
Abstract
Autophagy is a highly conserved catabolic process that mediates degradation of pernicious or dysfunctional cellular components, such as invasive pathogens, senescent proteins, and organelles. It can promote or suppress tumor development, so it is a “double-edged sword” in tumors that depends on the cell and tissue types and the stages of tumor. The epithelial-mesenchymal transition (EMT) is a complex biological trans-differentiation process that allows epithelial cells to transiently obtain mesenchymal features, including motility and metastatic potential. EMT is considered as an important contributor to the invasion and metastasis of cancers. Thus, clarifying the crosstalk between autophagy and EMT will provide novel targets for cancer therapy. It was reported that EMT-related signal pathways have an impact on autophagy; conversely, autophagy activation can suppress or strengthen EMT by regulating various signaling pathways. On one hand, autophagy activation provides energy and basic nutrients for EMT during metastatic spreading, which assists cells to survive in stressful environmental and intracellular conditions. On the other hand, autophagy, acting as a cancer-suppressive function, is inclined to hinder metastasis by selectively down-regulating critical transcription factors of EMT in the early phases. Therefore, the inhibition of EMT by autophagy inhibitors or activators might be a novel strategy that provides thought and enlightenment for the treatment of cancer. In this article, we discuss in detail the role of autophagy and EMT in the development of cancers, the regulatory mechanisms between autophagy and EMT, the effects of autophagy inhibition or activation on EMT, and the potential applications in anticancer therapy.
Similar content being viewed by others


Background
Autophagy can be stimulated by intracellular or environmental stresses, including nutrient deprivation, hypoxia, and damaged organelles. Generally, the complete macroautophagic process is divided into the following stages: induction, vesicle nucleation, vesicle elongation, docking and fusion, degradation, and recycling. The degraded and recycled metabolites can provide energy supplies and basic nutrients for cells growth [1]. Recent observations have shown that autophagy can suppress cancer development by eliminating potentially harmful components and mutant DNA and chromosomes or can promote cancer development by overcoming the stressful conditions and producing nutrients and adenosine triphosphate (ATP) to maintain protein synthesis and other metabolic functions, which depends on the cell/tissue types and the stages of cancer [2]. Thus, the effects of autophagy on anticancer treatment remain to be investigated in depth.
It is well-known that the epithelial-mesenchymal transition (EMT) is considered to be a major driver of cancer exacerbation from initiation to metastasis and plays a key part in the induction of cancer progression, metastasis, and drug resistance [3, 4]. The process of EMT contains adhesion junctions and loss of substrate polarity; acquisition of mesenchymal characteristics, such as spindle-shaped cell morphology and reorganization of actin stress fibers; enhancement of movement; and invasion and resistance to apoptosis [5].
As is well known, autophagy and EMT are major biological processes in the occurrence and development of cancer, and there is a complex relationship between autophagy-correlated and EMT-correlated signaling pathways. In previous studies, it has been found that EMT-related signaling pathways can trigger or repress autophagy. Significantly, autophagy is also involved in the induction and inhibition of EMT. On the one hand, EMT requires autophagy to support the viability of potentially metastasis of cancer cells. It has been indicated that an EMT-like phenotype corresponds to a higher autophagy flux, and the combination of an autophagy inhibitor (chloroquine) with the current therapeutic regimen could be more beneficial alongside the repressed EMT in renal cell carcinoma (RCC) [6]. On the other hand, a growing body of additional evidence indicates that autophagy acts to prevent EMT, and the activation of the autophagy may abate the acquisition of the EMT phenotype in cancer cells. It has been shown that induction of autophagy by nutrient deprivation or mechanistic target of rapamycin (mTOR) pathway inhibition leads to reduced migration and invasion in glioblastoma cells. Autophagy impairment determined by silencing of autophagy-related genes 5 (ATG5), ATG7, or Beclin-1 results in an increment of cell motility and invasiveness with the up-regulation of SNAIL and SLUG, two of the major transcription factors of the EMT process [7]. Because of the dual effects of autophagy on EMT, inhibiting EMT by targeting autophagy might be a novel strategy for anticancer therapy.
Some studies have demonstrated the effect of preclinical application of autophagy inhibitors or activators on anticancer treatment by regulating EMT. Collectively, in this review, we discuss in detail the role of autophagy and EMT in the development of cancers, the regulatory mechanisms between autophagy and EMT, the effects of autophagy inhibition or activation on EMT, and the potential applications in anticancer therapy.
The role of autophagy and EMT in the development of cancer
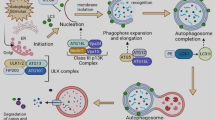
Autophagy is viewed as type II programmed cell death, namely, autophagic cell death, stimulated by cellular or environmental stresses in order to clear senescent organelles, protein aggregates, and intracellular pathogens through the formation of autophagosomes, subsequently targeting to lysosomal digestion, which maintains a steady state for cell survival by engulfing the metabolic waste, inhibiting the production of reactive oxygen species (ROS), eliminating damaged mitochondria and peroxisomes, and reducing DNA damage and chromosomal instability.
The macroautophagic process is triggered by the formation of the Atg1/ULK complex when the induction signals suppress mammalian target of rapamycin complex 1 (mTORC1). Then, the ULK complex binding to the phosphatidylinositol 3-kinase (PI3K) complex (Beclin1–hVps34–PI3K) establishes a putative mammalian preautophagosomal structure, probably together with vacuole membrane protein 1 (VMP1) and Atg9, where PI3K locally generates PI3P. Next, phagophore elongation bases on the Atg5-Atg12 and the microtubule-associated light chain 3 (MAP-LC3/Atg8/LC3) conjugation systems. Phagophore progressively engulfs a portion of the cytoplasm to form the double-membrane autophagosome after elongation and fusion. Finally, the fusion of an autophagosome with a lysosome triggers the formation of an autolysosome and degradation of the loads, and the recycles are released back into the cytosol for reuse [8, 9].
Normally, autophagy can prevent cancer initiation by removing intracellular mutants, damaged mitochondria, infectious pathogens, and misfolded proteins and inhibiting inflammation. However, during the advanced stages of cancer, autophagy promotes cancer development by producing nutrient substances and releasing ATP, which is beneficial for boosting a variety of biological metabolisms and satisfies the high demand for cancer cell proliferation, invasion, and metastasis [10]. Therefore, autophagy might exact the opposite effects on anticancer treatment in different tissue or development stages of cancer [11].
It was reported that autophagy not only can decrease the apoptosis of isolated cancer cells but also can inhibit the apoptosis of cancer cells in vivo during metastasis by regulating the cancer microenvironment, which indicates that autophagy plays an important role in promoting cancer metastasis, and the inhibition of autophagy might be an effective treatment strategy for malignant cancer [12]. According to current research, autophagy inhibition has been applied to a variety of cancer therapies, such as glioma, myeloma, breast cancer, rectal cancer, and prostate cancer [6]. As a “double-edged sword” in cancer, the regulatory mechanism of autophagy in cancer is complicated, but the observations about specific molecular markers of autophagy have attracted great attention in terms of current anticancer treatment. The choice of autophagy inhibitors and agonists during treatment deserves to be explored more deeply.
EMT is a dynamic multistep process that involves the loss of intercellular adhesion, the destruction of the cancer basement membrane and extracellular matrix, the reconstruction of the cytoskeleton, and the enhancement of cell motility [13, 14], which increased the difficulty of cancer treatment in clinical. There are three types of EMT that are based on the specific biological environment. Type 1 EMT is mainly related to embryo implantation, development, and organogenesis. Type 2 EMT is mainly associated with injury repair, tissue regeneration, and organ fibrosis. Type 3 EMT refers to phenotypic transformation associated with epithelial cell malignancy, which facilitates metastatic cancer cells to maintain a certain epithelial character while obtaining a mesenchymal phenotype, so EMT involving cancer metastasis refers to type 3.
EMT implies a complete transdifferentiation from a functional epithelial cell into a mesenchymal-like cell, which occurs along with the inhibition of senescence and anoikis as well as acquisition of immunosuppression and cancer stem cell (CSC)–like features, resistance to anticancer drugs, and apoptosis. These processes involve multiple signal transduction pathways and complicated molecular mechanisms [3]. Naturally, EMT is regulated by exosomes, extracellular matrix, oxygen deficit, and soluble factors such as hepatocyte growth factor, fibroblast growth factor (FGF), and members of transforming growth factor β (TGF-β) [15]. E-cadherin is essential for calcium-dependent cell-cell adhesion and signal transduction; its decrease or loss acts as a crucial role in EMT induction by promoting cell invasive movement and diffusion [16]. In addition, E-cadherin dysfunction is reflected by gene mutation, which will result in abnormal protein synthesis and hydrolysis.
During the EMT process, Zinc-finger E-box binding homeobox 1 (ZEB1) and SNAIL are regarded as the main EMT transcription factors to initialize and maintain the EMT process. ZEB1 is one of the most critical EMT conversion molecules, as a zinc finger structure of the DNA binding protein, which can prohibit E-cad gene expression by binding to E-cad promoter site. Furthermore, ZEB1 expression in human bronchial epithelial cells directly repressed epithelial splicing regulatory protein 1 (ESRP1), leading to increased expression of a mesenchymal splice variant of CD44 and increment of EMT in lung cancer [17]. SNAIL is regulated by various signals from the cancer microenvironment, is a prominent inducer of EMT, strongly repressing E-cadherin expression [18]. Mechanistically, in cholangiocarcinoma, atypical protein kinase C-iota (aPKC-ι) directly phosphorylates specificity protein 1 (Sp1) to up-regulate P-Sp1 that increased SNAIL expression by promoting Sp1 binding to the SNAIL promoter, resulting in EMT changes and immunosuppression [19].
It has been confirmed that EMT is a crucial step in the migration of cancer cells, which is controlled by different signaling transduction pathways and networks, such as autocrine IL11/IL6-gp130/JAK2/STAT, fibronectin-integrin, GAS6-Axl/Tyro3, PDGFR/FGFR/RET, and TGF-β R networks [20]. The current therapy strategies and drug development can focus on targeting these signal transduction pathways. However, there is a biological phenomenon of cancer cell dormancy when cancer enters into the stage of aggressive metastasis, which facilitates target cells to escape chemotherapy and radiotherapy, eventually leading to low survival rate and high cancer recurrence. Therefore, to find the best treatment period, further understanding the specific regulation mechanism between EMT and cancer metastasis is urgently needed.
Crosstalk between autophagy and EMT
Autophagy is mainly controlled by PI3K/AKT/mTOR, Beclin-1, p53, and JAK/STAT signaling pathways. These autophagy-related regulatory pathways have a dramatic impact on EMT. In the process of EMT, there are several signaling pathways, including integrin, WNTs, NF-kB, and TGF-β signaling pathways, that play a crucial role in autophagy. Furthermore, increasing observations have indicated that the functional interaction between cytoskeleton and mitochondria is also the crucial regulatory mechanism in the process of autophagy and EMT.
The PI3K/AKT/mTOR signaling pathway
mTOR is a serine-threonine kinase that controls several important aspects of mammalian cell function. Changes in mTOR activity have a dramatic impact on rates of translation, transcription, protein degradation, cytoskeleton dynamics, cell metabolism, and autophagy [21]. However, the upstream and downstream signal transduction pathways of mTOR are accurately complicated. The predominant upstream pathway is PI3K/AKT/mTOR that is significantly regulated by LKB1/adenosine monophosphate–activated protein kinase (AMPK) and Ras signaling pathways [22,23,24]. There are two main mTOR downstream signaling pathways. First, when mTOR is activated by various stresses, phosphorylated ribosomal protein S6 (P70S6) will promote mRNA translation, adhesion of ribosomes, and endoplasmic reticulum, inhibiting the delivery of endoplasmic reticulum and the formation of autophagic membrane [25]. Second, mTOR activity also inhibits the activity of eukaryotic initiation factor 4E (eIF4E)-binding proteins (4E-BPs) by phosphorylating 4E-BPs and releasing eIF4E [26]. Significantly, autophagy activation by inhibiting the mTOR pathway attenuates migration and invasion of gallbladder cancer via EMT inhibition [27]. For example, metformin can suppress the proliferation, migration, and EMT by inhibiting mTOR signaling and stimulating autophagy in thyroid cancer cell lines [28]. Likewise, water stress proteins (WSP1)-induced autophagy through down-regulating PI3K/AKT/mTOR pathway could degrade β catenin and inhibit EMT through increasing E-cadherin and decreasing N-cadherin, which inhibits cancer migration [29] (Fig. 1).

mTOR signaling pathway regulated autophagy and EMT. PI3K activation is induced by interaction with a growth factor receptor, direct binding to Ras, also induced by NF-κB and TGF-β activation. Activation of the PI3K/AKT signaling pathway blocks autophagy by prohibiting mTOR. The PI3K/Akt pathway positively regulates WNT/β-catenin through phosphorylating the serine at residue 552 in β-catenin and the serine at residue 9 in glycogen synthase kinase 3β (GSK3β), which increases intracellular β-catenin levels that combine with E-cadherin to promote EMT. Moreover, The PI3K/Akt pathway activity up-regulates nuclear factors SNAIL and SLUG, contributing to EMT activation. GSK-3β directly induces autophagy by activating LKB1/AMPK and in turn prohibiting the PI3K/AKT/mTOR pathway. It also indirectly triggers autophagy through promoting the hydrolysis of β-catenin protein. LKB1/AMPK activation plays a critical role in stimulating autophagy via decreasing the ratio of p-mTOR/mTOR and p-p70s6k/p70s6k. In addition, LKB1/AMPK hinders EMT by inhibiting Smad2/3 and TGF-β activity. Ras protein mutation not only activates the Ras/Rac1/Mkk7/JNK pathway (with JNK in turn binding to Atg5/Atg7) but also induces the Ras/Raf1/MEK1/2/ERK signaling pathway, which results in autophagy activation and EMT enhancement
PI3Ks activity can lead to cancer cell migration, adhesion, and malignant transformation, together with degradation of the extracellular matrix, which prohibit autophagy and enhance EMT by up-regulating SNAIL, SLUG, integrin-linked kinase (ILK), and WNT/β-catenin signaling. PI3K is activated by the interaction with a growth factor receptor or connexin with a phosphorylated tyrosine residue, causing a dimer conformational change. In addition, PI3K activation is induced by direct binding to Ras or by nuclear transcription factor κB (NF-κB) and TGF-β activation.
Activation of the PI3K/AKT/mTOR signaling pathway not only blocks autophagy but also plays an important role in regulating EMT [30]. It has been found that activation of the PI3K/AKT/mTOR signaling pathway can lead to EMT changes in tongue squamous cell carcinoma. Interestingly, P13K activation can facilitate the increment of ILK. ILK is a serine-threonine protein kinase that binds to the β-integrin cytoplasmic domain and down-regulates E-cadherin, leading to the EMT [31]. Moreover, the PI3K/Akt pathway positively regulates WNT/β-catenin through phosphorylating the serine at residue 552 in β-catenin and the serine at residue 9 in glycogen synthase kinase 3β (GSK3β), which increases levels of intracellular β-catenin that combines with E-cadherin to promote EMT [32]. In addition, the PI3K/AKT/mTOR signaling pathway is induced by growth factor, contributing to the promotion of cell metastasis and EMT by up-regulating nuclear factor, SNAIL, and SLUG and promoting matrix metalloproteinase (MMP) to degrade the cell matrix, which synergies with other EMT signaling pathways. For instance, TGF-β induces EMT by directly or indirectly activating the PI3K/Akt signaling pathway [33]. Invasion, migration, and TGF-β–induced EMT would be suppressed through the inhibition of the PI3K/Akt/mTOR signaling pathway [34]. EMT can also occur with sustained NF-κB activation, even in the absence of TGF-β [35]. Ras is a key tyrosine kinase receptor, and activation of the Ras/Akt signaling pathway not only is associated with malignancy in epithelial cells but also can promote EMT [32]. Thereby, highly specific, low-toxicity drugs that are targeted to the EMT-related PI3K/AKT/mTOR pathway must be developed.
Liver kinase B1 (LKB1) has been thought to act as a cancer suppressor [36]. AMPK is the energy sensor and signal transducer and mTOR is a central controller of cell growth and proliferation [37]. LKB1/AMPK activation plays a critical role in stimulating autophagy via decreasing the ratio of p-mTOR/mTOR and p-p70s6k/p70s6k and restraining PI3K/Akt/mTOR activity [38]. In addition, LKB1/AMPK hinders EMT by inhibiting Smad2/3 and TGF-β activity.
GSK-3β is a multifunctional protein kinase that can directly induce autophagy by activating LKB1/AMPK and in turn prohibiting the PI3K/Akt/mTOR pathway [39]. It also indirectly triggers autophagy through promoting the hydrolysis of β-catenin protein, followed by LKB1/AMPK activation and mTOR block. Theoretically, the knockdown of β-catenin can enhance apoptosis and autophagy through activating the LKB1/AMPK pathway and suppressing PI3K/Akt/mTOR signaling in head and neck squamous cell carcinoma [40].
Studies have shown that the LKB1/AMPK/mTORC1 pathway is involved in nesfatin-1/nucleobindin-2 (NUCB-2)–mediated EMT in colon cancer, and ZEB-1 is critical for regulation of NUCB-2-mediated migration and invasion [41]. The activation of LKB1/AMPK inhibits the migration of TGF-β–stimulated cancer cells by inhibiting Smad2/3 activity, which suggests that AMPK may be a target for cancer drug therapy [42].
Ras oncogene is a member of the GTPase gene family. The Ras signaling pathway induces EMT and has dual effects on autophagy. Ras promotes autophagy by Rac1/Mkk7/JNK and Ras/Raf1/MEK1/2/ERK signaling pathway and restrains autophagy by stimulating PI3K/AKT/mTORC1 pathway. Accumulating research has shown that the activated Ras protein will trigger multiple downstream pathways, causing abnormal cell proliferation and tumorigenesis.
Ras has dual effects on autophagy. First, Ras protein mutation does not only activate the Ras/Rac1/Mkk7/JNK pathway (with JNK in turn binding to Atg5/Atg7) [43], but also induces the Ras/Raf1/MEK1/2/ERK signaling pathway. The latter not only improves the transcriptional expression of BNIP and promotes BNIP-induced Bcl-2 release from Beclin1 but also induces the binding of Noxa to Mcl-1 and Beclin-1 dissociation from Mcl-1, which results in autophagy activation [44]. Second, autophagy is inhibited by activating Ras protein, boosting the Ras/PI3K/AKT/mTORC1 pathway and blocking ULK1/Atg13/FIP200 complex formation, which is essential for initiation of autophagy [45]. It is worth noting that a recent article suggested that activated Ras and mutant p53 may synergistically regulate autophagy [46].
Further, a report demonstrated that intracellular PD-L1 prominently activates the EMT process by interacting with H-Ras, which led to Ras/ERK/EMT activation [47]. It is distinct that the Nogo-B receptor (NgBR) is a specific receptor of Nogo-B that regulates vascular remodeling and angiogenesis, which triggers EMT based on the enhancement of EMT-related proteins and SNAIL1 protein expression via activation of the Ras/ERK pathway [48]. What’s more, the TGF-β–activated ERK pathway is necessary to mediate EMT in vitro [49]. Although Ras/Raf/MAPK activation alone cannot lead to prostate cancer initiation, it apparently accelerates progression caused by phosphatase and tensin homolog deleted from chromosome ten (PTEN) loss, accompanied by EMT and metastasis [50].
Currently, several PI3K/AKT/mTOR pathway-targeted drugs have been developed, such as curcumin, FTY720, and Bufalin [51,52,53]. However, these drugs need to be further improved because of the inevitable toxic side effects. Importantly, the PI3K/AKT/mTOR signaling pathway plays an irreplaceable role in resistance to radiotherapy and chemotherapy. The results have shown that BEZ235 can significantly enhance radiosensitivity by inhibiting the PI3K/AKT/mTOR signaling pathway [54, 55]. In addition, danusertib inhibits the PI3K/AKT/mTOR signaling pathway by inducing the activation of P38 MAPK, resulting in autophagy activation, which suggests that autophagy has a significant inhibitory effect on EMT [56, 57].
Beclin-1 signaling pathway
Beclin-1 is a homologous gene of the yeast gene Atg6/Vps30, which binds to VPS34 (Catalytic Subunit of ClassIIIPI3K) to construct a complex for inducing autophagy and prohibits EMT through down-regulating ZEB1, WNT1, and NF-κB. Profoundly, Beclin-1-induced autophagy accelerated EMT by up-regulating vimentin and Twist expression and decreasing E-cadherin expression (Fig. 2). It has been used as an independent biomarker for predicting overall survival and progression-free survival in patients with gastric and liver cancer [58].

Beclin-1 signaling pathway regulated autophagy and EMT. The Beclin-1 gene triggers autophagy by forming the PI3K complex and prohibiting EMT through down-regulating ZEB1, WNT1, and NF-κB. Additionally, Beclin-1-induced autophagy accelerated EMT by up-regulating vimentin and Twist expression and decreasing E-cadherin expression
It was reported that the Beclin-1 gene can trigger autophagy by forming the PI3K complex [59]. A growing body of research has shown that Beclin-1 knockout mice are more prone to spontaneous cancers. Clinic studies have confirmed that the cancer incidence increased after Beclin-1 was suppressed, such as ovarian, breast, and prostate cancers [60]. However, it has also been reported that overexpression of autophagy protein Beclin-1 in mammalian cells can cause cell death [61].
Current studies have shown that knockdown of Beclin-1 causes thyroid cancer cells to lose their epithelial properties and acquire mesenchymal characters consistent with EMT through stabilizing ZEB1 mRNA, and there is a negative correlation between Beclin-1 and ZEB1 in thyroid cancer [62]. In a further study, Beclin-1 gene knockout or low expression is involved in the activation of WNT1 and NF-κB, leading to cancer cell metastasis, which suggests that the knockout or low expression of the Beclin-1 gene may promote EMT and cancerogenesis by activating the WNT1 pathway, resulting in poor prognosis [63, 64]. Surprisingly, knockdown of Beclin-1 by small interfering RNA (siRNA) significantly inhibited the autophagy activation induced by rapamycin, consequentially suppressing EMT and the invasiveness of colon cancer cells via promoting vimentin and Twist down-regulation and E-cadherin up-regulation, suggesting that inhibiting Beclin-1-induced autophagy would an effective anticancer strategy [65].
P53 signaling pathway
P53 is a well-known cancer suppressor protein, which mediates cancer inhibition mainly through triggering autophagy dependence on autophagy-related gene expression and PI3K/AKT/mTOR inhibition and by blocking EMT based on decreased expression of ZEB1, ZEB2, and SNAIL. Interestingly, mutant P53 can trigger EMT and mitochondrial fission that in turn promote autophagy [46] (Fig. 3).

P53 signaling pathway regulated autophagy and EMT. Nucleus P53 promotes the up-regulation of autophagy by down-regulating PI3K/AKT/mTOR signaling and enhancing the expression of autophagy-related genes, including Ulk1/2, Atg4, Atg7, and Atg10. Nevertheless, P53 inhibits autophagy in the cytoplasm. The nucleus P53 reduced the expression of ZEB1, ZEB2, and SNAIL by activating the expression of miR-200a and miR-130b, resulting in EMT inhibition. Significantly, mutant P53 triggers EMT and mitochondrial fission that in turn strengthens autophagy
In the nucleus, P53 can down-regulate PI3K/AKT/mTOR signaling via interaction with PTEN, which promotes the up-regulation of autophagy. In addition, P53 enhances the expression of autophagy-related genes, including Ulk1/2, Atg4, Atg7, and Atg10 [66]. Increased autophagy continually contributes to p53-dependent apoptosis and cancer suppression. Nevertheless, P53 will inhibit autophagy in the cytoplasm [67], but the specific mechanism remains to be illustrated.
Furthermore, P53 can also simultaneously regulate EMT, and the nucleus P53 can reduce the expression of ZEB1, ZEB2, and SNAIL by activating the relevant microRNA, contributing to EMT inhibition [68, 69]. For instance, in pterygium, inactivation of p53 influences miR-200a expression, resulting in EMT progress through up-regulating ZEB1, ZEB2, and SNAIL gene expression [70]. Mutant P53 can bind to miR-130b promoter and inhibit its transcription, which induces the expression of ZEB1, promotes EMT occurrence, and enhances the ability of cell invasion [71]. Therefore, a targeted therapy strategy of the mutant p53 gene is likely to achieve excellent curative effects. In brief, increased stability and expression of p53 in the nucleus can induce autophagy and inhibit EMT, which suggests a wonderful strategy for anticancer therapy.
JAK/STAT signaling pathway
The JAK/STAT signaling pathway has a significant effect on essential cellular mechanisms such as proliferation, invasion, survival, inflammation, and immunity via inducing EMT and inhibiting autophagy, and autophagy induction hinders EMT through suppressing JAK/STAT signaling. It has been demonstrated that JAK/STAT signaling can transmit extracellular signals to the nucleus by activating receptor tyrosine kinase signaling and transcription activating factor–targeted genes (Fig. 4).

JAK/STAT signaling pathway regulated autophagy and EMT. Activation of JAK/STAT protein is stimulated by IL-6, leading to the up-regulation of the expression of MMP-2 and SNAIL and activation of EMT. On the other hand, the extracellular IL-6–mediated JAK/STAT signaling pathway accelerates the cancer process by prohibiting autophagy. Furthermore, autophagy induction hinders EMT through suppressing JAK/STAT signaling
At present, a number of observations about human solid tumors and hematological malignancies have found that JAK/STAT signaling pathway activation is closely involved in cancer cell proliferation, adjacent invasion, and distant metastasis. For example, activation of JAK/STAT protein stimulated by IL-6 up-regulates MMP-2 and SNAIL expression, which results in EMT [72, 73]. However, the JAK2/STAT3 inhibitor WP1066 prevents IL-6–induced activation of the JAK2/STAT3 pathway and EMT [74]. Furthermore, ovatodiolide can efficiently suppress nasopharyngeal cancer development by inducing apoptosis and inhibiting EMT and is consistent with repression of the JAK/STAT signaling pathway [75].
On the other hand, the extracellular IL-6–mediated JAK/STAT signaling pathway accelerates the cancer process by prohibiting autophagy [76]. Recent studies have shown that esveratrol can induce autophagy and hinder ovarian cancer cell migration by inhibiting the IL-6–mediated JAK/STAT signaling pathway. Likewise, quercetin induces autophagy by inhibiting the STAT3 pathway in primary effusion lymphoma [77, 78]. Previously, the report has shown that docetaxel-mediated autophagy significantly decreased castration-resistant prostate cancer (CRPC) cell viability and metastasis by inhibiting STAT3 [79]. Hence, autophagy activators might be used to hinder EMT through suppressing JAK/STAT signaling.
The integrin signaling pathway
Integrin-mediated signaling pathways have been found to control multiple mechanisms, such as cancer cell survival, proliferation, differentiation, and migration, by modifying the microenvironment. In brief, the integrin pathway inhibits autophagy. The integrin-regulated EMT is mainly mediated by focal adhesion kinase (FAK) and ILK; both FAK-Src–mediated and ILK-mediated integrin pathways induce EMT. Furthermore, autophagy promotes EMT via linking β-catenin and Smad signaling dependent on up-regulation of ILK (Fig. 5).

The integrin signaling pathway regulated autophagy and EMT. FAK-Src–mediated integrin pathway has been shown to inhibit autophagy and promote EMT. In addition, integrin enhances EMT by stimulating the EGFR-ERK/MAPK signaling pathway. ILK accelerates EMT by activating the WNT/β-catenin pathway. Similarly, ILK can promote EMT development by transferring β-catenin into the nucleus, causing down-regulation of E-cadherin. Additionally, integrin signaling promotes TGF-β1–dependent down-regulation of E-cadherin expression, which is essential for EMT induction. Furthermore, ILK inhibits autophagy by promoting the phosphorylation of AKT and activating mTOR. By contrast, autophagy stimulates β-catenin and Smad signaling to enhance ILK expression, resulting in EMT promotion
FAK is a linker molecule that aggregates different signaling proteins. The activation of the integrin-mediated FAK-Src pathway has been shown to inhibit autophagy and promote E-cadherin–dependent collective cell movement and EMT, leading to cancer development. Consequently, knockdown of FAK by siRNA or inhibition of Src kinase activity by dasatinib could inhibit E-cadherin–mediated cell-cell adhesions and EMT [80]. Epidermal growth factor (EGF) can also induce EMT in pancreatic cancer cells via stimulating the integrin/EGFR-ERK/MAPK signaling pathway [81].
The overexpression of ILK can promote EMT development by transferring β-catenin into the nucleus, causing down-regulation of E-cadherin [82]. In addition, integrin signaling promotes TGF-β1–dependent down-regulation of E-cadherin expression, which is essential for EMT induction in RCC. Therefore, the strategies targeted to the integrin–TGF-β1 interplay may represent a therapeutic target in RCC [83]. In addition, ILK acts as a downstream regulator of TGF-β, which accelerates EMT by activating the WNT/β-catenin pathway. Interestingly, TGF-β1 and Β-streptin potentiate the expression of ILK through the β-chain protein/Smad2 signaling pathway in renal fibrosis cells [84].
Furthermore, ILK inhibits autophagy by promoting the phosphorylation of AKT and activating mTOR. Knocking down ILK expression increases autophagy and protects cells from senescence induced by hyperphosphatemia [85]. By contrast, autophagy activity can stimulate β-catenin and Smad signaling by forming the p-β-catenin/p-Smad2 complex to enhance ILK expression, resulting in EMT promotion, indicating that autophagy inhibitors have the great capability to block EMT and cancer development by repressing β-catenin/Smad2 ILK activity [84].
WNTs signaling pathway
The WNTs pathway consists of the classical pathway and nonclassical WNT pathway, which accelerate EMT, and autophagy can down-regulate EMT by degrading the Twist1 protein and inhibiting WNTs pathway. Reduced WNTs signaling is correlated with loss of CSC viability (Fig. 6).

WNTs signaling pathway regulated autophagy and EMT. The WNTs pathway consists of the classical pathway and nonclassical WNT pathway. The classical pathway (WNT/β-catenin signaling pathway) directly leads to HIF-1α activation, which in turn results in overexpression of SLUG, SNAIL, and TWIST and induction of EMT. The nonclassical WNT pathway mainly contains two WNT proteins, WNT5A and WNT11, which facilitate EMT by inducing p38 (Mapk14) phosphorylation. Dishevelled (Dvl) is a basic and central component of WNT signaling, and it plays an important role in both β-catenin–mediated canonical and β-catenin–independent noncanonical WNT signaling. Dvl expression and stability are negatively controlled by autophagy in the late stages of cancer development, which in turn inhibits the WNT process. On the other hand, autophagy can decrease the stability of TWIST1 protein and hinder EMT
In the nonclassical WNT pathway, two WNT proteins, WNT5A and WNT11, facilitate EMT by inducing p38 (MAPK14) phosphorylation [86]. In the classical pathway, WNT/β-catenin, a typical WNT signaling pathway, directly leads to HIF-1α–induced EMT by combining with the intracellular domain of E-cadherin, subsequently connecting to the actin cytoskeleton and mediating intercellular adhesion. Here, we have shown that hypoxia or overexpression of HIF-1α promotes EMT and contributes to metastatic phenotypes. On the one side, HIF-1α up-regulates the expression of TWIST by directly binding to the hypoxia-response element in the TWIST proximal promoter [87]. On the other side, HIF-1α also results in overexpression of SLUG and SNAIL and the formation of EMT [88]. As for EGF-induced EMT, we focused on transcription repressors of E-cadherin, TWIST, SLUG, and SNAIL; the results showed that cancers express high levels of TWIST, which can be further enhanced by EGF [89].
There are cross-points between the WNT signaling pathway and the TGF-β, PI3K/AKt pathway. One report proved that osteopontin promotes the progression of hepatocellular carcinoma (HCC) via stimulating the PI3K/AKT/Twist signaling pathway, contributing to the enhancement of EMT [90]. Similarly, the TGF-β–induced ERK/MAPK pathway contributes to EMT induction, since ERK is required for removing cell adherens junctions to increase cell mobility [91].
Recently, we found that autophagy deficiency stabilizes the TWIST1 protein through accumulation of SQSTM1/p62. Mechanically, the Twist1 degradation is blocked by the interaction between SQSTM1 and TWIST1 in autophagosomes and proteasomes. Twist1 is a key downstream regulator of p62, which suggests that targeted p62-mediated TWIST1 stabilization is a promising therapeutic strategy for cancer prevention and treatment [92, 93]. In colon cancer cell lines, SNAIL overexpression increases the expression of the WNT signal target gene. The mechanism is that the interaction between SNAIL-N and β-catenin further activates the expression of the WNT downstream target gene, resulting in positive feedback of WNT signaling.
In eukaryotic cells, autophagy is a highly conserved self-digestive process that can produce nutritional substance, relieve metabolic stresses, and maintain cell survival. Dishevelled (Dvl) is a basic and central component of WNT signaling that plays an important role in both β-catenin–mediated canonical and β-catenin–independent noncanonical WNT signaling [94]. Dvl expression and stability are negatively controlled by autophagy in the late stages of cancer development, which in turn inhibits the WNT process [95, 96]. In a further study, the WNT signaling antagonist Dapper1 was induced by autophagy-accelerated Dvl2 degradation [97], which is mediated by GABA(A) receptor–associated protein like 1 (GABARAPL1), a cancer repressor, and p62 is required for the interaction of Dvl2 and GABARAPL1. GABARAPL1-mediated Dvl2 degradation is blocked when administered with 3-MA, a specific inhibitor of autophagy [98]. In addition, GABARAP is a cytoplasmic cadherin-6 (CDH6) conjugate. CDH6 is a type 2 cadherin, which drives EMT [99]. Previous studies have confirmed that the silence of CDH6 reverses EMT and reduces thyroid cancer cell metastasis, accompanied by autophagy induction [98, 100]. Therefore, the interplay between the degradation of Dvl, inhibition of WNT signal, and induction of autophagy provides a promising approach for anticancer treatment.
NF-κB signaling pathway
NF-κB is another important regulator of EMT. Its activation has been associated with aggressiveness and the metastatic potential of carcinomas [101], which inhibits autophagy by down-regulating Beclin-1 and promotes EMT by up-regulating related EMT markers. However, autophagy could suppress NF-κB signaling to inhibit EMT by down-regulating MMPs expression (Fig. 7).

NF-κB signaling pathway regulated autophagy and EMT. NF-κB activation increased transcriptional activation of EMT regulator genes expression via binding directly to the sites of EMT transcription factors, including SNAIL1, SLUG, TWIST1, and SIP1 promoter. In addition, TNF-α–mediated stability of SNAIL protein is strengthened by GSK3β activity, which is dependent on NF-κB activation. Furthermore, NF-κB binding to MMPs promotes SNAIL transcription. NF-κB can down-regulate autophagy by inhibiting Beclin-1, an initiator of autophagy. However, autophagy activation suppresses ROS–NF-κB signaling to down-regulate MMPs expression, contributing to EMT inhibition
In human breast cancer, NF-κB activation increased transcriptional activation of EMT regulator gene expression by binding directly to the sites of EMT transcription factors, including SNAIL1, SLUG, TWIST1 and SIP1 promoter, which promotes an aggressive phenotype of breast cancer cells [102]. Similarly, a study found that uric acid induces EMT in renal tubular epithelial cells by the activation of the TLR4/NF-κB signaling pathway [103]. In addition, NF-κB can regulate SNAIL-induced and ROS-dependent signaling pathways in response to MMP-3. The mechanism is that MMP-3 binding to p65 and cRel NF-κB subunits promotes SNAIL transcription. Specifically, MMPs are involved in several pathways that induce EMT, which provide a potential treatment for cancer [104]. Furthermore, tumor necrosis factor–α (TNF-α)–mediated stability of the SNAIL protein is strengthened by GSK3β activity, which is dependent on NF-κB activation [105].
NF-κB can stimulate or hinder autophagy by different mechanisms. Researchers have shown that NF-κB can down-regulate autophagy by inhibiting Beclin-1, an initiator of autophagy [106]. Notably, blocking NF-κB can significantly inhibit the proliferation of hepatocarcinoma cells, which is associated with autophagy enhancement [107]. On the other hand, ROS has an effect on cell transformation, metastasis, and response to therapy at different stages of cancer development, which stimulates NF-κB–dependent autophagy [105]. Inhibition of ROS–NF-κB–dependent autophagy could enhance brazilin-induced apoptosis in head and neck squamous cell carcinoma [108]. However, autophagy activation could suppress ROS–NF-κB signaling to down-regulate MMP-2 and MMP-9 expression, contributing to EMT inhibition [109]. Thus, autophagy activators might be used to prohibit NF-κB signaling, consequently repressing EMT and cancer progression.
TGF-β signaling pathway
TGF-β, secreted by cancer cells and stromal fibroblasts in the cancer microenvironment, is considered as a primary inducer of EMT through inducing SNAIL expression and cooperating with Smad2 and Smad3 and WNT/β-catenin signaling [110]. In addition, TGF-β triggers autophagy by stimulating expression of mRNA transcripts of several autophagy-related genes. Notably, autophagy enhances TGF-β1 expression by inducing activation of cyclic adenosine monophosphate (cAMP)/ protein kinase A (PKA)/cAMP response element binding (CREB) signalling, leading to EMT progression (Fig. 8).

TGF-β signaling pathway regulated autophagy and EMT. Activation of TGF-β/Smad3 in epithelial cells triggers EMT, and the activation of TGF-β/Smad2 signaling pathway also stimulates EMT. Moreover, TGF-β1 induces EMT and cancer metastasis by directly targeting the cytoplasmic domain of E-cadherin (CDH1) and activating WNT/β-catenin signaling. Sometimes, TGF-β cooperates with synergistic factors to induce EMT, such as Ras. Once TGF-β is stimulated, EMT-related transcription factor STAT3 interacts with Ras, which induces SNAIL expression and promotes EMT. Naturally, TGF-β can stimulate the expression of mRNA transcripts of several autophagy-related genes, such as Beclin-1, Atg5, Atg7, and death-associated protein kinase (Dapk), and it induces accumulation of autophagosomes and activation of autophagic flux, which potentiates the induction of the autophagy. It is worthy that autophagy induces TGF-β1 expression and TGF-β1-dependent EMT via triggering cAMP/PKA/CREB signaling, which relies on autophagy-dependent phosphodiesterase 4A (PDE4A) degradation
The TGF-β pathway contributes to EMT occurrence by mediating Smad and non-Smad signal transduction pathways. Activation of TGF-β/Smad3 in epithelial cells triggers significant morphological and functional phenotypical alterations [111], which indicates that TGF-β/Smad3–dependent signaling plays a key role in regulating autophagy-induced EMT [112]. In addition, M2 macrophages also induce EMT through stimulating the TGF-β/Smad2 signaling pathway [113]. Sometimes TGF-β cooperates with synergistic factors to induce EMT, such as Ras and β-catenin. Once TGF-β is stimulated, EMT-related transcription factor STAT3 interacts with Ras, which induces SNAIL expression and promotes EMT [114, 115]. Profoundly, miR-23a promotes TGF-β1–induced EMT and cancer metastasis in breast cancer cells by directly targeting the cytoplasmic domain of E-cadherin (CDH1) and activating WNT/β-catenin signaling [116]. More importantly, TGF-β1 has been identified as the most effective factor that can independently induce EMT [117].
Among these, TGF-β has dual effects on cancer occurrence and development, which depends on the cell type and environment [108]. First, TGF-β1 promotes human carcinoma cell invasion by inducing autophagy, and the autophagy inhibitor 3-MA could effectively reverse this process [118]. Second, TGF-β stimulates the expression of mRNA transcripts of several autophagy-related genes, such as Beclin-1, Atg5, Atg7, and death-associated protein kinase (Dapk), and it induces accumulation of autophagosomes and activation of autophagic flux, which potentiates the induction of the proapoptotic Bcl-2 family protein Bim and contributes to Bim-mediated apoptosis in hematopoietic cells [119, 120]. It is worthy that autophagy induces TGF-β1-dependent EMT in HCC via triggering cAMP/PKA/CREB signaling, which relies on autophagy-dependent phosphodiesterase 4A (PDE4A) degradation [121]. Hence, further researches could focus on developing the autophagy inhibitors to down-regulate PDE4A-activated cAMP/PKA/CREB signaling and TGF-β1 induced EMT.
Interplay between cytoskeleton and mitochondria
The cytoskeleton structures are essential for promoting cell movement and cytoskeleton remodeling, especially supporting EMT activation. Mitochondria are considerably multifunctional organelles, which prominently mediate energy conversion and are crucial regulators of a number of signaling pathways associated with cancer progression. In particular, the interaction of mitochondria and cytoskeleton plays a critical role in regulating autophagy and EMT (Fig. 9).

Interplay between cytoskeleton and mitochondria. Cytoskeleton polymerization induced by EMT, which in turn supports mitochondrial fission that are essential for further sustain EMT process by providing energy supplies, and depolymerization of actin cytoskeleton is sufficient for reversing EMT phenotype. Massive activation of autophagy induces mitochondrial fusion and the reconstitution of mitochondrial network, which subsequently reduces the number of available free mitochondria and counteracts EMT. Mitochondrial protein BNIP3 potentially supported mitochondrial fission and turnover through stimulating mitophagy by directly binding to both mitochondria and the autophagosomal protein LC3, but also enhanced cytoskeleton polymerization. The interaction between BNIP3 and cadherin-6 (CDH6) drives EMT, restrains autophagy and promotes mitochondrial fission
Accumulating evidence has indicated that actin cytoskeleton remodeling potentially drives EMT, invasion and metastasis [122]. Mechanically, hypoxia-induced phosphatidylinositol (4,5) bisphosphate (PIP2) level increased in cancer cells by activating the HIF-1α/RhoA/ROCK1 signaling pathway supported actin filament (F-actin) expression and attenuate the binding amounts of F-actin and capping actin protein of muscle Z-line alpha subunit 1 (CAPZA1), which in turn enhanced EMT [123, 124]. In addition, actin dynamics and membrane-cytoskeleton scaffolds are required for the early formation of autophagosome in starvation induced autophagy [125]. It was reported that the colocalization of actin filaments with important autophagy markers [126]. Mitochondria are the important energy resource for multiple biological metabolisms, such as autophagy, migration and invasiveness. Increased fission and loss of mitochondrial network have been identified as characteristics of oncogenic transformation and accelerates EMT and cancer migration [127, 128].
Notably, mitochondrial dynamics provide ATP for cytoskeleton remodeling to promote EMT during cancer progression, while autophagy regulates mitochondrial dynamics by eliminating the damaged mitochondria. The cytoskeleton is composed of three main types of polymers: actin filaments, microtubules and intermediate filaments, which have been associated with mitochondrial network properties and various mitochondrial functions [129]. For instance, cytoskeleton polymerization and remodeling induced by EMT, which in turn supports mitochondrial fission that are essential for further sustaining cell migration and EMT process by providing energy supplies, and depolymerization of actin cytoskeleton is sufficient for reversing EMT phenotype [129, 130]. Further findings have demonstrated that massive activation of autophagy induces mitochondrial fusion and the reconstitution of mitochondrial network, which subsequently reduces the number of available free mitochondria and counteracts cell migration and EMT [130]. Recently, it was reported that the mitochondrial protein, B-cell lymphoma 2 (BCL-2) interacting protein 3 (BNIP3) in melanoma potentially supported mitochondrial fission and turnover through stimulating mitophagy by directly binding to both mitochondria and the autophagosomal protein LC3, but also maintained cellular architecture and enhanced cytoskeleton polymerization [131], which revealed the pro-tumorigenic role of BNIP3 in driving EMT and melanoma cell’s migration. Indeed, BNIP3-silenced melanoma cells resulted in re-organization of focal adhesion sites and repressed cell–cell interaction [132]. Profoundly, the interaction between BNIP3 and CDH6 drives EMT, restrains autophagy and promotes mitochondrial fission through dynamin-related protein 1 (DRP1)-mediated mechanism [99]. Therefore, novel avenues enhancing autophagy to suppress mitochondrial fission and cytoskeleton polymerization have represented a promising strategy for anticancer therapy by prohibiting EMT.
Regulating EMT by targeting autophagy
Autophagy and EMT act as major biological processes in the occurrence and development of the cancer. Recent observations indicate that there is a complex link between the two processes. On the one hand, cells are dependent on autophagy activation to survive during the EMT. On the other hand, autophagy functions as the tumor-suppressive signal, which hinders the early phases of metastasis and activation of the EMT [130]. Therefore, regulating EMT by targeting autophagy is a very promising strategy for treating cancer (Fig. 10).

Regulating EMT by targeting autophagy. The autophagy attenuates EMT by inhibiting the overexpression of SNAIL and SLUG and activation of ROS-NF-κB-HIF-1α pathway. Moreover, ROS-induced NF-κB activation up-regulates SNAIL and MMPs expression to promote EMT. Additionally, autophagy accelerates lysosomal- mediated degradation of SNAIL and TWIST, resulting in EMT inhibition. On the other side, autophagy activation enhances EMT by increasing the expression of HMGB1, metastasis-associated protein oncostatin M and MMP-9, facilitating EMT markers expression in both RNA and protein levels accompany with promotion of TGF-β2/Smad signaling pathway activity, and up-regulating ILK by linking β-catenin and Smad signaling to induce EMT
It was reported that inhibition of autophagy may promote EMT through the ROS/heme oxygenase-1 (HO-1) pathway in ovarian cancer. N-acetylcysteine (NAC; ROS scavenging agent) and Znpp (HO-1 inhibitor) could impair the migration and invasion through decreasing the expression of HO-1 and reversing EMT [133]. Another report showed that an autophagy defect can promote cancer cell migration and EMT, enhance aerobic glycolysis, and convert cell phenotype toward malignant, which depends on the activation of the ROS–NF-κB–HIF-1α pathway. EMT and metastasis will be attenuated when ROS is cleaned by the antioxidant NAC [134]. What’s more, MMPs could promote EMT in mammary epithelial cells, which are stimulated by ROS-induced NF-κB–dependent activation of SNAIL [104]. In addition, Myriam Catalano et al. demonstrated that Beclin-1 silence and ATG7 down-regulation could enhance the EMT process by overexpression of SNAIL and SLUG in glioblastoma cells, and cell migration and invasion would be attenuated when autophagy is induced upon starvation and treatment with mTOR inhibitors [7]. Moreover, studies about breast and colon cancers have described that the death effector domain-containing DNA-binding protein (DEDD) not only significantly represses EMT by inducing autophagy through direct interaction with the class III PI-3 kinase (PI3KC3)/Beclin-1 but also promotes the autophagy-mediated lysosomal degradation of SNAIL and TWIST [135, 136]. Accordingly, autophagy exerts pro-survival functions by prohibiting EMT, and autophagy activators represent a potential strategy for therapeutic interventions.
However, autophagy helps cancer cells to survive in stressful environmental and intrinsic conditions and also exerts pro-death functions via heightening EMT, which depends on cell types and the stages of cancer [137]. For instance, high mobility group box 1 (HMGB1) induces EMT in association with increased autophagy through increasing the expression of discoidin domain receptor 1 (DDR1) and decreasing the phosphorylation of mTOR [138]. Moreover, autophagy is critical for activation of TGF-β/Smad3–dependent signaling, leading to EMT and cancer cell invasion in HCC [112]. Besides, TGF-β1–induced autophagy promotes EMT based on up-regulation of ILK by linking β-catenin and Smad signaling [84]. Significantly, Li et al. found that neutrophil autophagy activity can increase the expression of metastasis-associated protein oncostatin M and MMP-9 and contribute to cancer cell metastasis in HCC [139]. In the late development of cancer progression, cell motility and migration capacity is weakened by inhibiting autophagy-related genes, and cell invasion is restored by the activation of autophagy-related genes [140]. It is acknowledged that TGF-β2 could up-regulate autophagy and facilitate EMT marker expression in both RNA and protein levels accompanied by Smad signaling pathway activity. A further study observed that proinflammatory cytokines such as TNF-α, it can down-regulate autophagy and increase ROS levels, thus antagonizing TGF-β2–induced EMT, which suggests that autophagy plays a prometastatic role in facilitating EMT by regulating ROS levels and TNF-α can inhibit EMT by suppressing autophagy [141]. Consequently, these findings may provide novel avenues for therapeutic research by hindering autophagy, which may be beneficial to patients with cancer.
Cancer treatment has been complicated, which is mainly associated with autophagy and EMT. Therefore, inhibition of EMT by controlling autophagy must be beneficial for cancer treatment. However, the relevant therapeutic measures must depend on tissue/cell types (epithelial cell or interstitial cells) and the stages of cancer development (early or advanced); thus, the choice of inductors or inhibitors of autophagy need to be further analyzed.
Conclusion
Cell autophagy and EMT, which play indispensable and significant roles in the occurrence and development of cancer, bring great challenges to anticancer treatment. Based on the complicated link between autophagy and EMT, therapeutic avenues targeting autophagy have attracted lots of attention by hindering EMT and further suppressing cancer development. Hence, future studies should focus on profoundly exploring the regulation mechanisms between autophagy and EMT at the molecular and genetic levels. In addition, the discovery of various transcription factors that induce EMT and new EMT markers is urgently needed, as they will be beneficial for further understanding the mechanism of EMT and obtaining effective anticancer treatment strategies by blocking metastasis. It has been reported that autophagy has dual effects on EMT depending on the contextual need of cancer cells during metastases. Currently, autophagy inhibitors, such as chloroquine and 3-methyladenine, and autophagy activators, such as rapamycin, have translational applications in anticancer therapy by regulating EMT (Tables 1 and 2). However, there are few observations about the clinical application of autophagy regulators because of the inevitable side effects, such as high cytotoxicity and low specificity. Therefore, we should try our best to discover more potential and accurate approaches to stimulate or block autophagy, subsequently suppressing EMT and controlling cancer development, which might be a more promising approach for anticancer therapy when combined with other anticancer strategies.
Abbreviations
- 4E-BPs:
-
Eukaryotic initiation factor 4E (eIF4E)-binding proteins
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- aPKC-ι:
-
Atypical protein kinase C-iota
- ATG:
-
Autophagy-related genes
- BNIP3:
-
B-cell lymphoma 2 (BCL-2) interacting protein 3
- CDH1:
-
Cytoplasmic domain of E-cadherin
- CDH6:
-
Cadherin-6
- CDH6:
-
Cadherin-6
- CQ:
-
Chloroquine
- CSC:
-
Cancer stem cell
- DDR1:
-
Discoidin domain receptor 1
- DEDD:
-
Domain-containing DNA-binding protein
- DRP1:
-
Dynamin-related protein 1
- EMT:
-
Epithelial-mesenchymal transition
- EMT-TFs:
-
EMT-transcription factors
- ERK:
-
Extracellular signal-regulated protein kinase
- Erα:
-
Estrogen receptor-α
- ESRP1:
-
Epithelial splicing regulatory protein 1
- FAK:
-
Focal adhesion kinase
- FGF:
-
Fibroblast growth factor
- FGFR:
-
Fibroblast growth factor receptor
- GABARAPL1:
-
GABA(A) receptor-associated protein like 1
- GSK3β:
-
Glycogen synthase kinase 3β
- HGF:
-
Hepatocyte growth factor
- HMGB1:
-
High mobility group box 1
- HO-1:
-
Heme oxygenase-1
- HRE:
-
Hypoxia-response element
- IL:
-
Interleukin
- ILK:
-
Integrin-linked kinase
- JNK:
-
c-Jun NH2-terminal kinase
- LKB1:
-
Liver kinase B1
- MMP:
-
Matrix metalloproteinase
- mTOR:
-
Mammalian target of rapamycin
- NAC:
-
N-acetylcysteine
- NF-kB:
-
Nuclear transcription factor kB
- NgBR:
-
Nogo-B receptor
- P70S6:
-
Phosphorylated ribosomal protein S6
- PAS:
-
Pre-autophagosomal structure
- PDGFR:
-
Platelet-derived growth factor receptor
- PI3K:
-
Phosphatidylinositol 3-kinase
- PI3KC3:
-
Class III PI-3-kinase
- PKB/AKt:
-
Protein Kinase B
- PTEN:
-
Phosphatase and tensin homolog deleted from chromosome ten
- ROS:
-
Reactive oxygen species
- Sp1:
-
Specificity protein 1
- STAT:
-
Signal transducer and activator of transcription
- TGF-β:
-
Members of transforming growth factor-β
- TNF-α:
-
Tumor Necrosis Factor-α
- VMP1:
-
Vacuole membrane protein 1
- WSP1:
-
Water stress proteins
- ZEB1:
-
Zinc-finger E-box binding homeobox 1
References
Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;61(6):585–96.
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–42.
Saitoh M. Involvement of partial EMT in cancer progression. J Biochem. 2018;164(4):257–64.
Krebs AM, Mitschke J, Lasierra Losada M, et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. 2017;19(5):518–29.
Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342(6159):1234850.
Singla M, Bhattacharyya S. Autophagy as a potential therapeutic target during epithelial to mesenchymal transition in renal cell carcinoma: An in vitro study. Biomed Pharmacother. 2017;94:332–40.
Catalano M, D'Alessandro G, Lepore F, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9(8):1612–25.
Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24(1):24–41.
Wang J, Davis S, Zhu M, et al. Autophagosome formation: where the secretory and autophagy pathways meet. Autophagy. 2017;13(5):973–4.
Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–75.
Cheong H, Lu C, Lindsten T, et al. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30(7):671–8.
DeNardo DG, Barreto JB, Andreu P, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91–102.
Tanaka S, Hosokawa M, Yonezawa T, et al. Induction of epithelial-mesenchymal transition and down-regulation of miR-200c and miR-141 in oxaliplatin-resistant colorectal cancer cells. Biol Pharm Bull. 2015;38(3):435–40.
Xu W, Liu H, Liu ZG, et al. Histone deacetylase inhibitors upregulate snail via Smad2/3 phosphorylation and stabilization of snail to promote metastasis of hepatoma cells. Cancer Lett. 2018;420(2018):1–13.
Saitoh M. Epithelial-mesenchymal transition is regulated at post-transcriptional levels by transforming growth factor-beta signaling during tumor progression. Cancer Sci. 2015;106(5):481–8.
Matos ML, Lapyckyj L, Rosso M, et al. Identification of a novel human E-cadherin splice variant and assessment of its effects upon EMT-related events. J Cell Physiol. 2017;232(6):1368–86.
Larsen JE, Nathan V, Osborne JK, et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J Clin Invest. 2016;126(9):3219–35.
Wang Y, Shi J, Chai K, et al. The role of snail in EMT and tumorigenesis. Curr Cancer Drug Targets. 2013;13(9):963–72.
Qian Y, Yao W, Yang T, et al. aPKC-iota/P-Sp1/snail signaling induces epithelial-mesenchymal transition and immunosuppression in cholangiocarcinoma. Hepatology. 2017;66(4):1165–82.
Thomson S, Petti F, Sujka-Kwok I, et al. A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis. 2011;28(2):137–55.
Switon K, Kotulska K, Janusz-Kaminska A, et al. Molecular neurobiology of mTOR. Neuroscience. 2017;341:112–53.
Xue JF, Shi ZM, Zou J, et al. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed Pharmacother. 2017;89:1252–61.
Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314–41.
Mauro L, Naimo GD, Gelsomino L, et al. Uncoupling effects of estrogen receptor alpha on LKB1/AMPK interaction upon adiponectin exposure in breast cancer. FASEB J. 2018;32(8):4343–55.
Renard J, Loureiro M, Rosen LG, et al. Cannabidiol counteracts amphetamine-induced neuronal and behavioral sensitization of the mesolimbic dopamine pathway through a novel mTOR/p70S6 kinase signaling pathway. J Neurosci. 2016;36(18):5160–9.
So L, Lee J, Palafox M, et al. The 4E-BP-eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Sci Signal. 2016;9(430):ra57.
Zong H, Yin B, Zhou H, et al. Inhibition of mTOR pathway attenuates migration and invasion of gallbladder cancer via EMT inhibition. Mol Biol Rep. 2014;41(7):4507–12.
Han B, Cui H, Kang L, et al. Metformin inhibits thyroid cancer cell growth, migration, and EMT through the mTOR pathway. Tumour Biol. 2015;36(8):6295–304.
Guo SJ, Liang XX, Guo M, et al. Migration inhibition of water stress proteins from Nostoc commune Vauch via activation of autophagy in DLD-1 cells. Int J Biol Macromol. 2018;119:669–76.
Lunardi A, Webster KA, Papa A, et al. Role of aberrant PI3K pathway activation in gallbladder tumorigenesis. Oncotarget. 2014;5(4):894–900.
Li L, Pan XY, Shu J, et al. Ribonuclease inhibitor up-regulation inhibits the growth and induces apoptosis in murine melanoma cells through repression of angiogenin and ILK/PI3K/AKT signaling pathway. Biochimie. 2014;103:89–100.
Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhes Migr. 2015;9(4):317–24.
Wendt MK, Smith JA, Schiemann WP. Transforming growth factor-beta-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. 2010;29(49):6485–98.
Baek SH, Ko JH, Lee JH, et al. Ginkgolic acid inhibits invasion and migration and TGF-beta-induced EMT of lung Cancer cells through PI3K/Akt/mTOR inactivation. J Cell Physiol. 2017;232(2):346–54.
Maier HJ, Schmidt-Strassburger U, Huber MA, et al. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010;295(2):214–28.
Peart T, Ramos Valdes Y, Correa RJ, et al. Intact LKB1 activity is required for survival of dormant ovarian cancer spheroids. Oncotarget. 2015;6(26):22424–38.
Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381–400.
Wang P, Jiang L, Zhou N, et al. Resveratrol ameliorates autophagic flux to promote functional recovery in rats after spinal cord injury. Oncotarget. 2018;9(9):8427–40.
Sun A, Li C, Chen R, et al. GSK-3beta controls autophagy by modulating LKB1-AMPK pathway in prostate cancer cells. Prostate. 2016;76(2):172–83.
Chang HW, Lee YS, Nam HY, et al. Knockdown of beta-catenin controls both apoptotic and autophagic cell death through LKB1/AMPK signaling in head and neck squamous cell carcinoma cell lines. Cell Signal. 2013;25(4):839–47.
Kan JY, Yen MC, Wang JY, et al. Nesfatin-1/Nucleobindin-2 enhances cell migration, invasion, and epithelial-mesenchymal transition via LKB1/AMPK/TORC1/ZEB1 pathways in colon cancer. Oncotarget. 2016;7(21):31336–49.
Lin H, Li N, He H, et al. AMPK inhibits the stimulatory effects of TGF-beta on Smad2/3 activity, cell migration, and epithelial-to-mesenchymal transition. Mol Pharmacol. 2015;88(6):1062–71.
Byun JY, Yoon CH, An S, et al. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis. 2009;30(11):1880–8.
Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13(12):928–42.
Kochetkova EY, Blinova GI, Bystrova OA, et al. Suppression of mTORC1 activity in senescent Ras-transformed cells neither restores autophagy nor abrogates apoptotic death caused by inhibition of MEK/ERK kinases. Aging (Albany NY). 2018;10(11):3574–89.
Zhang X, Cheng Q, Yin H, et al. Regulation of autophagy and EMT by the interplay between p53 and RAS during cancer progression (review). Int J Oncol. 2017;51(1):18–24.
Qiu XY, Hu DX, Chen WQ, et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim Biophys Acta Mol basis Dis. 2018;1864(5 Pt A):1754–69.
Wu D, Zhao B, Qi X, et al. Nogo-B receptor promotes epithelial-mesenchymal transition in non-small cell lung cancer cells through the Ras/ERK/Snail1 pathway. Cancer Lett. 2018;418:135–46.
Xie L, Law BK, Chytil AM, et al. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia. 2004;6(5):603–10.
Mulholland DJ, Kobayashi N, Ruscetti M, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012;72(7):1878–89.
Zhang L, Wang H, Zhu J, et al. FTY720 reduces migration and invasion of human glioblastoma cell lines via inhibiting the PI3K/AKT/mTOR/p70S6K signaling pathway. Tumour Biol. 2014;35(11):10707–14.
Wang H, Zhang C, Xu L, et al. Bufalin suppresses hepatocellular carcinoma invasion and metastasis by targeting HIF-1alpha via the PI3K/AKT/mTOR pathway. Oncotarget. 2016;7(15):20193–208.
Jiao D, Wang J, Lu W, et al. Curcumin inhibited HGF-induced EMT and angiogenesis through regulating c-met dependent PI3K/Akt/mTOR signaling pathways in lung cancer. Mol Ther Oncolytics. 2016;3:16018.
Ni J, Cozzi P, Hao J, et al. Epithelial cell adhesion molecule (EpCAM) is associated with prostate cancer metastasis and chemo/radioresistance via the PI3K/Akt/mTOR signaling pathway. Int J Biochem Cell Biol. 2013;45(12):2736–48.
Chang L, Graham PH, Hao J, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875.
Zi D, Zhou ZW, Yang YJ, et al. Danusertib induces apoptosis, cell cycle arrest, and autophagy but inhibits epithelial to mesenchymal transition involving PI3K/Akt/mTOR signaling pathway in human ovarian cancer cells. Int J Mol Sci. 2015;16(11):27228–51.
Li JP, Yang YX, Liu QL, et al. The pan-inhibitor of Aurora kinases danusertib induces apoptosis and autophagy and suppresses epithelial-to-mesenchymal transition in human breast cancer cells. Drug Des Devel Ther. 2015;9:1027–62.
Yang M, Zhao H, Guo L, et al. Autophagy-based survival prognosis in human colorectal carcinoma. Oncotarget. 2015;6(9):7084–103.
Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–50.
Zhou WH, Tang F, Xu J, et al. Low expression of Beclin 1, associated with high Bcl-xL, predicts a malignant phenotype and poor prognosis of gastric cancer. Autophagy. 2012;8(3):389–400.
Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–39.
Li S, Zhang HY, Du ZX, et al. Induction of epithelial-mesenchymal transition (EMT) by Beclin 1 knockdown via posttranscriptional upregulation of ZEB1 in thyroid cancer cells. Oncotarget. 2016;7(43):70364–77.
Cicchini M, Chakrabarti R, Kongara S, et al. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy. 2014;10(11):2036–52.
Cai Z, Capoulade C, Moyret-Lalle C, et al. Resistance of MCF7 human breast carcinoma cells to TNF-induced cell death is associated with loss of p53 function. Oncogene. 1997;15(23):2817–26.
Shen H, Yin L, Deng G, et al. Knockdown of Beclin-1 impairs epithelial-mesenchymal transition of colon cancer cells. J Cell Biochem. 2018;119(8):7022–31.
Kenzelmann Broz D, Spano Mello S, Bieging KT, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27(9):1016–31.
Kumar A, Singh UK, Chaudhary A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med Chem. 2015;7(12):1535–42.
Chang CJ, Chao CH, Xia W, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13(3):317–23.
Kim T, Veronese A, Pichiorri F, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208(5):875–83.
Wu CW, Peng ML, Yeh KT, et al. Inactivation of p53 in pterygium influence miR-200a expression resulting in ZEB1/ZEB2 up-regulation and EMT processing. Exp Eye Res. 2016;146:206–11.
Dong P, Karaayvaz M, Jia N, et al. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013;32(27):3286–95.
Huang W, Yu LF, Zhong J, et al. Stat3 is involved in angiotensin II-induced expression of MMP2 in gastric cancer cells. Dig Dis Sci. 2009;54(10):2056–62.
Xie TX, Wei D, Liu M, et al. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23(20):3550–60.
Xiao J, Gong Y, Chen Y, et al. IL-6 promotes epithelial-to-mesenchymal transition of human peritoneal mesothelial cells possibly through the JAK2/STAT3 signaling pathway. Am J Physiol Renal Physiol. 2017;313(2):F310–8.
Liu SC, Huang CM, Bamodu OA, et al. Ovatodiolide suppresses nasopharyngeal cancer by targeting stem cell-like population, inducing apoptosis, inhibiting EMT and dysregulating JAK/STAT signaling pathway. Phytomedicine. 2018;56:269–78.
Maycotte P, Jones KL, Goodall ML, et al. Autophagy supports breast cancer stem cell maintenance by regulating IL6 secretion. Mol Cancer Res. 2015;13(4):651–8.
Granato M, Rizzello C, Gilardini Montani MS, et al. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J Nutr Biochem. 2017;41:124–36.
Ferraresi A, Phadngam S, Morani F, et al. Resveratrol inhibits IL-6-induced ovarian cancer cell migration through epigenetic up-regulation of autophagy. Mol Carcinog. 2017;56(3):1164–81.
Hu F, Zhao Y, Yu Y, et al. Docetaxel-mediated autophagy promotes chemoresistance in castration-resistant prostate cancer cells by inhibiting STAT3. Cancer Lett. 2018;416:24–30.
Canel M, Serrels A, Miller D, et al. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: effects on E-cadherin dynamics. Cancer Res. 2010;70(22):9413–22.
Sheng W, Chen C, Dong M, et al. Calreticulin promotes EGF-induced EMT in pancreatic cancer cells via integrin/EGFR-ERK/MAPK signaling pathway. Cell Death Dis. 2017;8(10):e3147.
Bravou V, Klironomos G, Papadaki E, et al. ILK over-expression in human colon cancer progression correlates with activation of beta-catenin, down-regulation of E-cadherin and activation of the Akt-FKHR pathway. J Pathol. 2006;208(1):91–9.
Feldkoren B, Hutchinson R, Rapoport Y, et al. Integrin signaling potentiates transforming growth factor-beta 1 (TGF-beta1) dependent down-regulation of E-cadherin expression - important implications for epithelial to mesenchymal transition (EMT) in renal cell carcinoma. Exp Cell Res. 2017;355(2):57–66.
Pang M, Wang H, Rao P, et al. Autophagy links beta-catenin and Smad signaling to promote epithelial-mesenchymal transition via upregulation of integrin linked kinase. Int J Biochem Cell Biol. 2016;76:123–34.
Sosa P, Alcalde-Estevez E, Plaza P, et al. Hyperphosphatemia promotes senescence of myoblasts by impairing autophagy through ilk overexpression, a possible mechanism involved in sarcopenia. Aging Dis. 2018;9(5):769–84.
Andre P, Song H, Kim W, et al. Wnt5a and Wnt11 regulate mammalian anterior-posterior axis elongation. Development. 2015;142(8):1516–27.
Shin S, Im HJ, Kwon YJ, et al. Human steroid sulfatase induces Wnt/beta-catenin signaling and epithelial-mesenchymal transition by upregulating Twist1 and HIF-1alpha in human prostate and cervical cancer cells. Oncotarget. 2017;8(37):61604–17.
Ha JH, Ward JD, Radhakrishnan R, et al. Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Galphai2, Src, and HIF1alpha signaling nexus. Oncotarget. 2016;7(25):37664–79.
Lo HW, Hsu SC, Xia W, et al. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007;67(19):9066–76.
Yu X, Zheng Y, Zhu X, et al. Osteopontin promotes hepatocellular carcinoma progression via the PI3K/AKT/Twist signaling pathway. Oncol Lett. 2018;16(4):5299–308.
Zhang J, Tian XJ, Xing J. Signal transduction pathways of EMT induced by TGF-beta, SHH, and WNT and their crosstalks. J Clin Med. 2016;5(4). https://doi.org/10.3390/jcm5040041.
Qiang L, Zhao B, Ming M, et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A. 2014;111(25):9241–6.
Qiang L, He YY. Autophagy deficiency stabilizes TWIST1 to promote epithelial-mesenchymal transition. Autophagy. 2014;10(10):1864–5.
Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205.
Cheng M, Xue H, Cao W, et al. Receptor for activated C kinase 1 (RACK1) promotes Dishevelled protein degradation via autophagy and antagonizes Wnt signaling. J Biol Chem. 2016;291(24):12871–9.
Gao C, Cao W, Bao L, et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol. 2010;12(8):781–90.
Ma B, Liu B, Cao W, et al. The Wnt signaling antagonist Dapper1 accelerates Dishevelled2 degradation via promoting its ubiquitination and aggregate-induced autophagy. J Biol Chem. 2015;290(19):12346–54.
Zhang Y, Wang F, Han L, et al. GABARAPL1 negatively regulates Wnt/beta-catenin signaling by mediating Dvl2 degradation through the autophagy pathway. Cell Physiol Biochem. 2011;27(5):503–12.
Gugnoni M, Sancisi V, Gandolfi G, et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene. 2017;36(5):667–77.
Miao Y, Zhang Y, Chen Y, et al. GABARAP is overexpressed in colorectal carcinoma and correlates with shortened patient survival. Hepatogastroenterology. 2010;57(98):257–61.
Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17(5):548–458.
Pires BR, Mencalha AL, Ferreira GM, et al. NF-kappaB is involved in the regulation of EMT genes in breast cancer cells. PLoS One. 2017;12(1):e0169622.
Liu H, Xiong J, He T, et al. High uric acid-induced epithelial-mesenchymal transition of renal tubular epithelial cells via the TLR4/NF-kB signaling pathway. Am J Nephrol. 2017;46(4):333–42.
Cichon MA, Radisky DC. ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-kB-dependent activation of snail. Oncotarget. 2014;5(9):2827–38.
Wu Y, Deng J, Rychahou PG, et al. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15(5):416–28.
Nopparat C, Sinjanakhom P, Govitrapong P. Melatonin reverses H2 O2 -induced senescence in SH-SY5Y cells by enhancing autophagy via sirtuin 1 deacetylation of the RelA/p65 subunit of NF-kappaB. J Pineal Res. 2017;63(1).
Sun X, Li L, Ma HG, et al. Bisindolylmaleimide alkaloid BMA-155Cl induces autophagy and apoptosis in human hepatocarcinoma HepG-2 cells through the NF-kappaB p65 pathway. Acta Pharmacol Sin. 2017;38(4):524–38.
He ZJ, Zhu FY, Li SS, et al. Inhibiting ROS-NF-kappaB-dependent autophagy enhanced brazilin-induced apoptosis in head and neck squamous cell carcinoma. Food Chem Toxicol. 2017;101:55–66.
Huang M, Xin W. Matrine inhibiting pancreatic cells epithelial-mesenchymal transition and invasion through ROS/NF-kappaB/MMPs pathway. Life Sci. 2018;192:55–61.
Katsuno Y, Lamouille S, Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25(1):76–84.
Miyazono K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85(8):314–23.
Li J, Yang B, Zhou Q, et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis. 2013;34(6):1343–51.
Zhu L, Fu X, Chen X, et al. M2 macrophages induce EMT through the TGF-beta/Smad2 signaling pathway. Cell Biol Int. 2017;41(9):960–8.
Carito V, Bonuccelli G, Martinez-Outschoorn UE, et al. Metabolic remodeling of the tumor microenvironment: migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle. 2012;11(18):3403–14.
Saitoh M, Endo K, Furuya S, et al. STAT3 integrates cooperative Ras and TGF-beta signals that induce snail expression. Oncogene. 2016;35(8):1049–57.
Ma F, Li W, Liu C, et al. MiR-23a promotes TGF-beta1-induced EMT and tumor metastasis in breast cancer cells by directly targeting CDH1 and activating Wnt/beta-catenin signaling. Oncotarget. 2017;8(41):69538–50.
Kong FF, Zhu YL, Yuan HH, et al. FOXM1 regulated by ERK pathway mediates TGF-beta1-induced EMT in NSCLC. Oncol Res. 2014;22(1):29–37.
Shen J, Zhao DS, Li MZ. TGF-beta1 promotes human gastric carcinoma SGC7901 cells invasion by inducing autophagy. Eur Rev Med Pharmacol Sci. 2017;21(5):1013–9.
Suzuki HI, Kiyono K, Miyazono K. Regulation of autophagy by transforming growth factor-beta (TGF-beta) signaling. Autophagy. 2010;6(5):645–7.
Zhang C, Zhang X, Xu R, et al. TGF-beta2 initiates autophagy via Smad and non-Smad pathway to promote glioma cells’ invasion. J Exp Clin Cancer Res. 2017;36(1):162.
Hu S, Wang L, Zhang X, et al. Autophagy induces transforming growth factor-beta-dependent epithelial-mesenchymal transition in hepatocarcinoma cells through cAMP response element binding signalling. J Cell Mol Med. 2018;22(11):5518–32.
Peng JM, Bera R, Chiou CY, et al. Actin cytoskeleton remodeling drives epithelial-mesenchymal transition for hepatoma invasion and metastasis in mice. Hepatology. 2018;67(6):2226–43.
Huang D, Cao L, Xiao L, et al. Hypoxia induces actin cytoskeleton remodeling by regulating the binding of CAPZA1 to F-actin via PIP2 to drive EMT in hepatocellular carcinoma. Cancer Lett. 2019;448:117–27.
Huang D, Cao L, Zheng S. CAPZA1 modulates EMT by regulating actin cytoskeleton remodelling in hepatocellular carcinoma. J Exp Clin Cancer Res. 2017;36(1):13.
Aguilera MO, Beron W, Colombo MI. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy. 2012;8(11):1590–603.
Kast DJ, Dominguez R. The cytoskeleton-autophagy connection. Curr Biol. 2017;27(8):R318–26.
Kashatus JA, Nascimento A, Myers LJ, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57(3):537–51.
Serasinghe MN, Wieder SY, Renault TT, et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell. 2015;57(3):521–36.
Bartolak-Suki E, Imsirovic J, Nishibori Y, et al. Regulation of mitochondrial structure and dynamics by the cytoskeleton and mechanical factors. Int J Mol Sci. 2017;18(8). https://doi.org/10.3390/ijms18081812.
Gugnoni M, Sancisi V, Manzotti G, et al. Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer. Cell Death Dis. 2016;7(12):e2520.
Shi C, Cai Y, Li Y, et al. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMK II pathways. Redox Biol. 2018;14:59–71.
Maes H, Van Eygen S, Krysko DV, et al. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 2014;5:e1127.
Zhao Z, Zhao J, Xue J, et al. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am J Cancer Res. 2016;6(10):2162–77.
Qin W, Li C, Zheng W, et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget. 2015;6(37):39839–54.
Lv Q, Wang W, Xue J, et al. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res. 2012;72(13):3238–50.
Lv Q, Hua F, Hu ZW. DEDD, a novel tumor repressor, reverses epithelial-mesenchymal transition by activating selective autophagy. Autophagy. 2012;8(11):1675–6.
Jiang GM, Tan Y, Wang H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019;18(1):17.
Ouyang F, Huang H, Zhang M, et al. HMGB1 induces apoptosis and EMT in association with increased autophagy following H/R injury in cardiomyocytes. Int J Mol Med. 2016;37(3):679–89.
Li XF, Chen DP, Ouyang FZ, et al. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J Hepatol. 2015;62(1):131–9.
Lock R, Kenific CM, Leidal AM, et al. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4(4):466–79.
Dash S, Sarashetti PM, Rajashekar B, et al. TGF-beta2-induced EMT is dampened by inhibition of autophagy and TNF-alpha treatment. Oncotarget. 2018;9(5):6433–49.
Su Z, Li G, Liu C, et al. Autophagy inhibition impairs the epithelial-mesenchymal transition and enhances cisplatin sensitivity in nasopharyngeal carcinoma. Oncol Lett. 2017;13(6):4147–54.
Tong H, Yin H, Hossain MA, et al. Starvation-induced autophagy promotes the invasion and migration of human bladder cancer cells via TGF-beta1/Smad3-mediated epithelial-mesenchymal transition activation. J Cell Biochem. 2019;120(4):5118–27.
Wang H, Zhang Y, Wu Q, et al. miR-16 mimics inhibit TGF-beta1-induced epithelial-to-mesenchymal transition via activation of autophagy in non-small cell lung carcinoma cells. Oncol Rep. 2018;39(1):247–54.
Jiang Y, Jiao Y, Liu Y, et al. Sinomenine hydrochloride inhibits the metastasis of human glioblastoma cells by suppressing the expression of matrix Metalloproteinase-2/−9 and reversing the endogenous and exogenous epithelial-mesenchymal transition. Int J Mol Sci. 2018;19(3). https://doi.org/10.3390/ijms19030844.
Wang Y, Liu Y, Lu J, et al. Rapamycin inhibits FBXW7 loss-induced epithelial-mesenchymal transition and cancer stem cell-like characteristics in colorectal cancer cells. Biochem Biophys Res Commun. 2013;434(2):352–6.
Reka AK, Kuick R, Kurapati H, et al. Identifying inhibitors of epithelial-mesenchymal transition by connectivity map-based systems approach. J Thorac Oncol. 2011;6(11):1784–92.
Ye R, Dai N, He Q, et al. Comprehensive anti-tumor effect of Brusatol through inhibition of cell viability and promotion of apoptosis caused by autophagy via the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Biomed Pharmacother. 2018;105:962–73.
Chen X, Chen Y, Lin X, et al. The drug combination of SB202190 and SP600125 significantly inhibit the growth and metastasis of Olaparib-resistant ovarian cancer cell. Curr Pharm Biotechnol. 2018;19(6):506–13.
Niu NK, Wang ZL, Pan ST, et al. Pro-apoptotic and pro-autophagic effects of the Aurora kinase a inhibitor alisertib (MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the activation of mitochondria-mediated pathway and inhibition of p38 MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther. 2015;9:1555–84.
Liao H, Huang Y, Guo B, et al. Dramatic antitumor effects of the dual mTORC1 and mTORC2 inhibitor AZD2014 in hepatocellular carcinoma. Am J Cancer Res. 2015;5(1):125–39.
Funding
This work was funded by the National Natural Science Foundation of China (No. 81502104; No. 81430041; No. 81620108017), National Program on Key Basic Research Project (No. 2018YFC0910600), Hunan Young Talent (No. 2017RS3051), Key Laboratory of Biomedical Imaging of Guangdong Province (2018B030322006).
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Author information
Authors and Affiliations
Contributions
Conception and design: GMJ, HS; Writing: HTC, HL, MJM, YT, XQM, XJM, MTC, CYZ, YL; review, and/or revision of the manuscript: GMJ, HS. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Authors’ information
As mentioned in the cover page.
Ethics approval and consent to participate
Not applicable’ for this section.
Consent for publication
The authors confirmed that we are consent for publishing the manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, HT., Liu, H., Mao, MJ. et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol Cancer 18, 101 (2019). https://doi.org/10.1186/s12943-019-1030-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-1030-2