
Abstract
Although solid tumors comprise malignant cells, they also contain many different non-malignant cell types in their micro-environment. The cellular components of the tumor stroma consist of immune and endothelial cells combined with a heterogeneous population of stromal cells which include cancer-associated fibroblasts. The bi-directional interactions between tumor and stromal cells therefore substantially affect tumor cell biology.
Herein, we discuss current available information on these interactions in breast cancer chemo-resistance. It is acknowledged that stromal cells extrinsically alter tumor cell drug responses with profound consequences for therapy efficiency, and it is therefore essential to understand the molecular mechanisms which contribute to these substantial alterations because they provide potential targets for improved cancer therapy. Although breast cancer patient survival has improved over the last decades, chemo-resistance still remains a significant obstacle to successful treatment.
Appreciating the important experimental evidence of mesenchymal stromal cells and cancer-associated fibroblast involvement in breast cancer clinical practice can therefore have important therapeutic implications.
Similar content being viewed by others
Introduction
Breast cancer is the most frequent cancer diagnosed in women, and is one of the greatest causes of global female death. In addition, the American Cancer Society reports this is 25% of all new cancer diagnoses in women world-wide (American Cancer Society, Cancer Facts and Figs. 2017). Breast cancer is a heterogeneous disease classified in the following three main groups based on immuno-histochemical analysis: (I) estrogen receptor ER(α)-positive, (II) human epidermal growth factor receptor Her2 positive and (III) triple negative (ER(α)-negative, progesterone receptor (PR) negative and Her2-negative. Further sub-typing is based on gene expression profiling which unraveled the gene cluster which is mostly expressed in luminal breast cells, myo-epithelial basal cells and cells associated with increased expression of Her2.
These sub-types are named “luminal-like, basal-like and Her2-enriched” [1], and the profiling also identified clinically important sub-types in these three molecular groups. For example, the luminal A and B subtypes induce different patient prognosis, where patients carrying the luminal B tumor type have worse prognosis [2, 3] and the basal-like and claudinlow subtype express mesenchymal markers such as vimentin. While this is present in epithelial tumors, it is not a component of normal breast tissue [4].
However, tumor cells alone do not drive tumor growth or progression. Despite early detection and increased knowledge of breast cancer biology, approximately 30% of patients with breast cancer experience recurrence. The relapse usually occurs in patients with adenocarcinoma cells with chemo-resistant phenotype; and while this was previously linked to tumor cell genetic alterations, it is now acknowledged that adjacent tissue surrounding tumor cells has an important role in tumor progression and resistance [5]. It is also evident that many “normal” cells add to tumor diversity by varying the micro-environment composition, stromal cell proportions and/or activation states.
In addition to malignant cells and various non-malignant cell populations, solid tumors also contain an extracellular matrix (ECM) which forms a complex tumor micro-environment (TME) or tumor stroma. These stromal cells, ECM, soluble factors and the physical state of the tumor microenvironment all affect solid tumor behavior in a complex manner [6]. Moreover, TME is now considered a hallmark of cancer biology [7], and researching the molecular characteristics and interactions between TME components and tumor cells is expected to produce important knowledge for developing new therapeutic approaches.
Tumor drug responses are not exclusively determined by the tumor cell’s intrinsic characteristics because tumor-associated stromal cells, including fibroblasts, mesenchymal stromal cells (MSCs), immuno-inflammatory cells, vascular endothelial cells and the ECM combine in response to anti-cancer treatment. These components influence the proliferation, invasion and metastasis of tumor cells [8], and the adjacent adipose tissue provides a rich source of MSCs which significantly contribute to stromal constituents in the breast cancer tumor micro-environment.
Many experimental studies have also confirmed that MSCs interact with breast cancer cells. They possess “homing ability” to breast cancer tissue and release growth factors which consequently promote migration and epithelial-to-mesenchymal transition (EMT). However, different reports on MSCs’ influence on chemotherapy response have produced contradictory findings, and while some studies have reported that MSCs contribute to increased breast cancer cell chemo-resistance [9,10,11,12], our results indicate that MSCs may even act as a drug sensitizer [13, 14].
To improve insight into tumor development and chemotherapeutic approaches, it is most important to understand the interplay between specific TME components, the associated cellular communication processes and resultant interactions of this network between cancer cells and the various tumor-associated cell populations. Here, we focus on the molecular communication between stromal cells, mainly MSCs and breast cancer cells, and the cell-to-cell signaling role and its effect on chemotherapy efficiency.
Cellular components of stroma in breast tumors
Tumor tissue is a heterogeneous mixture of cells, where cancer cells are surrounded by disorganized blood vessels formed by endothelial cells, lymphatic vessels, infiltrated immune cells (T cells, natural killers (NKs) and macrophages), adipocytes, fibroblasts and MSCs. Some of these cells exist in the tissue before tumor development and others are recruited to the micro-environment by the tumor cells [15, 16]. TME heterogeneity depends on location within the tumor, and TME cells located at the tumor periphery can significantly differ from cell types at the tumor core [17]. This is due to randomly generated mutations in the tumor cells, immune cell infiltration, tumor cell necrosis and interstitial pressure [18]. While each tumor has unique TME, critical TME components and their role in tumor progression remain similar in different cancers. Bi-directional communication between cells and their micro-environment is necessary for normal tissue homeostasis. However, it is also required for tumor growth, and therefore interaction between cancer cells and the surrounding stroma is an important relationship that alters all cell phenotypes, proliferation and metabolism. This communication also affects disease initiation and progression; and thus influences patient prognosis [19, 20].
This review specifically focuses on novel findings in the contribution of MSCs and cancer-associated fibroblasts (CAFs) in breast cancer chemo-resistance. Although the link between MSCs and CAFs remains undetermined, recent studies suggest they may have similar characteristics and pro-tumorigenic activity. In contrast, however, Su et al. found no overlap between these components [21, 22].
Mesenchymal stromal cells
Mesenchymal stem/stromal cells are multipotent spindle-shaped cells first described in the 1960’s as hematopoietic bone marrow supportive cells [23, 24]. Multiple populations of MSCs have now been derived from the plethora of adult and fetal tissues reviewed by Ullah and colleagues [25].
The term “mesenchymal stem cells” was popularized by Arnold Caplan many years later, in the belief that they can give rise to bone, cartilage, tendon, ligament, marrow stroma, adipocytes, dermis, muscle and connective tissue [26]. The International Society for Cellular Therapy (ISCT) recommended the term “multipotent mesenchymal stromal cells” because support for their “stemness” in vivo was lacking [27] and it further proposed minimum criteria to define MSCs [28]. The expression of negative surface marker CD34, however, remains controversial [29].
These characteristics are valid for all MSCs, but some differences still exist in isolates derived from various tissue types. Many studies reported additional MSC markers dependent on aspiration source. For example, stromal precursor antigen-1 (Stro-1) was identified as a “stemness” marker for the MSCs [30] and dental [31] and bone marrow-derived MSCs (BM-MSCs) [32] were reported to be Stro-1 positive while the adipose tissue-derived (AT-MSCs) are negative [33].
BM-MSCs and AT-MSCs share many important characteristics and few differences [34]. The AT-MSCs are more genetically stable in long-term culture, have lower senescence ratio, higher proliferative capacity and retain their differentiation potential for a longer period in culture than BM-MSCs [35]. In addition, AT-MSCs support haematopoiesis both in vitro and in vivo more efficiently than the BM-MSCs [36], and they also have significantly higher angiogenic potential [37]. Moreover, higher numbers of AT-MSCs are easily isolated from subcutaneous adipose tissue aspirate. This operation can be repetitive utilizing liposuction with minimal invasiveness, thus rendering this an attractive MSC source [38].
Defined by their ability to differentiate into multiple stromal cell lineages, the MSCs can be found in most body parts and they can migrate throughout the organism and into tumor tissue [39]. Therefore, tumors are sometimes considered “wounds that do not heal” because of chronic inflammation, immune cell infiltration and neo-vascularization [40]. Migration of MSCs to injury enables TME to recruit these cells by releasing inflammatory molecules, growth factors and cytokines. Although they preferentially “home and engraft” in tumors from the bone marrow, which is the major MSC reservoir, they also emanate from surrounding adipose tissue.
In addition, MSCs in TME can easily differentiate into CAFs [41], and MSC-like CAFs that express FSP and FAP [42] originate from BM-MSCs whereas the AT-MSCs mainly differentiate into vascular and fibro-vascular stromal cells [43, 44]. Here, it is also important to note that normal healthy tissues have almost no detectable FAP expression.
MSC migration to tumors leads to cellular interactions with tumor cells and TME components. This occurs both directly through gap junctions, membrane receptors and nanotubes and indirectly by soluble molecules [45]. The MSCs stimulate adjacent cells by releasing endocrine and paracrine signals. In turn, MSCs can be stimulated by tumor cells and develop an aberrant tumor-associated phenotype [46]. Consequently, they either promote or inhibit tumor cell growth [47, 48].
Reduction of tumor growth by MSCs can be mediated by inhibiting angiogenesis, suppressing the Wnt and AKT signaling pathways or inducing cell cycle arrest and apoptosis [46, 47, 49]. Thus, aberrant tumor-associated MSCs can acquire different functions after interaction with tumor cells. These include TGF-β secretion which contributes to both EMT and immune system suppression. Moreover, these MSCs release VEGF for neo-vascularization in the TME and produce CXCL12 to support tumor cell growth and survival [50]. While P2X signaling was recently identified as a pathway favoring MSCs-mediated breast cancer cell proliferation [51], high IFN-β expression suppresses human breast cancer cell growth [52]. Therefore, TME MSCs have either pro- or anti-tumorigenic properties depending on the cancer cell properties and the experimental settings [53].
Bartosh et al’s seminal research identified the remarkable phenomenon of cancer cell cannibalism and the acquired senescence-associated secretory phenotype (SASP). The authors discovered that breast cancer cells in 3D co-cultures entered dormancy after internalizing and degrading human BM-MSCs. The cannibalistic breast cancer cells then became highly resistant to chemotherapy and other stresses caused by nutrition deprivation. Most interestingly, these secreted SASP factors enabled dormant breast cancer cells to communicate with the various TME components [54].
MSCs provide a promising tool for many types of anti-tumor therapies because of their role in the TME; and this was comprehensively summarized in Valkenburg et al [55].
Fibroblasts in breast cancer
Fibroblasts are non-vascular, non-inflammatory, non-epithelial cells in connective tissue. They secrete the extracellular matrix (ECM) and basement membrane components, regulate epithelial cell differentiation, modulate immune system responses and maintain homeostasis [56]. Activated fibroblasts are termed “cancer-associated fibroblasts” (CAFs), and they are major stromal cells contributing to the TME. When activated by direct contact with leukocytes or secreted factors, including TGF-β, PDGF, FGF2, EGF and CXCL12 [57], CAFs promote tumor growth, increase angiogenesis, degrade ECM to release signaling molecules and promote EMT and metastasis [56]. Although CAFs were first considered tumor developmental elements lacking effect on cancer cells, they have since been identified as essential components of tumor progression [58].
The CAFs can be derived not only from normal fibroblasts, but also from other types of cells, including MSCs, epithelial cells, pericytes, adipocytes and endothelial cells [59]. An interaction between tumor-induced fibroblast activation and fibroblast-induced tumor proliferation and metastasis has been proven, thus it can be concluded that CAFs act as tumor supporters [60].
CAFs are present in the TME in aberrantly high numbers and differ from normal fibroblasts in many morphological and biological ways. CAFs are functionally defined by intensive proliferation and high ECM deposition, and further acknowledged as “activated myofibroblasts that cannot regress to an inactivated state” [61].
The CAFs exhibit differential gene expression of several factors compared to normal fibroblasts. Membrane protein FAPα, selectively expressed in activated CAFs, is one of the most important markers of these cells [62], and FSP-1, podoplanin-a, S100A4 protein, vimentin and PDGF receptors α and β are also highly expressed in CAFs [63]. Most recently, the IGFBP7 protein has been identified as a novel biomarker of tumor fibroblasts. IGFBP7-expressing CAFs have been demonstrated to promote colon cancer cell proliferation through paracrine tumor-stroma interactions in vitro [64]. In addition, TGF-β2 expression in CAFs was previously identified in metastatic colon cancer [65].
In summary, 46 differentially expressed genes regulated by the transforming growth factor (TGF)-β signaling pathway were identified in CAF cell lines compared to normal fibroblast cell lines [66]. All these genes encode for paracrine factors released into the TME. Moreover, numerous altered gene transcripts have been identified in breast CAFs, including ribosomal protein S6 kinase α3, FGF receptor 1, nardilysin and cyclin-dependent kinase inhibitor 1B [67].
Su et al. also recently identified the CD10 and GPR77 fibroblast-associated cell-surface molecules not previously described. These specifically define a CAF sub-population which promotes chemo-resistance and cancer formation in breast and lung cancer patients. CD10+GPR77+ CAFs secrete abundant interleukins IL-6 and IL-8 which provide a survival niche for cancer stem cells (CSCs) via continuous NF-κB signaling. Although most CAFs are relatively genetically stable, and therefore present a potential therapeutic target with lower risk of developing chemo-resistance [68], increasing data suggests that fibroblasts’ protective role enables cancer cells to evade the cytotoxic effects of chemotherapy. For example, HGF has been identified as an essential factor in the CAF-mediated resistance to lapatinib in HER2+ breast cancer [69]; and CAFs may also act as a physical barrier against anti-tumor drugs and decrease their availability to tumor cells.
Chemo-resistance in breast cancer and association with “stemness” phenotype
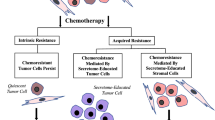
Chemo-resistance can be an intrinsic and inherent feature of tumor cells, where this is often associated with their quiescent state prior to treatment. In contrast, acquired resistance occurs despite initial positive response to therapy [70]. There are many mechanisms of resistance that include different involved cells and signaling pathways; dependent on the cancer type (Fig. 1). Up-regulation of the cancer stem cell phenotype can be critical in resistance to a variety of drugs in cancer treatments; including breast cancer treatment [71]. CSCs have increased capacity to actively export many drugs from cells by over-expressing ATP-binding cassette (ABC) drug transporter proteins. Moreover, CSCs have higher anti-apoptotic gene expression and a more effective DNA repair system [72].

Scheme of possible outcomes following chemotherapy treatment. There are three possible outcomes from chemotherapy treatment. a Intrinsic resistance to chemotherapy associated with the tumor cell quiescent state is usually defined by over-expression of ABC drug transporters, different anti-apoptotic genes and a more effective DNA repair system. b Acquired resistance that occurs despite initial positive response to therapy; here, different signaling pathways between secretome-educated cancer cells and MSCs are involved and c) Acquired chemo-resistance mediated by secretome-educated stromal cells through altered secretion profiles and different signaling pathways
The TME provides shelter for CSCs; thus inducing therapy resistance and tumor development. However, traditional cancer treatments, including the majority of chemotherapeutic agents and radiation, target actively dividing cells, and while they reduce tumor mass, they do not effectively remove quiescent cells such as CSCs. This can lead to tumor recurrence. For example, breast CSCs have paclitaxel resistance [73], and also decreased ROS expression which is critical in inducing DNA damage by ionizing radiation [74], but the TME blocks drug penetration and suppresses immune system responses [75]. Herein, we focus on the mechanisms involved in acquired chemo-resistance mediated by stromal cells in breast tumor TME. This is tightly linked to their mutual interactions and the “stemness-phenotype-support” exerted by MSCs and CAFs.
Chemo-resistance mediated by mesenchymal stromal cells
MSCs usually interact with breast CSCs through IL-6 and CXCL7 cytokine secretion. This signaling is responsible for the self-renewal potential of breast CSCs. Cytokines such as SDF-1 (CXCL12) produced by CAFs can also promote the proliferation of cancer cells carrying the SDF-1 receptor CXCR4; where SDF-1 expression-level correlates with breast cancer survival [76].
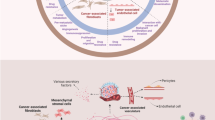
Multiple signaling pathways have been ascribed to the MSC- and CAF-mediated drug resistance in breast cancer, and these are often associated with induction of the “stemness” phenotype. Moreover, the protective effect of MSCs on breast cancer cells against cytotoxic drugs appears to need both secretory proteins and direct cell-to-cell interaction (Fig. 2). Here, IL-6 has an important role in acquired breast cancer chemo-resistance through its secretion by MSC which promotes great impact on the stimulation of ERα-positive breast cancer cell proliferation [77, 78]. In addition, IL-6 has proven protective effect against paclitaxel and doxorubicin in ERα-positive breast cancer [9, 79], and also against trastuzumab in Her-2 positive tumors [80]. However, IL-6 released by breast cancer cells mediates “homing” of MSCs into primary tumor sites, and then interacts with its MSC receptor to induce MSC CXCL7 secretion. These cytokines work together to provide chemokine networks that influence CSCs to promote resistance to anti-cancer drugs [81].

Chemo-resistance mechanisms mediated by MSCs in breast cancer. Communication between MSCs and breast cancer cells leading to resistance against cytotoxic drugs mostly involves secretory proteins. Following chemotherapy treatment, MSCs secrete increased levels of cytokines (IL-6, IL-8, CXCL1, CXCL7, TGFβ), PUFAs (not shown), plus hyaluronan, exososomes and other molecules. All these are involved in complex inter-cellular communication that ultimately manifests as a chemo-resistant cancer cell phenotype. Direct cell-to-cell interactions are also involved, especially through the functional gap junctions and connexin-interacting proteins and direct receptor communication
The MSC secreted CXCL1 cytokine enhances expression of ABCG2 by altered miR-106a expression in triple negative breast cancer cells. ABCG2 is also known as the breast cancer resistance protein (BRCP) and it is the ATP-binding cassette transporter protein responsible for the efflux of doxorubicin, and causes resistance to this drug [11]. Similarly, IL-8 secreted by MSCs increases ABCG2 expression. This results in reduced intracellular doxorubicin accumulation in triple negative breast cancer cells [10].
MSCs also produce abundant levels of transforming growth factor β (TGF-β) and this signaling pathway can trigger epithelial to mesenchymal transition (EMT). Thus, TGF-β contributes to the MSC drug protective effect by inducing EMT. MSCs can also promote EMT by the different mechanism of producing hyaluronan which enables MSCs to make CD44-expressing breast cancer cells produce lysyl oxidase (LOX). This then causes ECM stiffening by catalyzing collagen fiber cross-linking, and facilitates ECM-induced drug resistance [82]. Moreover, it also stimulates the expression of the Twist EMT-mediating transcription factor. In addition, hyaluronan can also be produced by breast cancer cells as a result of MSC secretion of basic fibroblast growth factor [83, 84]. Thus, the MSCs can foster hyaluronan accumulation; and hyaluronan excess in breast tumor stroma induces doxorubicin resistance [83].
The importance of cell-to-cell interaction between MSCs and breast cancer cells in acquired drug resistance is highlighted by MSC presence inducing over-expression of HER-2 and loss of PTEN. This indicates that MSCs regulate HER-2 receptor and PTEN tumor suppressor interaction in breast cancer cells by activating Src which induces subsequent PTEN inactivation. Therefore, Src and its downstream PI3K/Akt signaling pathway enhance resistance to trastuzumab [85].
Further recent study has shown that MSCs induce increased expression of PAG1/Cbp; a transmembrane adaptor protein which enhances resistance to adriamycin hydrochloride (ADMh) [86]. PAG1/Cbp is ubiquitously expressed in lipid rafts and is significantly involved in many signaling pathways that include Src-associated signaling and the AKT/mTOR pathway. Activation of Cbp leads to activation of Src and thus it can enhance resistance to ADMh; and most likely also to trastuzumab.
MSCs also secrete factors which protect carcinoma cells against platinum-based chemotherapeutics [87]. These include two types of polyunsaturated fatty acids (PUFAs), 12-oxo-5,8,10-heptadecatrienoic acid (KHT) and hexadeca-4,7,10,13-tetraenoic acid (16:4(n-3)). In minute quantities, these both induce resistance to a broad spectrum of chemotherapeutic agents. Central enzyme blocking involved in the production of these PUFAs (cyclooxygenase-1 and thromboxane synthase) prevents MSC-induced resistance. These combined findings show that MSCs are potent mediators of resistance to chemotherapy and important targets in enhancing patient treatment efficacy [12].
In addition to the above-stated molecular mechanisms, drug resistance is also mediated by exosomes derived from MSCs. These small cell-derived vesicles contain miR-23b; an miRNA inhibiting myristoylated alanine-rich C-kinase substrate (MARCKS). This is a prominent cellular substrate for protein kinase C, and its inhibition leads to breast CSC dormancy in the metastatic niche, and thereby to docetaxel treatment resistance [88].
However, there is also evidence that MSCs can not always protect tumor cells against cytotoxic drugs. Their protective activity depends on the type of the drug and also on the type of cancer cell. In addition, some reports suggest that MSCs can act as drug sensitizers. For example, BM-MSCs can sensitize breast cancer cell lines to kinase inhibitors [89], and AT-MSCs are able to render Her-2 positive breast cancer cells more sensitive to doxorubicin and 5-fluorouracil [13]. Interestingly, while some cancer cell lines react to MSCs by cell cycle arrest, others display higher proliferation activity in their presence [8, 90, 91]. Hence, cell cycle arrest is a potent mechanism enabling cancer cells to escape cytotoxic drug effects.
Conclusions
Future cancer therapy success depends on thoroughly understanding the many complex mechanisms involved, and establishing the pathways prominent in resistance to anti-cancer treatment. Developing methods of targeting them is then essential. There is also rapidly increasing research on the tumor micro-environment (TME) and its role in chemo-resistance acquisition, subsequent treatment failure and cancer recurrence. It is therefore critical that the TME is acknowledged as an important cancer-target strategy, and that further TME investigation is initiated.
While the TME in all breast cancer sub-types acts through a network of secreted molecules, adipose tissue is most important in mediating communication between the TME and breast cancer cells because especially in the breast it forms a major part of the tumor environment. Therefore mesenchymal stromal cells from adjacent adipose tissue, and especially the cancer-associated fibroblasts in the tumor-microenvironment, are of utmost importance in processes associated with cancer progression and resistance to therapy.
Finally, recent research stresses that stromal-cell-mediated protection against cytotoxic drugs requires both secretory proteins and direct cell-cell interactions. Therefore, further research into these processes is anticipated to provide better understanding of their effects on therapy resistance and hasten the design of effective therapeutic strategies and personalized regimens for breast-cancer patients.
Abbreviations
- ABCG2:
-
ATP-binding cassette super-family G member 2
- ADMh:
-
Adriamycin hydrochloride
- AT-MSCs:
-
Adipose tissue-derived mesenchymal stromal cells
- bFGF:
-
Basic fibroblast growth factor
- BM-MSCs:
-
Bone marrow-derived mesenchymal stromal cells
- BRCP:
-
Breast cancer resistance protein
- CAFs:
-
Cancer-associated fibroblasts
- CSCs:
-
Cancer stem cells
- CXCL:
-
C-X-C motif chemokine ligand
- CXCR4:
-
C-X-C chemokine receptor type 4
- ECM:
-
Extracellular matrix
- EGF:
-
Epidermal growth factor
- EMT:
-
Epithelial-to-mesenchymal transition
- ER:
-
Estrogen receptor
- FAPα:
-
Fibroblast activation protein-α
- FGF:
-
Fibroblastic growth factor
- FSP-1:
-
Fibroblast specific protein 1
- GPR77:
-
G protein-coupled receptor 77
- HER2:
-
Human epidermal growth factor receptor 2
- HGF:
-
Hepatocyte growth factor
- IFN-β:
-
Interferon beta
- IGFBP7:
-
Insulin-like growth factor-binding protein 7
- IL:
-
Interleukin
- ISCT:
-
International Society for Cellular Therapy
- LOX:
-
lysyl oxidase
- MARCKS:
-
Myristoylated alanine-rich C-kinase substrate
- MSCs:
-
Mesenchymal stromal cells
- NF-κB:
-
Nuclear factor kappa- B
- NKs:
-
Natural killers
- PAG1/Cbp:
-
C-terminal Src kinase (Csk)-binding protein (Cbp) encoded by PAG1
- PDGF:
-
Platelet-derived growth factor
- PR:
-
Progesterone receptor
- PTEN:
-
Phosphatase and tensin homolog
- PUFAs:
-
Polyunsaturated fatty acids
- SASP:
-
Senescence-associated secretory phenotype
- SDF-1:
-
Stromal cell-derived factor 1
- Stro-1:
-
Stromal precursor antigen-1
- TGF-β:
-
Transforming growth factor β
- TME:
-
Tumor microenvironment
- VEGF:
-
Vascular endothelial growth factor
References
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52 Epub 2000/08/30.
Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7 Epub 2009/02/11.
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–74 Epub 2001/09/13.
Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast cancer research : BCR. 2010;12(5):R68 Epub 2010/09/04.
Majidinia M, Yousefi B. Breast tumor stroma: a driving force in the development of resistance to therapies. Chem Biol Drug Des. 2017;89(3):309–18 Epub 2017/01/04.
McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov. 2013;12(3):217–28 Epub 2013/03/02.
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22 Epub 2012/03/24.
Dittmer J, Leyh B. The impact of tumor stroma on drug response in breast cancer. Semin Cancer Biol. 2015;31:3–15 Epub 2014/06/10.
Shi Z, Yang WM, Chen LP, Yang DH, Zhou Q, Zhu J, et al. Enhanced chemosensitization in multidrug-resistant human breast cancer cells by inhibition of IL-6 and IL-8 production. Breast Cancer Res Treat. 2012;135(3):737–47 Epub 2012/08/28.
Chen DR, Lu DY, Lin HY, Yeh WL. Mesenchymal stem cell-induced doxorubicin resistance in triple negative breast cancer. Biomed Res Int. 2014;2014:532161 Epub 2014/08/21.
Yeh WL, Tsai CF, Chen DR. Peri-foci adipose-derived stem cells promote chemoresistance in breast cancer. Stem Cell Res Ther. 2017;8(1):177 Epub 2017/07/29.
Roodhart JM, Daenen LG, Stigter EC, Prins HJ, Gerrits J, Houthuijzen JM, et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. 2011;20(3):370–83 Epub 2011/09/13.
Kucerova L, Skolekova S, Matuskova M, Bohac M, Kozovska Z. Altered features and increased chemosensitivity of human breast cancer cells mediated by adipose tissue-derived mesenchymal stromal cells. BMC Cancer. 2013;13:535 Epub 2013/11/12.
Kucerova L, Kovacovicova M, Polak S, Bohac M, Fedeles J, Palencar D, et al. Interaction of human adipose tissue-derived mesenchymal stromal cells with breast cancer cells. Neoplasma. 2011;58(5):361-70. Epub 2011/07/13.
Kucerova L, Skolekova S. Tumor microenvironment and the role of mesenchymal stromal cells. Neoplasma. 2013;60(1):1–10 Epub 2012/10/17.
Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18(1):84 Epub 2016/08/16.
Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral immune cell exploitation in colorectal Cancer metastases can be targeted effectively by anti-CCR5 therapy in Cancer patients. Cancer Cell. 2016;29(4):587–601 Epub 2016/04/14.
Orlando PA, Gatenby RA, Brown JS. Tumor evolution in space: the effects of competition colonization tradeoffs on tumor invasion dynamics. Front Oncol. 2013;3:45 Epub 2013/03/20.
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–52 Epub 2009/03/13.
Kucerova L, Durinikova E, Toro L, Cihova M, Miklikova S, Poturnajova M, et al. Targeted antitumor therapy mediated by prodrug-activating mesenchymal stromal cells. Cancer Lett. 2017;408:1–9 Epub 2017/08/26.
Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, et al. Cancer-associated fibroblasts share characteristics and Protumorigenic activity with mesenchymal stromal cells. Cancer Res. 2017;77(18):5142–57.
Su SC, Chen JN, Yao HR, Liu J, Yu SB, Lao LY, et al. CD10(+) GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell. 2018;172(4):841.
Friedenstein AJ, Deriglasova UF, Kulagina NN, Panasuk AF, Rudakowa SF, Luria EA, et al. Precursors for fibroblasts in different populations of hematopoietic cells as detected by the in vitro colony assay method. Exp Hematol. 1974;2(2):83–92 Epub 1974/01/01.
Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6(2):230–47 Epub 1968/03/01.
Ullah I, Subbarao RB, Rho GJ. Human mesenchymal stem cells - current trends and future prospective. Bioscience reports. 2015;35(2). Epub 2015/03/24.
Caplan AI. Mesenchymal stem cells. J Orthop Research. 1991;9(5):641–50 Epub 1991/09/01.
Edwin M, Horwitz MA. And Francesco Frassoni. Mesenchymal stromal cells. Curr Opin Hematol. 2006;13(6):419–25.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7 Epub 2006/08/23.
Lin CS, Ning H, Lin G, Lue TF. Is CD34 truly a negative marker for mesenchymal stromal cells? Cytotherapy. 2012;14(10):1159–63.
Park JC, Kim JM, Jung IH, Kim JC, Choi SH, Cho KS, et al. Isolation and characterization of human periodontal ligament (PDL) stem cells (PDLSCs) from the inflamed PDL tissue: in vitro and in vivo evaluations. J Clin Periodontol. 2011;38(8):721–31 Epub 2011/04/01.
Kadar K, Kiraly M, Porcsalmy B, Molnar B, Racz GZ, Blazsek J, et al. Differentiation potential of stem cells from human dental origin - promise for tissue engineering. J Physiol Pharmacol. 2009;60(Suppl 7):167–75 Epub 2010/04/24.
Stewart K, Walsh S, Screen J, Jefferiss CM, Chainey J, Jordan GR, et al. Further characterization of cells expressing STRO-1 in cultures of adult human bone marrow stromal cells. J Bone Miner Res. 1999;14(8):1345–56 Epub 1999/08/24.
Gronthos S, Franklin DM, Leddy HA, Robey PG, Storms RW, Gimble JM. Surface protein characterization of human adipose tissue-derived stromal cells. J Cell Physiol. 2001;189(1):54–63 Epub 2001/09/27.
Strioga M, Viswanathan S, Darinskas A, Slaby O, Michalek J. Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev. 2012;21(14):2724–52 Epub 2012/04/04.
Izadpanah R, Trygg C, Patel B, Kriedt C, Dufour J, Gimble JM, et al. Biologic properties of mesenchymal stem cells derived from bone marrow and adipose tissue. J Cell Biochem. 2006;99(5):1285–97 Epub 2006/06/24.
De Toni F, Poglio S, Youcef AB, Cousin B, Pflumio F, Bourin P, et al. Human adipose-derived stromal cells efficiently support hematopoiesis in vitro and in vivo: a key step for therapeutic studies. Stem Cells Dev. 2011;20(12):2127–38 Epub 2011/03/11.
Kim Y, Kim H, Cho H, Bae Y, Suh K, Jung J. Direct comparison of human mesenchymal stem cells derived from adipose tissues and bone marrow in mediating neovascularization in response to vascular ischemia. Cell Physiol Biochem. 2007;20(6):867–76 Epub 2007/11/06.
Puissant B, Barreau C, Bourin P, Clavel C, Corre J, Bousquet C, et al. Immunomodulatory effect of human adipose tissue-derived adult stem cells: comparison with bone marrow mesenchymal stem cells. Br J Haematol. 2005;129(1):118–29 Epub 2005/04/02.
Blonska M, Agarwal NK, Vega F. Shaping of the tumor microenvironment: stromal cells and vessels. Semin Cancer Biol. 2015;34:3–13 Epub 2015/03/22.
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–9 Epub 1986/12/25.
Grisendi G, Spano C, Rossignoli F, D Souza N, Golinelli G, Fiori A, et al. Tumor stroma manipulation by MSC. Curr Drug Targets. 2016;17(10):1111–26 Epub 2016/03/10.
Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J Natl Cancer Inst. 2012;104(17):1320–34 Epub 2012/08/23.
Guan J, Chen J. Mesenchymal stem cells in the tumor microenvironment. Biomedical reports. 2013;1(4):517–21 Epub 2014/03/22.
Kidd S, Spaeth E, Watson K, Burks J, Lu HB, Klopp A, et al. Origins of the Tumor Microenvironment: Quantitative Assessment of Adipose-Derived and Bone Marrow-Derived Stroma. PLoS One. 2012;7(2):e30563.
Melzer C, Yang Y, Hass R. Interaction of MSC with tumor cells. Cell Commun Signal. 2016;14(1):20 Epub 2016/09/10.
Hass R, Otte A. Mesenchymal stem cells as all-round supporters in a normal and neoplastic microenvironment. Cell Commun Signal. 2012;10(1):26 Epub 2012/09/05.
Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini F 3rd. Concise review: dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011;29(1):11–9 Epub 2011/02/01.
Hill BS, Pelagalli A, Passaro N, Zannetti A. Tumor-educated mesenchymal stem cells promote pro-metastatic phenotype. Oncotarget. 2017;8(42):73296–311 Epub 2017/10/27.
Rhee KJ, Lee JI, Eom YW. Mesenchymal stem cell-mediated effects of tumor support or suppression. Int J Mol Sci. 2015;16(12):30015–33 Epub 2015/12/24.
Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17(3):320–9 Epub 2011/03/09.
Maffey A, Storini C, Diceglie C, Martelli C, Sironi L, Calzarossa C, et al. Mesenchymal stem cells from tumor microenvironment favour breast cancer stem cell proliferation, cancerogenic and metastatic potential, via ionotropic purinergic signalling. Sci Rep. 2017;7(1):13162 Epub 2017/10/17.
Ryu H, Oh JE, Rhee KJ, Baik SK, Kim J, Kang SJ, et al. Adipose tissue-derived mesenchymal stem cells cultured at high density express IFN-beta and suppress the growth of MCF-7 human breast cancer cells. Cancer Lett. 2014;352(2):220–7 Epub 2014/07/13.
Kucerova L, Matuskova M, Hlubinova K, Altanerova V, Altaner C. Tumor cell behaviour modulation by mesenchymal stromal cells. Mol Cancer. 2010;9:129 Epub 2010/06/01.
Bartosh TJ, Ullah M, Zeitouni S, Beaver J, Prockop DJ. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). P Natl Acad Sci USA. 2016;113(42):E6447–E56.
Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15(6):366–81.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401 Epub 2006/03/31.
Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. 2010;316(17):2713–22 Epub 2010/05/11.
Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012;31(1–2):195–208 Epub 2011/11/22.
Anderberg C, Pietras K. On the origin of cancer-associated fibroblasts. Cell Cycle. 2009;8(10):1461–2 Epub 2009/04/28.
Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: an essential role in the tumor microenvironment. Oncol Lett. 2017;14(3):2611–20 Epub 2017/09/21.
Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001 Epub 2009/11/20.
Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ, Rettig WJ. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J Biol Chem. 1999;274(51):36505–12 Epub 1999/12/14.
Kim HM, Jung WH, Koo JS. Expression of cancer-associated fibroblast related proteins in metastatic breast cancer: an immunohistochemical analysis. J Transl Med. 2015;13:222 Epub 2015/07/15.
Rupp C, Scherzer M, Rudisch A, Unger C, Haslinger C, Schweifer N, et al. IGFBP7, a novel tumor stroma marker, with growth-promoting effects in colon cancer through a paracrine tumor-stroma interaction. Oncogene. 2015;34(7):815–25 Epub 2014/03/19.
Nakagawa H, Liyanarachchi S, Davuluri RV, Auer H, Martin EW Jr, de la Chapelle A, et al. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene. 2004;23(44):7366–77 Epub 2004/08/25.
Navab R, Strumpf D, Bandarchi B, Zhu CQ, Pintilie M, Ramnarine VR, et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc Natl Acad Sci U S A. 2011;108(17):7160–5 Epub 2011/04/09.
Rozenchan PB, Carraro DM, Brentani H, de Carvalho Mota LD, Bastos EP, E Ferreira EN, et al. Reciprocal changes in gene expression profiles of cocultured breast epithelial cells and primary fibroblasts. Int J Cancer. 2009;125(12):2767–77 Epub 2009/06/17.
Correia AL, Bissell MJ. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat. 2012;15(1–2):39–49 Epub 2012/02/18.
Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–9 Epub 2012/07/06.
Luqmani YA. Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract. 2005;14(Suppl 1):35–48 Epub 2005/08/17.
Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. 2008;68(9):3243–50 Epub 2008/05/03.
Sotiropoulou PA, Christodoulou MS, Silvani A, Herold-Mende C, Passarella D. Chemical approaches to targeting drug resistance in cancer stem cells. Drug Discov Today. 2014;19(10):1547–62 Epub 2014/05/14.
Farnie G, Sotgia F, Lisanti MP. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget. 2015;6(31):30472–86 Epub 2015/10/01.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–3 Epub 2009/02/06.
Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015;25(4):198–213 Epub 2014/12/30.
Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121(10):3804–9 Epub 2011/10/04.
Wilcken N, Zdenkowski N, White M, Snyder R, Pittman K, Mainwaring P, et al. Systemic treatment of HER2-positive metastatic breast cancer: a systematic review. Asia Pac J Clin Oncol. 2014;10(Suppl S4):1–14 Epub 2014/05/07.
Sasser AK, Sullivan NJ, Studebaker AW, Hendey LF, Axel AE, Hall BM. Interleukin-6 is a potent growth factor for ER-alpha-positive human breast cancer. FASEB J. 2007;21(13):3763–70 Epub 2007/06/26.
Conze D, Weiss L, Regen PS, Bhushan A, Weaver D, Johnson P, et al. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001;61(24):8851–8 Epub 2001/12/26.
Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47(4):570–84 Epub 2012/07/24.
Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011;71(2):614–24 Epub 2011/01/13.
Ng MR, Brugge JS. A stiff blow from the stroma: collagen crosslinking drives tumor progression. Cancer Cell. 2009;16(6):455–7 Epub 2009/12/08.
Bohrer LR, Chuntova P, Bade LK, Beadnell TC, Leon RP, Brady NJ, et al. Activation of the FGFR-STAT3 pathway in breast cancer cells induces a hyaluronan-rich microenvironment that licenses tumor formation. Cancer Res. 2014;74(1):374–86 Epub 2013/11/08.
Zimmerlin L, Donnenberg VS, Rubin JP, Donnenberg AD. Mesenchymal markers on human adipose stem/progenitor cells. Cytometry Part A. 2013;83(1):134–40 Epub 2012/11/28.
Daverey A, Drain AP, Kidambi S. Physical intimacy of breast Cancer cells with mesenchymal stem cells elicits Trastuzumab resistance through Src activation. Sci Rep. 2015;5:13744 Epub 2015/09/09.
Lu Y, Yang Y, Liu Y, Hao Y, Zhang Y, Hu Y, et al. Upregulation of PAG1/Cbp contributes to adipose-derived mesenchymal stem cells promoted tumor progression and chemoresistance in breast cancer. Biochem Biophys Res Commun. 2017;494(3–4):719–27 Epub 2017/10/29.
Skolekova S, Matuskova M, Bohac M, Toro L, Durinikova E, Tyciakova S, et al. Cisplatin-induced mesenchymal stromal cells-mediated mechanism contributing to decreased antitumor effect in breast cancer cells. Cell Commun Signal. 2016;14:4 Epub 2016/01/14.
Ono M, Kosaka N, Tominaga N, Yoshioka Y, Takeshita F, Takahashi RU, et al. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci Signaling. 2014;7(332):ra63 Epub 2014/07/06.
Dittmer A, Fuchs A, Oerlecke I, Leyh B, Kaiser S, Martens JW, et al. Mesenchymal stem cells and carcinoma-associated fibroblasts sensitize breast cancer cells in 3D cultures to kinase inhibitors. Int J Oncol. 2011;39(3):689–96 Epub 2011/06/15.
Rhodes LV, Muir SE, Elliott S, Guillot LM, Antoon JW, Penfornis P, et al. Adult human mesenchymal stem cells enhance breast tumorigenesis and promote hormone independence. Breast Cancer Res Treat. 2010;121(2):293–300 Epub 2009/07/15.
Qiao L, Xu ZL, Zhao TJ, Ye LH, Zhang XD. Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett. 2008;269(1):67–77 Epub 2008/06/24.
Acknowledgements
We are indebted to L.Rojikova and M.Dubrovcakova for excellent technical assistance with experiments cited in this review. The authors are indebted to all colleagues for their valuable suggestions and discussions.
Funding
Work cited in this review was supported by the Department of Molecular Oncology, Cancer Research Institute, Biomedical Research Center of Slovak Academy of Sciences, Bratislava. This research was supported by the Slovak Research and Development Agency under contract Nos. APVV-16-0010, APVV-15-0697, APVV-16-0178, the Scientific Grant Agency of The Ministry of Education, Science, Research and Sport of the Slovak Republic VEGA 1/0271/17, VEGA 2/0087/15 and by the 7FP platform ERA-NET programme EuroNanoMed II Innocent. The study experiments were made possible by the kind help and financial support from the Cancer Research Foundation and the League against Cancer.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
JP collected the related paper; LK, SM and JP drafted the manuscript; JP, LK, MC, MB, MM and SM discussed and wrote this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Plava, J., Cihova, M., Burikova, M. et al. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol Cancer 18, 67 (2019). https://doi.org/10.1186/s12943-019-0960-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-0960-z