Mucin1 and attenuates growth in pancreatic cancer model. Int J Mol Sci. 2018;19(7).
Bockhorn M, Tsuzuki Y, Xu L, Frilling A, Broelsch CE, Fukumura D. Differential vascular and transcriptional responses to anti-vascular endothelial growth factor antibody in orthotopic human pancreatic cancer xenografts. Clin Cancer Res. 2003;9(11):4221–6.
O'Sullivan D, Dowling P, Joyce H, McAuley E, McCann A, Henry M, McGovern B, Barham P, Kelleher FC, Murphy J, et al. A novel inhibitory anti-invasive MAb isolated using phenotypic screening highlights AnxA6 as a functionally relevant target protein in pancreatic cancer. Br J Cancer. 2017;117(9):1326–35.
Cubillo Gracian A, Dean A, Muñoz A, Hidalgo M, Pazo-Cid R, Martin M, Macarulla Mercade T, Lipton L, Harris M, Manzano-Mozo JL. 620PDYOSEMITE: A 3 arm double-blind randomized phase 2 study of gemcitabine, paclitaxel protein-bound particles for injectable suspension, and placebo (GAP) versus gemcitabine, paclitaxel protein-bound particles for injectable suspension and either 1 or 2 truncated courses of demcizumab (GAD). Ann Oncol. 2017;28(suppl_5).
Sahlin M, Bauden MP, Andersson R, Ansari D. Radioimmunotherapy-a potential novel tool for pancreatic cancer therapy? Tumour Biol. 2015;36(6):4053–62.
Sugyo A, Tsuji AB, Sudo H, Koizumi M, Ukai Y, Kurosawa G, Kurosawa Y, Saga T, Higashi T. Efficacy evaluation of combination treatment using gemcitabine and radioimmunotherapy with (90) Y-labeled fully human anti-CD147 monoclonal antibody 059–053 in a BxPC-3 xenograft mouse model of refractory pancreatic cancer. Int J Mol Sci. 2018;19(10).
Kasten BB, Gangrade A, Kim H, Fan J, Ferrone S, Ferrone CR, Zinn KR, Buchsbaum DJ. (212) Pb-labeled B7-H3-targeting antibody for pancreatic cancer therapy in mouse models. Nucl Med Biol. 2018;58:67–73.
Meng Q, Valentini D, Rao M, Maeurer M. KRAS RENAISSANCE(S) in tumor infiltrating B cells in pancreatic cancer. Front Oncol. 2018;8:384.
Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, Murugesan SR, Leach SD, Jaffee E, Yeo CJ, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. 2001;7(12):3862–8.
Avula LR, Rudloff M, El-Behaedi S, Arons D, Albalawy R, Chen X, Zhang X, Alewine C. Mesothelin enhances tumor vascularity in newly forming pancreatic peritoneal metastases. Mol Cancer Res. 2019; [Online ahead of print].
Montemagno C, Cassim S, Trichanh D, Savary C, Pouyssegur J, Pagès G, Fagret D, Broisat A, Ghezzi C. (99m)Tc-A1 as a novel imaging agent targeting mesothelin-expressing pancreatic ductal adenocarcinoma. Cancers. 2019;11(10):1531.
Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 2008;44(1):46–53.
Kelly RJ, Sharon E, Pastan I, Hassan R. Mesothelin-targeted agents in clinical trials and in preclinical development. Molc Cancer Ther. 2012;11(3):517–25.
Hassan R, Thomas A, Alewine C, Le DT, Jaffee EM, Pastan I. Mesothelin immunotherapy for cancer: ready for prime time? J Clin Oncol. 2016;34(34):4171–9.
Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immunotoxin treatment of cancer. Annu Rev Med. 2007;58:221–37.
Baldo P, Cecco S. Amatuximab and novel agents targeting mesothelin for solid tumors. Oncotargets Therapy. 2017;10:5337–53.
Hassan R, Cohen SJ, Phillips M, Pastan I, Sharon E, Kelly RJ, Schweizer C, Weil S, Laheru D. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin Cancer Res. 2010;16(24):6132–8.
Fujisaka Y, Kurata T, Tanaka K, Kudo T, Okamoto K, Tsurutani J, Kaneda H, Okamoto I, Namiki M, Kitamura C, et al. Phase I study of amatuximab, a novel monoclonal antibody to mesothelin, in Japanese patients with advanced solid tumors. Investig New Drugs. 2015;33(2):380–8.
Nath S, Mukherjee P. MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med. 2014;20(6):332–42.
Qu CF, Li Y, Song YJ, Rizvi SM, Raja C, Zhang D, Samra J, Smith R, Perkins AC, Apostolidis C, et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by (213) bi-C595 radioimmunoconjugate. Br J Cancer. 2004;91(12):2086–93.
Yang ZY, Yuan JQ, Di MY, Zheng DY, Chen JZ, Ding H, Wu XY, Huang YF, Mao C, Tang JL. Gemcitabine plus erlotinib for advanced pancreatic cancer: a systematic review with meta-analysis. PLoS One. 2013;8(3):e57528.
Wang Y, Hu GF, Zhang QQ, Tang N, Guo J, Liu LY, Han X, Wang X, Wang ZH. Efficacy and safety of gemcitabine plus erlotinib for locally advanced or metastatic pancreatic cancer: a systematic review and meta-analysis. Drug Des Dev Ther. 2016;10:1961–72.
Leca J, Martinez S, Lac S, Nigri J, Secq V, Rubis M, Bressy C, Serge A, Lavaut MN, Dusetti N, et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J Clin Invest. 2016;126(11):4140–56.
Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53(4):549–54.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, Stephens PJ, Daniels GA, Kurzrock R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–608.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–1.
Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–33.
Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, You L, Zheng L, Zhang T, Zhao Y. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017;407:57–65.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, Zheng L, Diaz LA Jr, Donehower RC, Jaffee EM, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36(7):382–9.
Ibrahim AM, Wang Y-h. Viro-immune therapy: a new strategy for treatment of pancreatic cancer. World J Gastroentero. 2016;22(2):748.
Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3(4):399–411.
Sindoni A, Minutoli F, Ascenti G, Pergolizzi S. Combination of immune checkpoint inhibitors and radiotherapy: review of the literature. Crit Rev Oncol Hematol. 2017;113:63–70.
Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, Nordquist E, Cruz-Monserrate Z, Yu L, Young G, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018;67(2):320–32.
Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. P Natl Acad Sci USA. 2013;110(50):20212–7.
Chen N, Morello A, Tano Z, Adusumilli PS. CAR T-cell intrinsic PD-1 checkpoint blockade: a two-in-one approach for solid tumor immunotherapy. OncoImmunology. 2017;6(2):e1273302.
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–44.
Turk JL, Parker D. Effect of cyclophosphamide on immunological control mechanisms. Immunol Rev. 1982;65:99–113.
Madondo MT, Quinn M, Plebanski M. Low dose cyclophosphamide: mechanisms of T cell modulation. Cancer Treat Rev. 2016;42:3–9.
Heylmann D, Bauer M, Becker H, van Gool S, Bacher N, Steinbrink K, Kaina B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: implications for the immune response. PLoS One. 2013;8(12):e83384.
Dimeloe S, Frick C, Fischer M, Gubser PM, Razik L, Bantug GR, Ravon M, Langenkamp A, Hess C. Human regulatory T cells lack the cyclophosphamide-extruding transporter ABCB1 and are more susceptible to cyclophosphamide-induced apoptosis. Eur J Immunol. 2014;44(12):3614–20.
Lutz E, Wu A, Bigelow E, Sharma R, Mo G, Soares K, Solt S, Dorman A, Wamwea A, Yager A. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–31.
Keenan BP, Saenger Y, Kafrouni MI, Leubner A, Lauer P, Maitra A, Rucki AA, Gunderson AJ, Coussens LM, Brockstedt DG, et al. A Listeria vaccine and depletion of T-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology. 2014;146(7):1784–1794.e6.
Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbe C, Maio M, Binder M, Bohnsack O, Nichol G, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–20.
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–41.
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6.
Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, Clendenin C, Gladney WL, Knoblock DM, Guirnalda PD, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6C(low) F4/80(+) extratumoral macrophages. Gastroenterology. 2015;149(1):201–10.
Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, Troxel AB, Sun W, Teitelbaum UR, Vonderheide RH, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19(22):6286–95.
DeSelm CJ, Tano ZE, Varghese AM, Adusumilli PS. CAR T-cell therapy for pancreatic cancer. J Surg Oncol. 2017;116(1):63–74.
Akce M, Zaidi MY, Waller EK, El-Rayes BF, Lesinski GB. The potential of CAR T cell therapy in pancreatic cancer. Front Immunol. 2018;9:2166.
Liu F, Saif MW. T cell optimization for the treatment of pancreatic cancer. Expert Opin Biol Th. 2017;17(12):1493–501.
Alrifai D, Sarker D, Maher J. Prospects for adoptive immunotherapy of pancreatic cancer using chimeric antigen receptor-engineered T-cells. Immunopharm Immunot. 2016;38(1):50–60.
Jindal V, Arora E, Masab M, Gupta S. Chimeric antigen receptor T cell therapy in pancreatic cancer: from research to practice. Med Oncol. 2018;35(6):84.
Rataj F, Kraus FBT, Chaloupka M, Grassmann S, Heise C, Cadilha BL, Duewell P, Endres S, Kobold S. PD1-CD28 fusion protein enables CD4+ T cell help for adoptive T cell therapy in models of pancreatic cancer and Non-hodgkin lymphoma. Front Immunol. 2018;9:1955.
Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, Brenner MK, Fisher WE, Leen AM, Vera JF. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25(1):249–58.
Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, Cuevas C, Dotson AM, Tan X, Hotes JL, Greenberg PD, et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell. 2015;28(5):638–52.
Hall M, Liu H, Malafa M, Centeno B, Hodul PJ, Pimiento J, Pilon-Thomas S, Sarnaik AA. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J Immunother Cancer. 2016;4(1):61.
Meng Q, Liu Z, Rangelova E, Poiret T, Ambati A, Rane L, Xie S, Verbeke C, Dodoo E, Del Chiaro M, et al. Expansion of tumor-reactive T cells from patients with pancreatic cancer. J Immunother. 2016;39(2):81–9.
Meng Q, Valentini D, Rao M, Moro CF, Paraschoudi G, Jager E, Dodoo E, Rangelova E, Del Chiaro M, Maeurer M. Neoepitope targets of tumour-infiltrating lymphocytes from patients with pancreatic cancer. Br J Cancer. 2018;120(1):97–108.
Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 2018;24(6):724–30.
Okamoto M, Kobayashi M, Yonemitsu Y, Koido S, Homma S. Dendritic cell-based vaccine for pancreatic cancer in Japan. World J Gastrointest Pharmacol Ther. 2016;7(1):133–8.
Deicher A, Andersson R, Tingstedt B, Lindell G, Bauden M, Ansari D. Targeting dendritic cells in pancreatic ductal adenocarcinoma. Cancer Cell Int. 2018;18:85.
Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, Goemann M, Coleman J, Grochow L, Donehower RC, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19(1):145–56.
Lutz E, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, Herman J, Sugar E, Piantadosi S, Cameron JL, Solt S, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A phase II trial of safety, efficacy, and immune activation. Ann Surg. 2011;253(2):328–35.
Hardacre JM, Mulcahy M, Small W, Talamonti M, Obel J, Krishnamurthi S, Rocha-Lima CS, Safran H, Lenz HJ, Chiorean EG. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: a phase 2 study. J Gastrointest Surg. 2013;17(1):94–100 discussion p. 100–101.
Macher BA, Galili U. The Galalpha1,3Galbeta1,4GlcNAc-R (alpha-gal) epitope: a carbohydrate of unique evolution and clinical relevance. Biochim Biophys Acta. 2008;1780(2):75–88.
Galili U, Anaraki F, Thall A, Hill-Black C, Radic M. One percent of human circulating B lymphocytes are capable of producing the natural anti-gal antibody. Blood. 1993;82(8):2485–93.
Deguchi T, Tanemura M, Miyoshi E, Nagano H, Machida T, Ohmura Y, Kobayashi S, Marubashi S, Eguchi H, Takeda Y, et al. Increased immunogenicity of tumor-associated antigen, mucin 1, engineered to express alpha-gal epitopes: a novel approach to immunotherapy in pancreatic cancer. Cancer Res. 2010;70(13):5259–69.
Tanida T, Tanemura M, Miyoshi E, Nagano H, Furukawa K, Nonaka Y, Akita H, Hama N, Wada H, Kawamoto K, et al. Pancreatic cancer immunotherapy using a tumor lysate vaccine, engineered to express alpha-gal epitopes, targets pancreatic cancer stem cells. Int J Oncol. 2015;46(1):78–90.
Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, Nemunaitis J, Sterman DH, Hassan R, Lutz E, Moyer B, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: phase I studies of safety and immune induction. Clin Cancer Res. 2012;18(3):858–68.
Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, Morse M, Zeh H, Cohen D, Fine RL, et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33(12):1325–33.
Nishikawa H, Kato T, Tanida K, Hiasa A, Tawara I, Ikeda H, Ikarashi Y, Wakasugi H, Kronenberg M, Nakayama T, et al. CD4+ CD25+ T cells responding to serologically defined autoantigens suppress antitumor immune responses. P Natl Acad Sci USA. 2003;100(19):10902–6.
Wang HY, Lee DA, Peng G, Guo Z, Li Y, Kiniwa Y, Shevach EM, Wang RF. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity. 2004;20(1):107–18.
Wang HY, Wang RF. Antigen-specific CD4+ regulatory T cells in cancer: implications for immunotherapy. Microbes Infect. 2005;7(7–8):1056–62.
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–5.
Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, Douek DC, Howie B, Robins H, Parkhurst MR, et al. Tumor- and neoantigen-reactive T-cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunol Res. 2016;4(9):734–43.
Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, Gartner JJ, Prickett TD, Cafri G, Robbins PF, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight. 2018;3(19):e122467.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Institute of Health (grants R01AI121180, R01CA221867 and R21AI109239 to J.S.), the National Natural Science Foundation of China (grants 81373875 and 81873156) and the Educational Department of Liaoning Province of China (grant LZ2019001 to Jia-Qiao Fan).
Author information
Authors and Affiliations
Contributions
Jia-Qiao Fan and Meng-Fei Wang contributed equally to this work. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fan, Jq., Wang, MF., Chen, HL. et al. Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol Cancer 19, 32 (2020). https://doi.org/10.1186/s12943-020-01151-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-020-01151-3