
Abstract
Cancer Stem cells (CSCs) are a unipotent cell population present within the tumour cell mass. CSCs are known to be highly chemo-resistant, and in recent years, they have gained intense interest as key tumour initiating cells that may also play an integral role in tumour recurrence following chemotherapy. Cancer cells have the ability to alter their metabolism in order to fulfil bio-energetic and biosynthetic requirements. They are largely dependent on aerobic glycolysis for their energy production and also are associated with increased fatty acid synthesis and increased rates of glutamine utilisation. Emerging evidence has shown that therapeutic resistance to cancer treatment may arise due to dysregulation in glucose metabolism, fatty acid synthesis, and glutaminolysis. To propagate their lethal effects and maintain survival, tumour cells alter their metabolic requirements to ensure optimal nutrient use for their survival, evasion from host immune attack, and proliferation. It is now evident that cancer cells metabolise glutamine to grow rapidly because it provides the metabolic stimulus for required energy and precursors for synthesis of proteins, lipids, and nucleic acids. It can also regulate the activities of some of the signalling pathways that control the proliferation of cancer cells.
This review describes the key metabolic pathways required by CSCs to maintain a survival advantage and highlights how a combined approach of targeting cellular metabolism in conjunction with the use of chemotherapeutic drugs may provide a promising strategy to overcome therapeutic resistance and therefore aid in cancer therapy.
Similar content being viewed by others


Background
Chemotherapy, along with radiotherapy and hormone therapy, is the most common treatment for cancer. Due to the side effects of treatment and chemo-resistance of the tumour cells, researchers have shifted their focus to more site-specific treatments in order to achieve better results [1].
Over the past decade, a critical role of a small subset of tumour cells, known as cancer stem cells (CSCs), was established in tumour relapse and propagation [2, 3]. Most solid tumours, including breast, brain, prostate, ovary, mesothelioma, and colon cancer contain this small subset of self-renewing tumour initiating cells [4]. Conventional anti-cancer therapies inhibit/kill the bulk of the heterogeneous tumour mass, resulting in tumour shrinkage. However, it has been suggested that later, the CSCs differentiate into tumour cells and are responsible for tumour relapse (Fig. 1). The identification of novel therapies to target CSCs has been the goal of many cancer research laboratories, and recent studies suggest the CSCs undergo metabolic alterations that include low mitochondrial respiration and high glycolytic activity. Exploiting the CSCs' metabolic alterations may provide new effective therapies and diminish the risk of recurrence and metastasis [5, 6].

Cancer stem cells (CSCs) are a small sub-population of tumour cells that are highly chemo-resistant and play a prominent role in tumour relapse. The chemo-resistant property of CSCs is believed to contribute to poor prognosis of conventional tumour treatments. Therefore, we rationalise that therapy targeting CSCs would enable chemo-sensitization by affecting downstream cellular signalling pathways of tumour cells and enable the drugs to destroy the tumour bulk.
Tumour cell metabolism
To induce their lethal effects and maintain survival, tumour cells alter their metabolism to ensure survival, evade host immune attack, and proliferate [7]. This clever strategy of tumour cells was exposed by Otto Warburg in the 1920s when he proved that, in spite of the presence of abundant oxygen, tumour cells metabolise glucose via glycolysis to produce lactate. They adopt this pathway in order to produce ATP through a fermentation process that is much faster compared to the conventional oxidative phosphorylation (respiration), and also avoids the requirement for mitochondrial oxidative phosphorylation. This meets the requirement for the tricarboxylic acid (TCA) cycle activity to be directed towards biosynthesis rather than ATP production. Inner regions of tumours are known to be hypoxic [7–10]. However, the application of anaerobic glycolysis for energy supply is just one part of the metabolic transformation of tumour cells. In order to multiply and survive, the cell must be able to replicate its genome, protein and lipid content, and other important constituents, and also pass on important biomolecules to daughter cells. To accomplish this, the tumour cells enhance the expression of glucose transporters (GLUTs) and monocarboxylate transporters (lactate/pyruvate) to ensure that glucose is delivered and that lactate is transported out of the cell [7, 11] (Fig. 2). Glutamine (via glutamate) and some of the pyruvate enters the TCA cycle to initiate the precursor supply towards biosynthetic reactions. The theoretical significance of the Warburg effect can be illustrated by the glucose uptake and solvent capacity of the cell cytoplasm, i.e. the maximum number of macromolecules that can be accommodated in the intracellular space. Thus, when the glucose uptake rate is low, glucose uptake capacity is the limiting factor and mitochondrial respiration becomes the preferred source for ATP generation. At a high glucose uptake rate, the cell identifies the solvent capacity as its prime source for generating ATP, which in turn activates aerobic glycolysis and lessens mitochondrial respiration (Fig. 2). Hence, the Warburg effect is the amicable catabolic choice for proliferating tumour cells [12]. The other interesting outcome elicited by the Warburg effect is the creation of a tumour environment that facilitates survival and proliferation of the tumours. In the process of their expansion, the tumours stretch the diffusion limits of their oxygenated blood supply and thus induce hypoxia and stabilize the transcription factor HIF. HIF triggers angiogenesis by regulating various associated factors, especially vascular endothelial growth factor [13, 14]. The other strategy adopted by these tumour cells to maximize their survival and proliferation is to increase their glutamine use for supply of biosynthetic precursors. Glutamine acts as a source of carbon and nitrogen for biosynthetic reactions of cancer cells. It gets converted to glutamate, enters into the TCA cycle, and acts as a precursor for the synthesis of important intermediates such as NADPH, anti-oxidants and amino acids such as α-ketoglutarate, aspartate, glutathione, and nucleic acids. The glutamine is converted to glutamate by the mitochondrial enzyme glutaminase. Glutaminase is highly expressed in rapidly growing tumour cells. Another link between oncogenic activation and tumour cell metabolism was determined when a study established that c-Myc increased glutaminase expression by suppressing miR-23a/b [7, 15, 16]. Glutamine may be partially or fully oxidised by tumour cells [17]. It acts as an energy source through catabolism or as a building block via anabolism in the body.

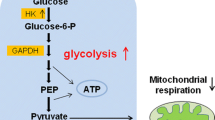
Impact of glucose utilisation by CSCs and non CSCs highlights the difference in their metabolic profiles. Pyruvate enters the TCA cycle to initiate the precursor or supply towards biosynthetic reactions. The Warburg effect in turn activates aerobic glycolysis and lessens mitochondrial respiration, suggesting a preferred choice for proliferation.
Cancer stem cells
The origin of CSCs is still unclear and further studies are required in each type of cancer. CSCs are known to remain in G0 phase [18, 19], the resting phase of the cell cycle, and express high drug efflux transport systems. CSCs, being in a dormant state, make it difficult for most anti-cancer drugs that target only proliferative tumour cells. CSCs exhibit specific characteristics such as self-renewal and heterogeneous differentiation capacity, small population (0.001–0.1 %), resistance to chemo/radiotherapy, high metastatic ability, sphere forming ability, and high ABC transporter expression [20, 21]. CSCs are also known to have a high migratory capacity [22], enabling their spread from the primary tumour to secondary sites [23, 24]. Various techniques have been established to isolate CSCs from the tumour mass and characterise them. CSCs are niche forming cells enriched with growth factors, and growing them in serum-free conditions containing growth factors, such as epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF), maintains the undifferentiated stem cell state and induces the proliferation of self-renewing, unipotent CSCs from parental cell lines [4, 25, 26]. CSCs are characterised by specific surface markers such as CD133+/CXCR4+, CD24+/CD44+, CD24+/CD44+/ESA+, c-Met+/CD44+, and ALDH1+/CD133+ in pancreatic cancer [27, 28]; CD24−/low/CD44+ in breast cancer; CD44+ in colon/ gastric/ head and neck/ovarian cancer; CD34+/CD38− in leukaemia cells; CD13/CD45/CD90 in liver cancer; CD117/CD90/EpCAM in lung cancer; CD20/CD166/Nestin in melanoma cancer; and CD133+/ABCG2+ in Glioblastoma Multiforme [29, 30]. CSCs also express various markers such as CXCR4/ ESA and Nestin [27]. CD44 is one of the most important CSC markers for its role in promoting tumour metastasis and invasion. CD44 has the capability to bind to its primary ligand hyaluronic acid (HA), which initiates CSC attachment to the extracellular matrix and contributes to tumour cell migration [31]. ONCOFID™-S is a conjugate of HA with SN38 (7-ethyl-10-hydroxycamptothecin) and studies have demonstrated that it showed higher anti-proliferative in-vitro activity compared to free SN38 when used against colon, gastric, breast, oesophageal, lung, and ovarian cancer cells, which overexpress CD44 [32, 33]. Therefore, a CD44-targeted therapeutic approach could be utilised for better anti-tumour drug delivery.
The CSCs with CD44+High and CD133+High expression are highly radio-resistant in colon cancer, and they also have higher expression of AKT (AKT1/2) compared to CD44Low and CD133Low cells, indicating their capacity for higher DNA repair and the ability to escape cell death/apoptosis post radiotherapy [34]. Therefore, selective targeting of these markers can be an effective way to deliver cytotoxic drugs to CSCs.
CSCs and their metabolic alterations
Although much is known regarding metabolic pathways important for tumour survival, the potential for therapeutic metabolic alteration of CSCs still remains under investigation [35, 36]. Recent studies indicate that CSCs have different metabolic properties when compared to the tumour bulk. One such study on brain tumour CSCs revealed that these cells show a low activity of mitochondrial respiration [37]. This finding triggered the need to study the effect of glucose in the microenvironment of CSCs because glucose was estimated to be critical for the CSCs. It was found that CSCs have higher glycolytic rates than other tumour cells [38]. Glucose induced the expression of specific genes in CSCs associated with glucose metabolism and the Akt pathway (c-Myc, GLUT1, HK-1, HK-2, and PDK-1), which contributes to the rise in the CSC population [38]. Glucose utilisation by CSCs and non CSCs was compared by measuring their glucose consumption and lactate production rates in order to establish evidence for the difference in metabolic profiles of CSCs and the bulk of the tumour. It has been observed that glucose uptake, lactate production, and ATP content are elevated in CSCs as compared to the non CSCs (Fig. 2) [39–41]. Many crucial molecules involved in glucose metabolism have been studied in relation to the metabolism of CSCs, such as hexokinase-1 (HK1), hexokinase-2 (HK2), pyruvate dehydrogenase kinase 1 (PDK1), and glyceraldehyde-3-phosphate dehydrogenase. The PDK1 levels are high in the CSC population (Fig. 2). PDK-1, via the TCA cycle, phosphorylates pyruvate dehydrogenase and suppresses the pyruvate to acetyl-CoA conversion. Furthermore, suppressing the metabolic flow of pyruvate in mitochondria induces the conversion of pyruvate to lactate in the cytosol [38, 42]. HK-1 and HK-2 both catalyse the conversion of glucose to glucose-6-phosphate in glycolysis, but the levels of HK-2 are lower in CSCs while that of HK-1 are higher, suggesting that HK-1 maintains CSCs’ glycolytic activity. Interestingly, HIF-1α and c-Myc expression (affects HK-2 expression) didn’t change in CSCs and tumour cells. The increase in expression of proteins in the Akt signalling pathway bestows CSCs with a longer life span [8, 38].
Palorini et al. [41] studied the effect of glycolysis inhibition and glucose deprivation on the CSC cell line 3AB-OS, which was derived from the human osteosarcoma cell line MG63. They reported that the 3AB-OS cells require glucose for survival and proliferation. The absence of glucose caused death of the CSC cell line. Glutamine deprivation led to a decline in the MG63 population, which suggested that the 3AB-OS population was not greatly affected by withdrawing glutamine.
Hence, incorporating these features into therapeutic strategies to treat cancer can produce an extensively efficient treatment for various cancers. Also, combining glycolytic inhibition strategies with existing chemotherapy can also help eliminate tumour load completely because the CSCs will also be targeted [41].
Targeting metabolic regulators
Understanding the mechanism by which CSCs are chemo-resistant and initiate tumour relapse is very important in order to address cancer therapy and to understand CSC biology (Fig. 1). B-cell lymphoma (Bcl-2) protein and its family members are known metabolic regulators, and it is recognised as a crucial mediator of mitochondrial apoptotic signalling. Its metabolic role was confirmed by the presence of Bcl-2 associated death promotor (BAD) in complex with glucokinase [43]. Glucokinase has a low affinity for glucose transporter proteins and is purely substrate driven, making it an ideal substrate sensor to detect glucose in pancreatic Beta cells and hepatocytes [43]. The activation of glucokinase is driven by phosphorylation of BAD by kinases such as Akt. The BAD`s pro-apoptotic capacity is inhibited when bound to glucokinase. However, dephosphorylated BAD, on dissociation with glucokinase, will bind to Bcl-2/xl, initiating apoptosis. Furthermore, it has been shown in some cancers that inhibition of BAD phosphorylation decreases cancer cell survival [44, 45].
The glucokinase complex and BAD accumulation will also promote glycolysis, which favours proliferation and CSC biosynthesis. However, dephosphorylation of BAD shifts the balance towards cell death and inhibits the metabolic signals necessary for high glucose flux to enable cell survival regulation [46].
The Bcl-2 protein family impairs the cell’s ability to release apoptogenic protein cytochrome c from the mitochondria by mediation of the balance between cell survival and apoptosis. It achieves this by binding to the pro-apoptotic proteins Bcl-2 associated X protein (BAX) and Bcl-2 homologous antagonist killer [33, 47]. While the mechanism of Bcl-2 expression in CSC chemo/drug resistance is still unclear, and it might be due to chromosomal translocation or another pathway, it was demonstrated that leukaemia CD34+ cells expressed Bcl-2 and Bcl-X [48], and Bcl-2 was highly expressed in breast CD44+/CD24−/low CSCs [49]. To further understand the role of the Bcl-2 protein family, it was demonstrated that Bcl-2 expression in CD133+ human hepatocellular carcinoma cells (HCC) can be regulated by the Akt signalling pathways, the inhibitor specific for AKT1 reduced this cell survival protein expression significantly, indicating that CD133+ HCC contribute to chemo-resistance through preferential activation of AKT/PKB and Bcl-2 cell survival response [50].
One of the mechanisms for CSCs to achieve their metabolic shift is through modifications of metabolic and apoptotic roles of Bcl-2 family proteins. The metabolic alterations of this family of proteins may prove potent in increasing cancer cells susceptibility to apoptosis and affecting tumorigenic metabolic reprogramming.
Targeting drug transporters
CSCs are known to possess a high efflux system that disables the chemo-therapeutic drugs' activity, resulting in the formation of highly drug-resistant tumours [31]. CSCs have been found to express several Adenosine triphosphate–binding cassette (ABC) transporters such as ABCB1/P-gp/MDR1, ABCG2/ BCRP/MXR, and ABCB5 [51, 52]. The ABC transporters are highly dependent on ATP generation in CSCs; thus, targeting CSC metabolism/glycolysis would lead to depleted ATP production and inhibition of ABC transporters. ABCG2 is considered as a high capacity transporter of various substrates including chemotherapeutic drugs [53]. With this in mind, it is has been suggested that ABCG2+ tumour cells can represent CSCs, which are known for their drug-resistance. Higher ABCG2 expression has been observed in various CSCs from lung [54], pancreas [55], and liver [56], and is co-expressed with CD133 in melanoma and pancreatic cancer cell lines [57, 58]. It is suggested that ABCG2 expression is upregulated by hypoxia via hypoxia-inducible transcription factor complex HIF-1α and HIF-2α signalling [59].
ABCB1/ P-glycoprotein (P-gp) /MDR1 are known to be expressed in the majority of drug resistant tumours. Being a product of the multidrug resistance (MDR1) gene, it acts as an ATP-dependent efflux pump to various anti-cancer drugs [60]. CSCs derived from pancreatic tumour cells have higher expression of ABCB1 and ABCG2 [61]. Furthermore, the first generation inhibitors (FGI) verapamil and PSC833 were unable to efficiently inhibit mitoxantrone efflux in leukaemic CSCs, showing that high expression of ABCB1 would lead to the development of chemo-resistant cells [62]. Second generation inhibitors (SGI) were structurally modified for more potency, low cell toxicity, and higher specificity, and include dexverapamil [63] and Valspodar (PSC833) [64]. Another SGI, PSC833, showed higher potency compared to the FGI`s, although this is also an inhibitor of cytochrome P-450 and caused drug-drug interaction associated with anti-cancer drugs [65]. Third generation inhibitors (TGI) utilise nano-molar concentrations to have more potency at reversing MDR compared to TGI and SGI. Zosuquidar (LY3359) [66], an oral P-gp inhibitor used in treating acute myeloid leukaemia, significantly increases the uptake of daunorubicin, idarubicin, and mitoxantrone [67]. Another inhibitor, Tariquidar [68], which is used at very low concentrations (25–80 nM), has a high P-gp affinity that inhibits its ATPase activity [69]. Although it has been used as a potent P-gp inhibitor in clinical trials [70], recent studies have shown that Tariquidar is both a substrate and inhibitor of P-gp, depending on its in-vivo dosage [71]. Fourth generation inhibitors (FGI) are natural compounds or plant extracts exhibiting less cytotoxicity and better oral bioavailability. In-vitro analysis showed MDR reversal of ABC drug transporters when treated with extracts of Chinese herbal plants such as flavonoids or stilbenes [72]. Some natural compounds such as trabectedin, cytarabin, and halaven, have been approved for clinical use based on their strong MDR reversal activity by impacting on ABC drug transporters [73–76].
ABCB5β (a half-transporter) has been found in malignant melanoma and breast cancer, and is known to mediate doxorubicin resistance [77]. The ABCG5+ cells represent 2–20 % of the melanoma tumours and have been shown to successfully recapitulate the tumour in immuno-deficient mice; however, these tumours were unable to regenerate ABCG5+ cells, suggesting their limited stemness capacity [20].
Inhibition of ABC transporters can also cause toxicity to a patient’s normal stem cells, since these have an enhanced DNA repair mechanism, particularly bone marrow-derived stem cells. In addition, ABCG2 and ABCB1 play a pivotal role in maintaining the blood brain barrier, and interfering with their normal function could have drastic consequences [78].
Targeting the tumour microenvironment
Tumour progression is due to adaptive cellular responses such as dormancy, invasiveness, and chemo-resistance in the tumour metabolic microenvironment [79]. Adaptive behaviour of CSCs in this heterogeneous microenvironment is one of the characteristics of CSCs [80]. The tumour microenvironment plays a pivotal role in cancer cell progression, particularly for CSCs, and it mostly involves hypoxia, nutrition, and low pH [81].
Hypoxia in the tumour microenvironment allows pro-angiogenic factors to stimulate new vessel growth within the solid tumour, although the vessels tend to be immature and exhibit poor perfusion [82]. Hypoxia, due to its spatial and temporal heterogeneity in tumours, is difficult to treat [83]. The hypoxic response within the microenvironment is regulated by Hypoxia inducible transcription factors, HIF-1α/ HIF-2α. The migration, glycolytic, angiogenic, and cell survival pathways constitute the transcription targets of HIF1α [84]. Hence, targeting HIF1α is a potential therapy for cancer treatment.
In hypoxic stress, the endoplasmic reticulum (ER) is inhibited, activating the Unfolder protein response (UPR). The UPR maintains ER homeostasis and its disruption initiates apoptosis. Aberrant activation by the UPR in the absence of the two ER membrane proteins PERK (PKR-like ER kinase) and IRE-1 (inositol-requiring) results in increased hypoxia and reduced growth rates [85, 86]. The UPR is an important cellular response mechanism in cancer, playing a role in calcium homeostasis, redox status, and glucose deprivation within the tumour.
Another potential target within the microenvironment is mammalian target of rapamycin (mTOR). During cell stress, nutrient and energy depletion within the solid tumour, mTOR activates the signalling cascades responsible for metabolism and cell survival mechanisms [87, 88]. The anti-diabetic drug metformin has exhibited potential anti-tumour activity; it reduces blood glucose levels, thereby inhibiting gluconeogenesis, and initiates AMPK (AMP-activated protein kinase) activation [89]. AMPK regulates the mTOR activity through activation of the tuberous sclerosis protein 1 complex (TSC1/2) [90].
The microenvironment of tumours is more acidic (pH 6.5–6.9) compared to normal tissues (pH 7.2–7.5), resulting in tumours having poor vascular perfusion and increased glycolytic flux [91, 92]. Knowing that tumour invasiveness is more active in an acidic microenvironment [93, 94], manipulating the tumour microenvironment pH by orally distributed systemic buffers is an effective way to increase the extracellular pH of tumours [95, 96].
Targeting glycolytic enzymes to reduce chemo-resistance in CSCs
Most cells satisfy their energy demands through glucose catabolism, which is subject to complex regulation. To inhibit glucose catabolism through the central pathway of glycolysis, various glycolytic enzymes or transporters must be targeted such as GLUT 1 - 4 [10], hexokinase [97], pyruvate kinase M2, and lactate dehydrogenase A [98].
Cancer cells have the ability to alter their metabolism in order to fulfil bioenergetic and biosynthetic requirements. The extracellular environment can be acidified by what is known as the ‘Warburg effect’ (a term referring to high levels of glycolytic pathway flux, even under aerobic conditions). When HIF-1α induces the expression of carbonic anhydrases, and there is an interaction with extracellular acidification, the pH ratio between the intracellular and extracellular environment is altered [99–102].The resultant pH shift affects drug absorption within the cell. At the same time, glycolytic adenosine triphosphate (ATP) production and the transporter induced over-expression of HIF-1α contribute to a decrease in the cytoplasmic retention of anti-cancer agents [37, 103].
Targeting mitochondrial respiration
The distinct metabolic profile of CSCs has been reported in a few types of cancer, demonstrating CSCs to be more dependent on mitochondrial respiration and less on glycolysis [104]. CSCs prefer oxidative phosphorylation (OXPHOS) for energy production in lung cancer [105], glioblastoma [106], pancreatic ductal adenocarcinoma (PDAC) [104, 107], and leukemic stem cells [108]. The finding in PDAC cells and PDAC-CSCs demonstrates that unlike other highly glycolytic tumour cells, the PDAC-CSCs do not depend on lactate production to generate NAD+ for anabolic respiration to support continued glycolysis and are more dependent on mitochondrial respiration [104]. OXPHOS inhibition impacts directly to the CSCs’ sphere formation capacity and tumorigenic potential, indicating extreme sensitivity to mitochondrial function inhibition [104, 107]. The CSCs’ strong dependence on mitochondrial electron transport chain activity on autophagic and catabolic processes makes them more resistant towards nutrient and environmental factors [104, 107]. In normal and leukaemic stem cells, a dependence on OXPHOS for energy production demonstrates the importance of mitochondrial respiration [108–111]. These findings imply an alternative approach to target tumour relapse by targeting OXPHOS in association with oncogenic pathway inhibitors in pancreatic cancer [104].
Glutaminolysis in cancer metabolism
Cancer cells metabolise glutamine, as well as glucose, to grow rapidly because it provides the required ATP and essential biomolecules such as proteins, lipids, and nucleic acids [112]. Glutamine influences the signalling pathways required for cancer cell proliferation, survival, and metabolism through regulation of mitochondrial reactive oxygen species (ROS) production [113, 114]. Activation of the PI3-Kinase-Akt pathway results in increased production of ROS in mitochondria through metabolic pathways [115]. Glutamine is first converted to glutamate by the enzyme glutaminase, and then glutamate is converted to α-ketoglutarate (αKG) by the action of glutamate dehydrogenase (or an aminotransferase). The rapidly growing tumour cells use glutamine as a carbon source for energy production and for the replenishment of TCA cycle intermediates such as pyruvate, oxaloacetate, and αKG to make up for the constant loss of citrate, which is exported out of the mitochondria for lipid synthesis. It has been observed that glutamine withdrawal in cells with increased c-Myc expression led to the death of the oncogenic cells [16]. Thus, it can be confirmed that cancer cells employ glutamine to provide substrates for the TCA cycle [113, 116]. Further studies have also demonstrated that the oncogene c-Myc impacts glutamine metabolism, thus stimulating the glutamine transporters SLC5A1 and SLC7A1 and, as a result, promoting the expression of glutaminase 1 by suppressing the expression of miR-23A and miR-23B [39].
These data provide a concrete platform to include glutamine metabolism in cancer as an integral part of cancer therapeutic strategies. Glutamine analogues such as 6-diazo-5-oxo-L-norleucine (L-DON), acivicin, and azaserine were found to demonstrate anti-cancer activities but were not formulated into drugs due to their neuro- and gastrotoxicity [117]. However, it has been shown that inhibition of glutamine metabolism via L-DON was able to reduce cancer metastasis in a VM-M3 mouse model [39, 118]. Zhou et al. [119] performed a proteomic analysis in pancreatic ductal adenocarcinoma that revealed the role of glutamine metabolism in cancer. They found that the level of glutaminase in the cancer cells was much higher compared to the normal ductal cells. In addition, the concentration of other enzymes such as cytidine triphosphate synthase, guanine monophosphate synthetase, and asparagine synthetase, which use glutamine as substrates, was found to be elevated in pancreatic cancer. This indicates that the high utilisation rates of glutamine by cancer cells are required to satisfy their need of nitrogen and energy for uninterrupted, fast growth. Paediatric acute leukaemia has been successfully treated by L-asparaginase, which catalyses the hydrolysis of asparagine to aspartic acid. This enzyme is also capable of hydrolysing glutamine to glutamic acid and ammonia, thus reducing blood glutamine levels [39].
Histone deacetylase (HDAC) inhibitors such as phenylbutyrate have been used pharmacologically to inhibit the invasive properties of breast and prostate cancer by inducing apoptosis and depleting the blood glutamine levels [120, 121]. It is generally used to treat hyperammonemia in urea cycle disorders, but it also brings down the level of glutamine in the plasma by forming a conjugate and thus helps curb tumour growth [122]. The glutamine transporters SLC1A5 (ASCT2) and SLC1A7, which are over-expressed in various human cancers such as colon, liver, colorectal adenocarcinomas, glioblastoma multiforme, and melanoma, have been attractive targets due to their role in cell survival signalling and also being a major source of glutamine delivery [123]. IL-γ-glutamyl-p-nitroanilide has been shown to inhibit SLC1A5 (ASCT2) and cause autophagy in cancer cells [39, 116]. A chemical compound termed 968 exhibited anti-glutaminase activity, which in turn suppressed the oncogenic transformation by c-Myc via down regulation of miR-23, which has been seen in prostate cancer and human B-cell lymphoma [39]. Another compound, Bis-2-(5-phenylacetamido-1, 2, 4-thiadiazol-2-yl) ethyl sulphide, also exhibited inhibitory effects on glutaminase, thus repressing glutamine availability to the cancer cells [117]. Ongoing and future work would aim at presenting a more detailed picture of glutamine metabolism and its involvement in cancer, which would help develop safe and effective glutamine inhibitors.
Conclusion
The targeting of CSCs is emerging as a novel therapy to eradicate the progression of various cancers. The inefficiency of traditional anti-cancer therapies lay the stepping stone for studying the metabolism of cancer cells and the pathways controlling and regulating their growth and proliferation, and converting them into formidable treatment options. Targeting the special metabolic traits of CSCs would enable the basis for the development of new therapeutic strategies to inhibit the bulk of the tumour. Clinically, targeting the CSCs resistant towards therapy and metastasis would enable long term disease free survival for the patients.
Though, drug development for CSC metabolism is gaining wide interest, it is still controversial issue as there are studies contradicting the glycolytic phenotype of CSC and oxidative state of CSCs. On the other hand, cancer cell metabolism has emerged to be one of the most fascinating and promising areas in cancer therapy research. The current research focuses on trying to understand the metabolic demands and profile of cancer cells, and design drugs accordingly in order to add a new exciting chapter to cancer treatment. Also, drugs targeting cancer metabolism can be employed for multiple cancers, which can possess a broad spectrum of activity, and are indeed under clinical trials that will likely result in new treatment options in the future [124]. Despite the limited research on the role of metabolism in CSCs and their ability to self-renew, tumour initiation, differentiation capacity, chemo-resistance and survive therapy, targeting CSCs metabolism holds great promise in translating cancer treatments. Though, combinatorial treatments involving both standard chemotherapeutic drugs and chemo-sensitizing agents on CSCs would probably be the most efficient CSC-targeted therapy (Fig. 1).
Abbreviations
- ABC:
-
ATP binding cassette
- Bcl-2:
-
B-cell lymphoma
- CSCs:
-
Cancer stem cells
- HK:
-
Hexokinase
- OXPHOS:
-
Oxidative phosphorylation
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PDK1:
-
Pyruvate dehydrogenase kinase 1
- PTCH1:
-
Patched homologue
- SDF1:
-
Stromal cell derived factor 1
- TCA:
-
Tricarboxylic acid cycle
- αKG:
-
α-ketoglutarate
References
Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62:220–41.
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401.
Tu LC, Foltz G, Lin E, Hood L, Tian Q. Targeting stem cells-clinical implications for cancer therapy. Curr Stem Cell Res Ther. 2009;4:147–53.
Yuan S, Wang F, Chen G, Zhang H, Feng L, Wang L, Colman H, Keating MJ, Li X, Xu RH, et al. Effective elimination of cancer stem cells by a novel drug combination strategy. Stem Cells. 2013;31:23–34.
Villalba M, Rathore MG, Lopez-Royuela N, Krzywinska E, Garaude J, Allende-Vega N. From tumor cell metabolism to tumor immune escape. Int J Biochem Cell Biol. 2012;45:106–13.
Pecqueur C, Oliver L, Oizel K, Lalier L, Vallette FM. Targeting metabolism to induce cell death in cancer cells and cancer stem cells. Int J Cell Biol. 2013;2013:805975.
Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS Journal. 2007;274:1393–418.
Annibaldi A, Widmann C. Glucose metabolism in cancer cells. Curr Opin Clin Nutr Metab Care. 2010;13:466–70.
Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61.
Dolfi SC, Chan LL-Y, Qiu J, Tedeschi PM, Bertino JR, Hirshfield KM, Oltvai ZN, Vazquez A. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer & metabolism. 2013;1:20.
Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–7.
Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608.
Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–90.
Lu W, Pelicano H, Huang P. Cancer metabolism: Is glutamine sweeter than glucose? Cancer Cell. 2010;18:199–200.
Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10:671–84.
Cheong HT, Park TM, Ikeda K, Takahashi Y. Cell cycle analysis of bovine cultured somatic cells by flow cytometry. Jpn J Vet Res. 2003;51:95–103.
Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013; 14: 10.1038/nrm3591
Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–9.
Adams JM, Strasser A. Is tumor growth sustained by rare cancer stem cells or dominant clones? Cancer Res. 2008;68:4018–21.
Bao B, Ali S, Banerjee S, Wang Z, Logna F, Azmi AS, Kong D, Ahmad A, Li Y, Padhye S, Sarkar FH. Curcumin analogue CDF inhibits pancreatic tumor growth by switching on suppressor microRNAs and attenuating EZH2 expression. Cancer Res. 2012;72:335–45.
Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–9.
Moriyama T, Ohuchida K, Mizumoto K, Cui L, Ikenaga N, Sato N, Tanaka M. Enhanced cell migration and invasion of CD133+ pancreatic cancer cells cocultured with pancreatic stromal cells. Cancer. 2010;116:3357–68.
Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, Kuperwasser C. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A. 2010;107:21737–42.
Reynolds BA, Weiss S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol. 1996;175:1–13.
Matsuda Y, Ishiwata T, Yoshimura H, Yamashita S, Ushijima T, Arai T. Systemic administration of small interfering RNA targeting human nestin inhibits pancreatic cancer cell proliferation and metastasis. Pancreas. 2016;45:93–100.
Warrier S, Bhuvanalakshmi G, Arfuso F, Rajan G, Millward M, Dharmarajan A. Cancer stem-like cells from head and neck cancers are chemosensitized by the Wnt antagonist, sFRP4, by inducing apoptosis, decreasing stemness, drug resistance and epithelial to mesenchymal transition. Cancer Gene Ther. 2014;21:381–8.
Karsten U, Goletz S. What makes cancer stem cell markers different? Springerplus. 2013;2:301.
Tomao F, Papa A, Rossi L, Strudel M, Vici P, Lo Russo G, Tomao S. Emerging role of cancer stem cells in the biology and treatment of ovarian cancer: basic knowledge and therapeutic possibilities for an innovative approach. J Exp Clin Cancer Res. 2013;32:48.
Moitra K, Lou H, Dean M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther. 2011;89:491–502.
Serafino A, Zonfrillo M, Andreola F, Psaila R, Mercuri L, Moroni N, Renier D, Campisi M, Secchieri C, Pierimarchi P. CD44-targeting for antitumor drug delivery: a new SN-38-hyaluronan bioconjugate for locoregional treatment of peritoneal carcinomatosis. Curr Cancer Drug Targets. 2011;11:572–85.
Mattheolabakis G, Milane L, Singh A, Amiji MM. Hyaluronic acid targeting of CD44 for cancer therapy: from receptor biology to nanomedicine. J Drug Target. 2015;23:605–18.
Sahlberg SH, Spiegelberg D, Glimelius B, Stenerlow B, Nestor M. Evaluation of cancer stem cell markers CD133, CD44, CD24: association with AKT isoforms and radiation resistance in colon cancer cells. PLoS One. 2014;9, e94621.
Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016;114:1305–12.
Peiris-Pages M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. 2016;18:55.
Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–36.
Liu PP, Liao J, Tang ZJ, Wu WJ, Yang J, Zeng ZL, Hu Y, Wang P, Ju HQ, Xu RH, Huang P. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014;21:124–35.
Hammoudi N, Ahmed KBR, Garcia-Prieto C, Huang P. Metabolic alterations in cancer cells and therapeutic implications. Chin J Cancer. 2011;30:508.
Liu Y, Meyer C, Xu C, Weng H, Hellerbrand C, ten Dijke P, Dooley S. Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol. 2013;304:G449–68.
Palorini R, Votta G, Balestrieri C, Monestiroli A, Olivieri S, Vento R, Chiaradonna F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J Cell Biochem. 2014;115:368–79.
Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31:1143–51.
Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–6.
Winter PS, Sarosiek KA, Lin KH, Meggendorfer M, Schnittger S, Letai A, Wood KC. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci Signal. 2014;7:ra122.
Chang CY, Shen CC, Su HL, Chen CJ. Gefitinib induces apoptosis in human glioma cells by targeting Bad phosphorylation. J Neurooncol. 2011;105:507–22.
Sastry KS, Al-Muftah MA, Li P, Al-Kowari MK, Wang E, Ismail Chouchane A, Kizhakayil D, Kulik G, Marincola FM, Haoudi A, Chouchane L. Targeting proapoptotic protein BAD inhibits survival and self-renewal of cancer stem cells. Cell Death Differ. 2014;21:1936–49.
Kim R, Emi M, Tanabe K. Role of mitochondria as the gardens of cell death. Cancer Chemother Pharmacol. 2006;57:545–53.
Konopleva M, Zhao S, Hu W, Jiang S, Snell V, Weidner D, Jackson CE, Zhang X, Champlin R, Estey E, et al. The anti-apoptotic genes Bcl-X(L) and Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+ cells. Br J Haematol. 2002;118:521–34.
Madjd Z, Mehrjerdi AZ, Sharifi AM, Molanaei S, Shahzadi SZ, Asadi-Lari M. CD44+ cancer cells express higher levels of the anti-apoptotic protein Bcl-2 in breast tumours. Cancer Immun. 2009;9:4.
Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–58.
Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34:732–40.
Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene. 2007;26:1357–60.
Robey RW, Polgar O, Deeken J, To KW, Bates SE. ABCG2: determining its relevance in clinical drug resistance. Cancer Metast Rev. 2007;26:39–57.
Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67:4827–33.
Wang YH, Li F, Luo B, Wang XH, Sun HC, Liu S, Cui YQ, Xu XX. A side population of cells from a human pancreatic carcinoma cell line harbors cancer stem cell characteristics. Neoplasma. 2009;56:371–8.
Shi GM, Xu Y, Fan J, Zhou J, Yang XR, Qiu SJ, Liao Y, Wu WZ, Ji Y, Ke AW, et al. Identification of side population cells in human hepatocellular carcinoma cell lines with stepwise metastatic potentials. J Cancer Res Clin Oncol. 2008;134:1155–63.
Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, Gritti A, Piccinini A, Porro D, Santinami M, et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007;43:935–46.
Olempska M, Eisenach PA, Ammerpohl O, Ungefroren H, Fandrich F, Kalthoff H. Detection of tumor stem cell markers in pancreatic carcinoma cell lines. Hepatobiliary Pancreat Dis Int. 2007;6:92–7.
Martin CM, Ferdous A, Gallardo T, Humphries C, Sadek H, Caprioli A, Garcia JA, Szweda LI, Garry MG, Garry DJ. Hypoxia-inducible factor-2alpha transactivates Abcg2 and promotes cytoprotection in cardiac side population cells. Circ Res. 2008;102:1075–81.
Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–98.
Zhou J, Wang CY, Liu T, Wu B, Zhou F, Xiong JX, Wu HS, Tao J, Zhao G, Yang M, Gou SM. Persistence of side population cells with high drug efflux capacity in pancreatic cancer. World J Gastroenterol. 2008;14:925–30.
Raaijmakers MH, de Grouw EP, van der Reijden BA, de Witte TJ, Jansen JH, Raymakers RA. ABCB1 modulation does not circumvent drug extrusion from primitive leukemic progenitor cells and may preferentially target residual normal cells in acute myelogenous leukemia. Clin Cancer Res. 2006;12:3452–8.
Wilson WH, Jamis-Dow C, Bryant G, Balis FM, Klecker RW, Bates SE, Chabner BA, Steinberg SM, Kohler DR, Wittes RE. Phase I and pharmacokinetic study of the multidrug resistance modulator dexverapamil with EPOCH chemotherapy. J Clin Oncol. 1995;13:1985–94.
Tidefelt U, Liliemark J, Gruber A, Liliemark E, Sundman-Engberg B, Juliusson G, Stenke L, Elmhorn-Rosenborg A, Mollgard L, Lehman S, et al. P-Glycoprotein inhibitor valspodar (PSC 833) increases the intracellular concentrations of daunorubicin in vivo in patients with P-glycoprotein-positive acute myeloid leukemia. J Clin Oncol. 2000;18:1837–44.
Kathawala RJ, Gupta P, Ashby Jr CR, Chen ZS. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat. 2015;18:1–17.
Kemper EM, Cleypool C, Boogerd W, Beijnen JH, van Tellingen O. The influence of the P-glycoprotein inhibitor zosuquidar trihydrochloride (LY335979) on the brain penetration of paclitaxel in mice. Cancer Chemother Pharmacol. 2004;53:173–8.
Tang R, Faussat AM, Perrot JY, Marjanovic Z, Cohen S, Storme T, Morjani H, Legrand O, Marie JP. Zosuquidar restores drug sensitivity in P-glycoprotein expressing acute myeloid leukemia (AML). BMC Cancer. 2008;8:51.
Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447–59.
Lubelski J, van Merkerk R, Konings WN, Driessen AJ. Nucleotide-binding sites of the heterodimeric LmrCD ABC-multidrug transporter of Lactococcus lactis are asymmetric. Biochemistry. 2006;45:648–56.
Kannan P, Telu S, Shukla S, Ambudkar SV, Pike VW, Halldin C, Gottesman MM, Innis RB, Hall MD. The "specific" P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem Neurosci. 2011;2:82–9.
Kakarla P, Inupakutika M, Devireddy AR, Gunda SK, Willmon TM, Ranjana KC, Shrestha U, Ranaweera I, Hernandez AJ, Barr S, Varela MF. 3D-Qsar and contour map analysis of Tariquidar analogues as multidrug resistance protein-1 (MRP1) inhibitors. Int J Pharm Sci Res. 2016;7:554–72.
Michalak K, Wesolowska O. Polyphenols counteract tumor cell chemoresistance conferred by multidrug resistance proteins. Anticancer Agents Med Chem. 2012;12:880–90.
Abraham I, Jain S, Wu C-P, Khanfar MA, Kuang Y, Dai C-L, Shi Z, Chen X, Fu L, Ambudkar SV, et al. Marine sponge-derived sipholane triterpenoids reverse P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Biochem Pharmacol. 2010;80:1497–506.
Huang X-C, Xiao X, Zhang Y-K, Talele TT, Salim AA, Chen Z-S, Capon RJ. Lamellarin O, a pyrrole alkaloid from an Australian marine sponge, Ianthella sp., reverses BCRP mediated drug resistance in cancer cells. Mar Drugs. 2014;12:3818–37.
Lopez D, Martinez-Luis S. Marine natural products with P-glycoprotein inhibitor properties. Mar Drugs. 2014;12:525–46.
Shi Z, Jain S, Kim IW, Peng XX, Abraham I, Youssef DT, Fu LW, El Sayed K, Ambudkar SV, Chen ZS. Sipholenol A, a marine-derived sipholane triterpene, potently reverses P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Cancer Sci. 2007;98:1373–80.
Frank NY, Pendse SS, Lapchak PH, Margaryan A, Shlain D, Doeing C, Sayegh MH, Frank MH. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J Biol Chem. 2003;278:47156–65.
Hill CR, Jamieson D, Thomas HD, Brown CDA, Boddy AV, Veal GJ. Characterisation of the roles of ABCB1, ABCC1, ABCC2 and ABCG2 in the transport and pharmacokinetics of actinomycin D in vitro and in vivo. Biochem Pharmacol. 2013;85:29–37.
Yeom CJ, Goto Y, Zhu Y, Hiraoka M, Harada H. Microenvironments and cellular characteristics in the micro tumor cords of malignant solid tumors. Int J Mol Sci. 2012;13:13949–65.
Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, Liu A, Miu K, Watnick RS, Reinhardt F, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73.
Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010;102:789–95.
Gillies RJ, Schornack PA, Secomb TW, Raghunand N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia. 1999;1:197–207.
Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425–37.
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32.
Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. Embo j. 2005;24:3470–81.
Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–7.
Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol. 2011;23:744–55.
Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–95.
Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF, Landau BR, Shulman GI. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–9.
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90.
Griffiths JR. Are cancer cells acidic? Br J Cancer. 1991;64:425–7.
Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol. 1984;2:343–66.
Moellering RE, Black KC, Krishnamurty C, Baggett BK, Stafford P, Rain M, Gatenby RA, Gillies RJ. Acid treatment of melanoma cells selects for invasive phenotypes. Clin Exp Metastasis. 2008;25:411–25.
Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006;66:6699–707.
Silva AS, Yunes JA, Gillies RJ, Gatenby RA. The potential role of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer Res. 2009;69:2677–84.
Bailey KM, Wojtkowiak JW, Hashim AI, Gillies RJ. Targeting the metabolic microenvironment of tumors. Adv Pharmacol. 2012;65:63–107.
Krasnov GS, Dmitriev AA, Snezhkina AV, Kudryavtseva AV. Deregulation of glycolysis in cancer: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a therapeutic target. Expert Opin Ther Targets. 2013;17(6):681-93.
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–64.
Sawayama H, Ishimoto T, Sugihara H, Miyamoto Y, Baba Y, Yoshida N, Baba H, Miyanari N. Clinical impact of the Warburg effect in gastrointestinal cancer (Review). Int J Oncol. 2014;45:1345–54.
Greijer AE, de Jong MC, Scheffer GL, Shvarts A, van Diest PJ, van der Wall E. Hypoxia-induced acidification causes mitoxantrone resistance not mediated by drug transporters in human breast cancer cells. Cell Oncol. 2005;27:43–9.
Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–83.
Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387–94.
Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE, Sarkadi B, Sorrentino BP, Schuetz JD. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279:24218–25.
Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–32.
Ye XQ, Li Q, Wang GH, Sun FF, Huang GJ, Bian XW, Yu SC, Qian GS. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int J Cancer. 2011;129:820–31.
Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, Radovanovic I, Rheinbay E, Provero P, Stamenkovic I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26:1926–44.
Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Grana O, et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22:590–605.
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O'Dwyer KM, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–41.
Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–88.
Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–8.
Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, Andreeff M. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–56.
Newsholme EA, Crabtree B, Ardawi MS. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci Rep. 1985;5:393–400.
Daye D, Wellen KE. Metabolic reprogramming in cancer: unraveling the role of glutamine in tumorigenesis. Semin Cell Dev Biol. 2012;23:362–9.
Phan LM, Yeung S-CJ, Lee M-H. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med. 2014;11:1.
Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–13.
Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4, e532.
Hensley CT, Wasti AT, Deberardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678.
Shelton LM, Huysentruyt LC, Seyfried TN. Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. Int J Cancer. 2010;127:2478–85.
Zhou W, Fredolini C, Liotta LA, Petricoin EF, Capello M, Novelli F, Racanicchi L, Piemonti L. Proteomic analysis reveals warburg effect and anomalous metabolism of glutamine in pancreatic cancer cells. J Proteome Res. 2012;11:554–63.
Carducci MA, Nelson JB, Chan-Tack KM, Ayyagari SR, Sweatt WH, Campbell PA, Nelson WG, Simons JW. Phenylbutyrate induces apoptosis in human prostate cancer and is more potent than phenylacetate. Clin Cancer Res. 1996;2:379–87.
Dyer ES, Paulsen MT, Markwart SM, Goh M, Livant DL, Ljungman M. Phenylbutyrate inhibits the invasive properties of prostate and breast cancer cell lines in the sea urchin embryo basement membrane invasion assay. Int J Cancer. 2002;101:496–9.
Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–33.
Fuchs BC, Bode BP. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol. 2005;15:254–66.
Jang M, Kim SS, Lee J. Cancer cell metabolism: Implications for therapeutic targets. Exp Mol Med. 2013;45.
Acknowledgement
Abhijeet Deshmukh is supported by a scholarship from the Office of Research & Development, the School of Biomedical Sciences and Faculty of Health Sciences, Curtin University. The authors additionally thank the School of Biomedical Sciences, Curtin University for research and postgraduate student support.
Funding
Not applicable.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Authors’ contributions
AD drafted the outline and generated the figures. AD wrote the manuscript. KD contributed to the manuscript. AD conceived of the study and AD, FA, and PN critically reviewed, revised, and approved the final manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
No ethics approval was required for the purpose of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Deshmukh, A., Deshpande, K., Arfuso, F. et al. Cancer stem cell metabolism: a potential target for cancer therapy. Mol Cancer 15, 69 (2016). https://doi.org/10.1186/s12943-016-0555-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-016-0555-x