
Abstract
Cerebral malaria (CM), results from Plasmodium falciparum infection, and has a high mortality rate. CM survivors can retain life-long post CM sequelae, including seizures and neurocognitive deficits profoundly affecting their quality of life. As the Plasmodium parasite does not enter the brain, but resides inside erythrocytes and are confined to the lumen of the brain’s vasculature, the neuropathogenesis leading to these neurologic sequelae is unclear and under-investigated. Interestingly, postmortem CM pathology differs in brain regions, such as the appearance of haemorragic punctae in white versus gray matter. Various host and parasite factors contribute to the risk of CM, including exposure at a young age, parasite- and host-related genetics, parasite sequestration and the extent of host inflammatory responses. Thus far, several proposed adjunctive treatments have not been successful in the treatment of CM but are highly needed. The region-specific CM neuro-pathogenesis leading to neurologic sequelae is intriguing, but not sufficiently addressed in research. More attention to this may lead to the development of effective adjunctive treatments to address CM neurologic sequelae.
Similar content being viewed by others


Background
Malaria is transmitted through the bite of Plasmodium-infected female Anopheles mosquitoes. It remains one of the most common vector-transmitted diseases, leading to a high disease morbidity and mortality. Although there are several Plasmodium species with the potential to cause disease, Plasmodium falciparum and Plasmodium vivax are the main two species responsible for most complications in humans, with P. vivax more prevalent in South East Asian countries, and India [1,2,3]. In 2018, there were roughly 228 million cases of malaria world-wide resulting in 405,000 deaths [1]. Of these deaths, 67% (272,000) were in children under the age of 5 years [1].
Multiple complications can occur as a result of P. falciparum infection, with cerebral malaria (CM) causing some of the highest mortality rates [1, 4, 5]. Furthermore, patients that survive CM can remain with life-long post CM sequelae, especially neurological deficits, affecting quality of life [6]. Severe malaria, due to P. falciparum infection, presents differently in children than adults, especially regarding the onset of CM. Whereas paediatric CM mortality is reportedly lower than adult CM mortality, paediatric CM is associated with a higher rate of seizures and post-CM neurocognitive deficits [7, 8]. These variances in CM disease presentation may arise due to differences in the immature brain, including differences in host responses of the cerebral vasculature in different brain regions to sequestration and the magnitude of inflammation. This review focuses on the underlying immunopathophysiological mechanisms of paediatric P. falciparum malaria and subsequent neurological sequelae as seen in sub-Saharan Africa.
Host genetic susceptibility and resistance
Given that more than one million children per year were dying from P. falciparum in Africa alone prior to the twenty-first century [4], malaria is, from a genetic standpoint, the evolutionary driver resulting in genetic erythrocyte diseases such as sickle-cell, thalassaemia and glucose-6-phosphate dehydrogenase deficiency. This is supported by the observations that, despite homozygote mortality, the HbS allele has a high prevalence in areas endemic with malaria as well as the observation that independent genetic mutations have developed in different ethnic and geographical populations [9]. Other host genetic factors contributing to CM susceptibility include inflammatory factors and regulatory regions, such as Type 1 Interferon receptor variants in Malawi [10], IL17 in Nigeria and IL4 and IL22 in populations in Mali [11, 12]. In addition, earlier reports showed a role for intercellular adhesion molecular -1 (ICAM-1) Kilifi variants in CM [13]. A recent study in Kilifi, Kenya, identified 15 genes associated with increased paediatric malaria [14], and an Indian study identified TNF polymorphisms [15]. In addition, epidemiological studies reported association of outcomes of malarial infections with age and previous exposure to epigenetic modifications [16,17,18,19]. This arises from a recent discovery that the production of the citric acid cycle metabolites succinate and fumarate increased during severe malaria, including CM. These metabolites can serve as modulators of epigenetic enzymes, such as histone and DNA demethylases [20]. There is growing evidence that recurrent parasite infections, by invoking hyper responsiveness of the Toll-like Receptors (TLR) ligand stimulation, can result in epigenetic modifications with phenotypes showing resistance to malaria [21]. Indeed, these epigenetic modifications were reported among Plasmodium infected Kenyan children [16]. Coinfections in paediatric CM patients, such as HIV, are considered independent risk factors for death. Autopsy studies have demonstrated a two-fold increase in intravascular monocytes and platelets in HIV infected children who died from CM [22]. In addition, increased T cell presence was observed in human CM brains with HIV co-infection [23, 24]. It is likely that in co-infected patients, the HIV associated immune dysregulation further amplifies the pathological damage of CM, leading to increased T cell influx into the brain [22, 24, 25]. Taken together, various host factors contribute to susceptibility to severe malaria and, although there are differences among regions, factors associated with strong host-immune responses appear key.
Clinical characteristics
CM is the most severe neurological complication of the infection by P. falciparum and is a clinical syndrome whose hallmark is impaired consciousness, with coma being the most severe manifestation [26]. Clinical features of pediatric malaria, including CM, involve a relapsing diurnal fever, which is produced after parasite release upon rupture of Plasmodium infected red blood cells (PRBC), secondary to asexual replication and cytokine release [9, 27]. Patients with acute infection can present with a diffuse CM encephalopathy, a rapid progressive coma, and/or seizures without return to consciousness. In some cases, focal neurologic signs are present [28]. At the end stages of disease, children often display signs of brainstem dysfunction, such as abnormal pupillary and corneal reflexes, a dysconjugate gaze and irregular breathing patterns [28,29,30,31,32]. Although some sequelae, such as cortical blindness, improve with time, long-term clinical follow-up assessments in paediatric CM survivors showed elevated persistence of neurological sequelae, including hemiplegia, ataxia, paresis, seizure disorders, language deficits, altered behaviour, severe cerebral palsy and cognitive impairments [28, 29, 32, 33]. These neurologic sequelae may lead to an impaired quality of life and loss of disability adjusted life years. The exact underlying factors that play a role in the neuropathogenesis leading to poor neurological outcome in children are unclear. However, autopsy findings have ascertained that intravascular sequestration of Plasmodium-infected red blood cells is associated with perivascular damage, including axonal injury, myelin loss and breakdown of the blood brain barrier (BBB) [34], as reflected in Fig. 1. How exactly sequestration leads to BBB breakdown is unclear. As discussed later, sequestration together with soluble Plasmodium factors, may have both direct and indirect effects on BBB integrity, which may be amplified by the cytokine storm and influx of plasma factors, including albumin, that are toxic to neurons. Interestingly, in paediatric African CM patients, sequestration reportedly occurs in the brain vasculature irrespective of the region, however postmortem pathology revealed different host vascular responses [34, 35]. A predominance of multiple haemorrhagic punctate lesions is observed in white matter areas and corpus callosum, but not visible in other brain regions, such as gray matter or basal ganglia [35]. Moreover, adult P. falciparum CM also features a predominance of punctate white matter damage, as shown by postmortem pathology and magnetic resonance imaging (MRI) [36, 37]. Taken together, this suggests that potential phenotypic heterogeneity in the local host vasculature [38] that can attract differential PRBC sequestration can lead to alternate host responses [39] (Fig. 1).

Graphical abstract of cerebral malaria pathogenesis. Cerebral malaria pathology manifests itself differently in white matter and gray matter of the brain. Whereas haemorragic punctae are abundant in white matter, they are not obvious in gray matter. The cerebral vasculature in these brain areas is different, which may lead to differential attachment of PRBC—as guided by var gene expression—of PfEMP1 and resulting activation of alternate signalling pathways in the brain endothelial vasculature in these regions. The release of chemokines and cytokines from the inflamed BBB endothelium towards the brain, in conjunction with the opening of the blood brain barrier that allows ingress of both neurotoxic plasma substances and soluble Plasmodium factors into the brain, leads to astroglial activation. This, together with an influx of immune cells, causes neurological damage that is responsible for the post CM neurologic sequelae
The clinical World Health Organization (WHO) diagnostic criteria for CM, (P. falciparum on blood smear, coma and no other known cause of coma) [40], has the potential to result in misdiagnoses. Using these criteria, an autopsy study conducted in Malawi [35] reported that 23% of clinically diagnosed CM cases were, in fact, a different pathology entirely. This has the potential to skew the results of post CM cognitive studies in that some children might already have pre-existing neurocognitive problems. Fundoscopy and diagnosis of retinopathy has been shown to improve the specificity of the clinical diagnosis of CM, although the retinopathy appears to be less specific in adults [8, 41, 42]. CM-retinopathy is a constellation of ocular changes that includes retinal whitening, retinal haemorrhages, vascular changes and papilledema, and increased expression of vascular cell adhesion molecule-1 (VCAM-1) [41, 43, 44]. The severity of malaria retinopathy is also positively correlated with an increased risk of death [41, 43, 44]. Even after resolution of Plasmodium infection, neurologic symptoms persist in almost one-fourth of children with retinopathy-positive CM [28]. In addition, retinopathy negative CM patients were found to have pre-existing neurologic conditions, which subsequently allows for the possibility of inaccurate post CM neurological assessments [8]. Unfortunately, not all clinicians have access to funduscopes, as such instrumentation is relatively expensive for LIMC. Increased access to affordable funduscopy adaptations, e.g. by using adapted cell phones in combination with appropriate algorithms and training will improve appropriate diagnosis of CM and may provide predictions on risk of neurologic sequelae.
Access to other non-invasive imaging modalities, such as MRI, is extremely limited in low and middle income countries. Nevertheless, MRI studies have provided further insight into the pathogenesis and cause of death of sub-Saharan paediatric CM patients. MRI findings in 120 children with retinopathy-positive CM demonstrated increased cerebral volume (50%) and T2 brain abnormalities, suggesting inflammation in the basal ganglia (84.2%), white matter (71,7%), brain stem, thalamus (40%), corpus callosum (49,2%) and cerebellum (49,2%) 30. A subsequent 1.5 Tesla MRI study performed on 16 retinopathy-positive CM children in Zambia indicated that diffusion weighted imaging abnormalities spared the gray matter, indicating a likely vasogenic, as opposed to cytotoxic, oedema. Micro-haemorrhages and parasite sequestration occurred in the same white matter regions. The diffusion weighted imaging results are consistent with micro-haemorrhages and parasite sequestration co-occurring in white matter regions with vascular congestion [45]. These regional differences in MRI findings suggest potential differences in host vasculature between these white- and gray matter regions (Fig. 1).
MRI findings that have been associated with poor to fatal CM outcomes include signs of elevated intracranial pressure, cerebral oedema, decreased cerebrospinal fluid (CSF) volume posterior cerebral involvement, thalamic and supratentorial gray matter lesions and patchy areas of lobular involvement [31, 45]. MRI studies have also demonstrated a clear link between cerebral oedema, depth of coma, and increased mortality [30].
Neuropathogenesis of cerebral malaria
CM pathogenesis is multifaceted and, until recently, has been complicated by disease heterogeneity, often inaccurate clinical case classifications and a lack of large, clinical prospective studies [35]. Although already adopted by some groups with access to funduscopy, an overall standard use of CM-retinopathy as an inclusion criterion for studies will likely result in more precise follow up for assessing the late sequelae of CM. The enigma of CM neuropathogenesis and resultant coma has confounded scientists for decades as the pathogen itself, residing inside PRBC’s, does not directly or physically enter the central nervous system (CNS) due to the BBB, but remains inside the vascular lumen (Fig. 1). Yet, severe neurological symptoms, including coma, are a hallmark in CM and pathological evidence of neuronal injury have also been demonstrated by elevated tau levels in the CSF of children with CM [46]. This highlights the vital role of the BBB endothelium in CM, as the BBB is at the interface of PRBC intravascular sequestration and underlying neuronal damage (Fig. 1).
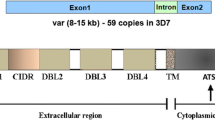
Two main theories (1) the “mechanical hypothesis” and (2) the “cytokine storm” hypothesis provide an explanation underlying CM-neuropathogenesis. The mechanical hypothesis is based on the contribution of intravascular sequestration of PRBCs that results in multiple consequences, including vascular congestion, hypo-perfusion and localized hypoxia [47, 48]. In addition, differences in local blood flow may contribute to increased intracranial pressure in CM and lobular differences. This may be due to either differences in vascular supply, e.g. occipital lobe via posterior cerebral artery versus other lobes via the circle of Willis, due to PRBC sequestration or due to a combination of these factors. Together, these factors ultimately lead to a breakdown of the BBB, cerebral oedema and a pro-thrombotic state [49,50,51]. The parasite-encoded P. falciparum erythrocyte membrane protein-1 (PfEMP-1) is expressed on PRBC surfaces and interacts with host receptors. PfEMP-1 is devised to save PRBCs from clearance by the spleen and is responsible for intravascular PRBC sequestration. PfEMP-1 is encoded by a variable gene (var-gene) and, depending on which var-gene is expressed, interacts with various host adhesion receptors, such as ICAM-1, EPCR and CD36 [52,53,54,55]. Binding of PRBC expressing differential PfEMP-1, as encoded by the var gene family, to its respective receptor leads to downstream host signalling, including activation of inflammatory and coagulatory pathways, eventually leading to loss of BBB integrity and encephalopathy (Fig. 1). In addition, as assessed in in vitro experiments with brain endothelial cells, differential host endothelial responsiveness may affect development of CM in patients [56]. Post-mortem data, animal models for CM and in vitro data, demonstrate that PRBC sequestration correlates with brain vascular activation. This is shown by the presence of large vesicular nuclei, endothelial destruction, activation of transcription factor NF-κB, increased expression of cell adhesion molecules, such as ICAM-1, VCAM-1, E-selectin, cytokine release and BBB breakdown [35, 57,58,59,60,61,62]. Endothelial damage in CM is also demonstrated by changes in the endothelial glycocalix upon exposure to PRBC, both in vitro [63] and in vivo in human CM [64] and murine experimental CM (eCM) [65] and release of endothelial vesicles into the circulation [66].
Although several studies correlate the degree of PRBC sequestration in the brain to increased CM severity [34, 67, 68], the extent to which this correlates with clinical symptoms, coma development and mortality in CM is debated [69]. Therefore, as proposed by the “cytokine storm hypothesis”, peripheral inflammation, neutrophil activation [60] and increased circulations of multiple serum cytokines such as TNF, IFNγ, and IL-2, IL-6, IL-8, and IL-10 contribute to the CM pathogenesis [12, 70]. When compared with patients with uncomplicated malaria, circulating IL-6, MCP-1 and vascular endothelial expression of CD61 are upregulated [56]. The increase in inflammatory markers are indicative of both immune cell and endothelial inflammation and associated with PRBC sequestration [57, 61]. Increased levels of soluble plasma neutrophil proteins and impaired neutrophil chemotaxis were found in retinopathy positive paediatric CM [60], indicative of neutrophil activation. These activated neutrophils may, similar to the intravascular localized monocytes [22], contribute to vascular activation. In addition, CD8 T cells have been found associated with the brain vasculature, both intravascular and perivascular and with the endothelial basal lamina where they can contribute to cerebral vascular activation, both in human and murine studies. Murine eCM studies indicated that those T cells that transmigrated further into the neuropil can damage neurons through release of Granzyme B and/or perforin [71, 72]. At this moment, it is not clear if the CD8+T cells preferentially invade specific white matter or gray matter brain regions. Additional mechanisms whereby neuronal damage occurs may involve caspases in select neurons, as shown in human CM [73].
Vascular integrity and lymphocyte transmigration can also be affected by sphingolipid alterations. For example, therapeutic sphingosine-1-phosphate (S1P) blocking agents, such as FTY720 decreased lymphocyte trafficking into the brain and lowered peripheral IFNγ levels [74]. Not all studies have found a relation between peripheral cytokines and cerebral oedema in CM [75].
Taken together, sequestration and inflammation, in conjunction with elevation of coagulation factors and alterations in blood metabolites, all contribute to CM neuropathogenesis [46, 70, 76,77,78,79,80,81], which may occur in a region-specific manner, e.g. white matter or gray matter. Regardless of either the cytokine storm- or sequestration-mediated CM hypothesis, the effects of endothelial activation can be seen incrementally even in subclinical presentations of parasitaemia as indicated by elevated serum levels of von Willebrand factor (VWF), soluble ICAM-1, and soluble-VCAM-1 [82, 83]. This signifies the paramount importance of the contribution of the BBB endothelium in the pathogenesis of CM and while the exact relationship between cerebral oedema and peripheral inflammation has yet to be fully elucidated, it is likely that they are also correlated with downstream neural stem cell repair processes.
Post CM neuro-sequelae and potential mechanisms
Post CM, persistent neurologic sequelae, including cognitive impairment, motor skills, visual coordination, seizures and attention deficit hyperactivity disorder, occur in up to 25% of paediatric survivors [28, 33, 84,85,86]. The highest risk for deficits in motor, language, and social development was for children under 5 years of age [87]. Between 3 and 6 months post infection, cognitive deficits specific to working memory can intensify with language development being the most consistently affected in paediatric survivors [7, 32, 88, 89]. Cognitive impairments, including memory and attention, can persist for as long as nine years post CM episodes [84, 85]. Ten percent of paediatric CM survivors in one study had at least one mental health sequela with onset ranging from six to twelve months’ post infection and a median of 21 months follow up. The three top mental health disorders in this group were attention deficit hyperactivity disorder, conduct disorder and oppositional defiant disorder [90]. Post-CM, patients with increased externalizing behaviours (i.e. poor attention and aggression) were also reported [87, 91]. However, accurate assessment of neurologic sequelae can be challenging. Most studies done, thus far, on the long term sequelae of CM have used the 2000 clinical WHO definition of CM [7, 84, 85, 88, 91, 92]. This could mean that part of these children did not have true CM, or another underlying infectious condition as well. This opens the possibility that children were included who already had pre-existing neurocognitive conditions or that this was due to the co-infection and not CM. Regardless, the presence of neurologic sequelae in both retinopathy positive and negative children has been well documented [29]. The retinopathy negative children display a different clinical pattern and presentation. Although retinopathy positive patients display more abnormalities on MRI in a variety of different brain regions, the percentage of patients with neurologic sequelae is similar [30]. The lower mortality in the retinopathy negative patients may be due to the presence of potentially protective- co-infections, skewing the immune responses 30,45,93. Interestingly various brain regions appear to be differentially affected. Due to diverse study cohorts in the different countries and assessment methodologies, these studies cannot be directly compared for behavioural assessment. However, it is clear that, as a result of complicated Plasmodium infections, there is a degree of behavioural difficulties in many cases that satisfy the criteria for a mental health disorder diagnosis.
Seizures are common in children with CM and, as a long-term consequence, sustained seizure disorders, often refractory to at least one antiepileptic medication may develop, even months after a CM episode [28, 29, 33, 84, 85, 94,95,96]. The presence of seizures could contribute to development of other post CM neurological sequelae, including developmental delays [7, 28]. In particular, acute seizures during CM episodes was an indicator for future developmental disabilities raising the question of whether acute seizures might promote epileptogenesis and increased risk for chronic epilepsy [28, 89, 93].
Elucidation of the mechanisms underlying the development of behavioural disorders in CM is challenging, especially as complex human behaviours are difficult to recapitulate in animal models. There are several animal models that are used to study CM pathogenesis, but there are ethical concerns in using primate models [97, 98] and these are very expensive. The animal model that is mostly used to study CM is the Plasmodium berghei ANKA strain in a murine model, which recreates a number of features observed in human CM: including vascular inflammation, haemorrhagic punctae in parts of the brain and a set of neurologic sequelae that can be measured in behavioural tests [99,100,101]. Like any model system for a disease, the P. berghei ANKA model is not ideal and has received critiques. Critiques on the murine eCM model revolved around the absence of a PfEMP-1 analogue in the parasite, limited sequestration, the findings of effective therapeutics in the eCM models versus their limited success in human CM [102] and perceived differences in T cell involvement in murine eCM versus human CM. Although authors rightfully caution against blindly extrapolating murine model data, caution to their stance is warranted as addressed at several scientific meetings and in publications [103,104,105,106,107]. Importantly, although previous work failed to show the presence of CD8+T cells in human CM brains [34], recently and due to updated and highly sensitive methodologies, their presence was confirmed in human CM brains; these were both intravascular and perivascular, transmigrated into the neuropil and associated with the choroid plexus, an important entry point into the ventricles and cerebrospinal fluid [23,24,25, 108]. In addition, differences in the measured outcomes of a study may also contribute to, e.g. the survival or incidence of neurologic sequelae in survivors. It is, therefore, imperative to compare results obtained in animal models with human pathology. Due to ethical issues and regulatory guidelines, availability of pathological human CM specimens for CM research is highly limited. Improved patient participation/consent from family members and improved access to the wider CM research community could greatly benefit future CM patients. Moreover, other outcomes than just survival should be taken into account, such as reductions in neurologic sequelae when evaluating new adjunctive treatments for effectiveness.
Several studies using the murine P. berghei ANKA eCM model demonstrated an increase in anxiety-like behaviours following resolution of P. berghei ANKA infection [109]. Inflammatory signalling, such as increased brain-cortical TNF levels in conjunction with either increased IL-6 or IL-1β, and alterations in growth factor levels, e.g. brain derived neurotrophic factor and neuregulin, can contribute to the development of anxiety in eCM and behavioural sequelae [109,110,111,112]. Endothelin family peptide ET-1 (Pre-proendothelin) also plays an important role in inducing BBB damage and produces a CM-like picture in rodent models [113]. Selective endothelin receptor antagonists also improved outcome in cognitive decline and decreased brain hemorrhages in mice [113,114,115].
In response to increased inflammation in eCM, Darling et al. showed the relevance of the receptor tyrosine kinase EphA at the BBB endothelium [116]. Upregulation of EphA2 was shown to be required for the loss of BBB junction proteins both in eCM and in human brain microvascular endothelial cells and for infiltration of CD8+T cell into the brain in the eCM model.
Taken together, these studies suggest that CM episodes, both in humans and in animal models can lead to neurological deficits. The presence of specific neuronal repair processes after resolution of Plasmodium infection is implicated, although the nature and efficiency of these underlying mechanisms is unclear. The neuronal damage inflicted during Plasmodium infection could be related to the extent of the inflammatory responses, the amount of PRBC sequestration or, post-infection, to the efficiency of the neurological repair mechanisms. Very little is known about post CM repair processes. Given the extent of the neuronal damage, neuro-progenitors will be migrating in and will need to replace these damaged neurons. Especially due to the still highly inflammatory environment in the brain, this process can be skewed and contribute to the neuro-sequelae. More research into the underlying molecular pathogenic and repair mechanisms of CM is needed. This research should focus on several key areas, including ameliorating brain vascular inflammation, breach of barrier function and neuronal damage. In addition, a focus on the neuronal repair processes in CM is needed. This may lead to identification of targets for adjunctive therapies targeting the brain vasculature, neuroprotection and neuro repair. Study outcomes should focus not only on survival, but also target neurologic sequelae and behaviour.
Quest for novel adjunctive therapies
Effective adjunctive neuro-protective therapies are currently not available. Numerous attempts to develop therapies targeting neurological sequelae have failed or have even increased the incidence of adverse sequelae, as reviewed by Varo et al. [117]. These attempts have mainly focused on efforts to decrease sequestration, inflammation, coagulation, and oxidative stress and have thus far proven ineffective in reducing mortality and preventing adverse neurological outcomes [118,119,120,121,122]. For example, treatment of CM patients with dexamethasone, commonly used in other neurological disorders to reduce inflammation and combat vasogenic oedema, actually resulted in longer coma and increased neurological complications [123]. Adjunctive therapies attempting to counteract brain oedema with the osmotic diuretic mannitol, known to lower intracranial pressure, showed no beneficial effect in CM patients [124]. Also targeting inflammation, monoclonal anti-TNF antibody therapy worsened CM outcome and increased the incidence of post-CM neurologic sequelae [125]. Although pharmacologic reduction of TNF levels using pentoxifylline, a phosphodiesterase inhibitor, showed slight improvements in survival for humans in some studies [126, 127], others showed no benefit on clinical outcomes [128, 129], or actually increased mortality when studied in children [130]. Interestingly, decreases in hippocampal TNF and IL-6 levels were observed in a murine eCM model with cannabidiol treatment, which also increased brain derived neurotrophic factor levels, promoted eCM survival and prevented post-CM anxiety-like behaviours [111]. The dual role of TNF in neurogenesis could be due to differential downstream signalling effects, dependent on which TNF-receptor is involved. While signalling through TNFR1 significantly hinders neurogenesis, activation of TNFR2 contributes to survival and proliferation of neural stem cells [131]. Neurogenesis, as driven by neural stem cells in the sub granular zone of the hippocampus, is important in learning and memory function [132, 133]. In the murine eCM model, increased expression of TNFR1 and, to a higher degree, TNFR2 is seen [134, 135]. Consequently, a full blockade of TNF signalling may hinder its neurogenic actions and, thus, lead to higher neurocognitive deficits. Targeting other cytokines might show promise for possible interventional strategies. A study examining the post mortem serum and CSF of children in Ghana with CM showed elevation of IP-10, IL-8, MIP-1β, PDGFbb, IL-1ra, Fas-L, sTNF-R1, and sTNF-R2 [136]. Neuregulin, a neurotrophic growth factor was shown to be protective of eCM and was postulated as an effective adjunctive therapy to reduce CNS tissue injury [112]. Administration of neuregulin-1 resulted in a 73% increased survival in eCM, as well as a decrease in systemic and CNS cytokines; TNF, IL-6, IL-1α and CXCL10 [137].
Further attempts to elucidate the role of other potential contributors to improving survival include targeting the complement system with a C5/C5a receptor blockade [138] and blood flow/vascular health using inhaled nitric oxide [139] Nitric oxide has a role in the pathogenesis of CM with low peripheral concentrations contributing to the pathology [140, 141]. Nitric oxide based adjunctive therapy is also effective in eCM through a variety of mechanisms, indicating that direct nitric oxide administration or through dietary supplementation of citrulline may be beneficial as adjunct therapy [142].
Other adjunctive therapies that target inflammation and immune responses and show promise in eCM studies include the peroxisome proliferator-activated receptor gamma agonist rosiglitazone, which improves both survival and neurocognitive outcome [143, 144]. The cholesterol lowering drug atorvastatin [145] also dampens inflammation and thereby reduces both brain endothelial damage and BBB opening [146]. Moreover, rosglitazone boosted neuroprotective pathways and, in human studies, showed promise for treatment of uncomplicated malaria [117], but its effects in human CM are, thus far, unclear. Erythropoietin, a hormone produced by the kidneys, has been considered for use in CM as also has antioxidant and anti-inflammatory components in animal models. Murine studies have demonstrated positive, neuroprotective results, either alone [147] or when paired with artesunate [148]. However, clinical studies demonstrated that high erythropoietin levels are associated with extended coma and increased mortality [149]. As the BBB inflammation and loss of integrity also plays a central role in CM, a potential therapeutic approach of blocking EphA2 to protect the BBB from breakdown was suggested in recent eCM studies [116].
It is clear from both animal and clinical studies that, although high levels of inflammation and brain oedema have been associated with CM mortality and neuro-sequelae, addressing this pharmacologically still presents significant challenges. Completely suppressing inflammation is detrimental, but ameliorating inflammation appears beneficial in CM survival outcome and may likely be neuroprotective as well.
Many of these studies on adjunctive therapies in mice have examined mortality outcomes and did not assess neurocognitive impairment. Thus, it is possible that neurocognitive effects may have been missed. These types of studies could have strengthened the argument for adjunctive therapy. Additional research is needed to elucidate the complex neuropathology leading to long-term neurological deficits, to identify biomarkers predictive of the severity of the neuro-sequelae and how this can be addressed clinically with adjunctive neuroprotective treatments. This requires testing additional outcomes beyond survival, both short and long term and to include a focus on reduction of neurologic sequelae.
Concluding remarks
CM is a devastating disease with complex neuro-pathophysiology that can lead to a variety of neurologic sequelae affecting a person throughout life. The heterogeneity of clinical symptoms and outcomes range from full recovery, to various neurologic sequelae and, often, to death. With improvements in healthcare in sub-Saharan Africa more paediatric CM patients are expected to survive. This population may develop lasting neurologic sequelae, thus contributing to a growing global health concern. Several clinical studies have identified specific characteristics of these neurological sequelae, including cognitive and, behavioural deficits and seizure disorders. Human post-mortem and imaging studies have increased understanding of the neuropathology. Murine eCM studies can address mechanisms involved in eCM development and genesis of neurologic sequelae. Although current research suggests a significant role for inflammation in eliciting neuronal damage and the development of post CM sequelae, more research is needed to address the more specific underlying molecular pathophysiological drivers and signalling mechanisms. This underlines the importance of collaboration among different fields, including clinical research, animal research, and exchange of samples as key to advancing existing knowledge of CM neuropathogenesis. Together, this may lead to identification of novel targets for adjunctive treatments to ameliorate post-CM neuro-sequelae.
Availability of data and materials
Not applicable.
Abbreviations
- BBB:
-
Blood brain barrier
- CM:
-
Cerebral malaria
- CSF:
-
Cerebrospinal fluid
- eCM:
-
Experimental cerebral malaria
- ICAM-1:
-
Intercellular cell adhesion molecule-1
- MRI:
-
Magnetic resonance imaging
- PRBC:
-
Plasmodium infected red blood cells
- PfEMP-1:
-
Plasmodium falciparum erythrocyte membrane protein-1
- PRBC:
-
Plasmodium infected red blood cells
- VCAM-1:
-
Vascular cell adhesion molecule-1
- WHO:
-
World Health Organization
References
WHO. Malaria Key Facts. Geneva, World Health Organization, 2019. https://www.who.int/news-room/fact-sheets/detail/malaria. Accessed 08 May 2019.
Kochar DK, Das A, Kochar SK, Saxena V, Sirohi P, Garg S, et al. Severe Plasmodium vivax malaria: a report on serial cases from Bikaner in northwestern India. Am J Trop Med Hyg. 2009;80:194–8.
Battle KE, Lucas TCD, Nguyen M, Howes RE, Nandi AK, Twohig KA, et al. Mapping the global endemicity and clinical burden of Plasmodium vivax, 2000-17: a spatial and temporal modelling study. Lancet. 2019;394:332–43.
Gething PW, Casey DC, Weiss DJ, Bisanzio D, Bhatt S, Cameron E, et al. Mapping Plasmodium falciparum mortality in Africa between 1990 and 2015. N Engl J Med. 2016;375:2435–45.
Weiss DJ, Lucas TCD, Nguyen M, Nandi AK, Bisanzio D, Battle KE, et al. Mapping the global prevalence, incidence, and mortality of Plasmodium falciparum, 2000–17: a spatial and temporal modelling study. Lancet. 2019;394:322–31.
Severe malaria. Trop Med Int Health. 2014;19 Suppl 1:7-131.
Boivin MJ, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, et al. Cognitive impairment after cerebral malaria in children: a prospective study. Pediatrics. 2007;119:e360–6.
Birbeck GL, Beare N, Lewallen S, Glover SJ, Molyneux ME, Kaplan PW, et al. Identification of malaria retinopathy improves the specificity of the clinical diagnosis of cerebral malaria: findings from a prospective cohort study. Am J Trop Med Hyg. 2010;82:231–4.
Phillips MA, Burrows JN, Manyando C, van Huijsduijnen RH, Van Voorhis WC, Wells TNC. Malaria. Nat Rev Dis Primers. 2017;3:17050.
Feintuch CM, Tare A, Cusumano LR, Benayoun J, Ryu S, Sixpence A, et al. Type I interferon receptor variants in gene regulatory regions are associated with susceptibility to cerebral malaria in Malawi. Am J Trop Med Hyg. 2018;98:1692–8.
Marquet S, Conte I, Poudiougou B, Argiro L, Dessein H, Couturier C, et al. A functional IL22 polymorphism (rs2227473) is associated with predisposition to childhood cerebral malaria. Sci Rep. 2017;7:41636.
Hunt NH, Ball HJ, Hansen AM, Khaw LT, Guo J, Bakmiwewa S, et al. Cerebral malaria: gamma-interferon redux. Front Cell Infect Microbiol. 2014;4:113.
Jenkins NE, Mwangi TW, Kortok M, Marsh K, Craig AG, Williams TN. A polymorphism of intercellular adhesion molecule-1 is associated with a reduced incidence of nonmalarial febrile illness in Kenyan children. Clin Infect Dis. 2005;41:1817–9.
Ndila CM, Uyoga S, Macharia AW, Nyutu G, Peshu N, Ojal J, et al. Human candidate gene polymorphisms and risk of severe malaria in children in Kilifi, Kenya: a case-control association study. Lancet Haematol. 2018;5:e333–45.
Mohanty S, Singh US, Mohanty S, Mohanty AK, Pande V, Das A. Evolutionary interplay of single nucleotide polymorphisms at the promoter region of TNF-alpha gene in different clinical outcomes of malaria in India. Infect Genet Evol. 2019;69:107–16.
Schrum JE, Crabtree JN, Dobbs KR, Kiritsy MC, Reed GW, Gazzinelli RT, et al. Cutting edge: plasmodium falciparum induces trained innate immunity. J Immunol. 2018;200:1243–8.
Qidwai T, Jamal F, Singh S. Exploring putative molecular mechanisms of human pyruvate kinase enzyme deficiency and its role in resistance against Plasmodium falciparum malaria. Interdiscip Sci. 2014;6:158–66.
Quin JE, Bujila I, Cherif M, Sanou GS, Qu Y, Vafa Homann M, et al. Major transcriptional changes observed in the fulani, an ethnic group less susceptible to malaria. Elife. 2017;6:e29156.
Gupta H, Chaudhari S, Rai A, Bhat S, Sahu PK, Hande MH, et al. Genetic and epigenetic changes in host ABCB1 influences malaria susceptibility to Plasmodium falciparum. PLoS ONE. 2017;12:e0175702.
Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24:807–19.
Franklin BS, Parroche P, Ataide MA, Lauw F, Ropert C, de Oliveira RB, et al. Malaria primes the innate immune response due to interferon-gamma induced enhancement of toll-like receptor expression and function. Proc Natl Acad Sci USA. 2009;106:5789–94.
Hochman SE, Madaline TF, Wassmer SC, Mbale E, Choi N, Seydel KB, et al. Fatal pediatric cerebral malaria is associated with intravascular monocytes and platelets that are increased with HIV coinfection. MBio. 2015;6:e01390.
Swanson PA, Hart GT, Russo MV, Nayak D, Yazew T, Pena M, et al. CD8+T cells induce fatal brainstem pathology during cerebral malaria via luminal antigen-specific engagement of brain vasculature. PLoS Pathog. 2016;12:e1006022.
Riggle BA, Manglani M, Maric D, Johnson KR, Lee MH, Neto OLA, et al. CD8+T cells target cerebrovasculature in children with cerebral malaria. J Clin Invest. 2020;130:1128–38.
Barrera V, Haley MJ, Strangward P, Attree E, Kamiza S, Seydel KB, et al. Comparison of CD8(+) T cell accumulation in the brain during human and murine cerebral malaria. Front Immunol. 2019;10:1747.
Idro R, Marsh K, John CC, Newton CR. Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr Res. 2010;68:267–74.
Karunaweera ND, Grau GE, Gamage P, Carter R, Mendis KN. Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivax malaria. Proc Natl Acad Sci USA. 1992;89:3200–3.
Birbeck GL, Molyneux ME, Kaplan PW, Seydel KB, Chimalizeni YF, Kawaza K, et al. Blantyre malaria project epilepsy study (BMPES) of neurological outcomes in retinopathy-positive paediatric cerebral malaria survivors: a prospective cohort study. Lancet Neurol. 2010;9:1173–81.
Postels DG, Taylor TE, Molyneux M, Mannor K, Kaplan PW, Seydel KB, et al. Neurologic outcomes in retinopathy-negative cerebral malaria survivors. Neurology. 2012;79:1268–72.
Potchen MJ, Kampondeni SD, Seydel KB, Birbeck GL, Hammond CA, Bradley WG, et al. Acute brain MRI findings in 120 Malawian children with cerebral malaria: new insights into an ancient disease. Am J Neuroradiol. 2012;33:1740–6.
Seydel KB, Kampondeni SD, Valim C, Potchen MJ, Milner DA, Muwalo FW, et al. Brain swelling and death in children with cerebral malaria. N Engl J Med. 2015;372:1126–37.
Idro R, Carter JA, Fegan G, Neville BG, Newton CR. Risk factors for persisting neurological and cognitive impairments following cerebral malaria. Arch Dis Child. 2006;91:142–8.
Kariuki SM, Abubakar A, Newton CR, Kihara M. Impairment of executive function in Kenyan children exposed to severe falciparum malaria with neurological involvement. Malar J. 2014;13:365.
Dorovini-Zis K, Schmidt K, Huynh H, Fu W, Whitten RO, Milner D, et al. The neuropathology of fatal cerebral malaria in Malawian children. Am J Pathol. 2011;178:2146–58.
Taylor TE, Fu WJ, Carr RA, Whitten RO, Mueller JS, Fosiko NG, et al. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat Med. 2004;10:143–5.
Nagatake T, Hoang VT, Tegoshi T, Rabbege J, Ann TK, Aikawa M. Pathology of falciparum malaria in Vietnam. Am J Trop Med Hyg. 1992;47:259–64.
Wijdicks EFM, Park JG. Surviving cerebral malaria. Neurology. 2018;91:978–9.
Villabona-Rueda A, Erice C, Pardo CA, Stins MF. The evolving concept of the blood brain barrier (BBB): from a single static barrier to a heterogeneous and dynamic relay center. Front Cell Neurosci. 2019;13:405.
Claessens A, Rowe JA. Selection of plasmodium falciparum parasites for cytoadhesion to human brain endothelial cells. J Vis Exp. 2012;59:e3122.
World Health Organization. Communicable Diseases Cluster Severe falciparum malaria. Trans R Soc Trop Med Hyg. 2000;94(Suppl 1):S1–90.
Barrera V, Hiscott PS, Craig AG, White VA, Milner DA, Beare NA, et al. Severity of retinopathy parallels the degree of parasite sequestration in the eyes and brains of Malawian children with fatal cerebral malaria. J Infect Dis. 2015;211:1977–86.
Kochar DK, Shubhakaran A, Kumawat BL, Thanvi I, Joshi A, Vyas SP. Ophthalmoscopic abnormalities in adults with falciparum malaria. QJM. 1998;91:845–52.
Beare NA, Southern C, Chalira C, Taylor TE, Molyneux ME, Harding SP. Prognostic significance and course of retinopathy in children with severe malaria. Arch Ophthalmol. 2004;122:1141–7.
MacCormick IJ, Beare NA, Taylor TE, Barrera V, White VA, Hiscott P, et al. Cerebral malaria in children: using the retina to study the brain. Brain. 2014;137:2119–42.
Potchen MJ, Kampondeni SD, Seydel KB, Haacke EM, Sinyangwe SS, Mwenechanya M, et al. 1.5 tesla magnetic resonance imaging to investigate potential etiologies of brain swelling in pediatric cerebral malaria. Am J Trop Med Hyg. 2018;98:497–504.
Medana IM, Idro R, Newton CR. Axonal and astrocyte injury markers in the cerebrospinal fluid of Kenyan children with severe malaria. J Neurol Sci. 2007;258:93–8.
White NJ, Warrell DA, Looareesuwan S, Chanthavanich P, Phillips RE, Pongpaew P. Pathophysiological and prognostic significance of cerebrospinal-fluid lactate in cerebral malaria. Lancet. 1985;1:776–8.
Macherson GG, Warrell MJ, White NJ, Looareesuwan S, Warrell DA. Human cerebral malaria A quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol. 1985;119:385–401.
Hemmer CJ, Kern P, Holst FG, Radtke KP, Egbring R, Bierhaus A, et al. Activation of the host response in human Plasmodium falciparum malaria: relation of parasitemia to tumor necrosis factor/cachectin, thrombin-antithrombin III, and protein C levels. Am J Med. 1991;91:37–44.
Jimmy EO, Saliu I, Ademowo O. Fibrinopeptide-A and fibrinogen interactions in acute, Plasmodium falciparum malaria. Ann Trop Med Parasitol. 2003;97:879–81.
Clemens R, Pramoolsinsap C, Lorenz R, Pukrittayakamee S, Bock HL, White NJ. Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. Br J Haematol. 1994;87:100–5.
Avril M, Bernabeu M, Benjamin M, Brazier AJ, Smith JD. Interaction between endothelial protein C receptor and intercellular adhesion molecule 1 to mediate binding of Plasmodium falciparum-infected erythrocytes to endothelial cells. MBio. 2016;7:e00615–6.
Brown A, Turner L, Christoffersen S, Andrews KA, Szestak T, Zhao Y, et al. Molecular architecture of a complex between an adhesion protein from the malaria parasite and intracellular adhesion molecule 1. J Biol Chem. 2013;288:5992–6003.
Lau CK, Turner L, Jespersen JS, Lowe ED, Petersen B, Wang CW, et al. Structural conservation despite huge sequence diversity allows EPCR binding by the PfEMP1 family implicated in severe childhood malaria. Cell Host Microbe. 2015;17:118–29.
Shabani E, Hanisch B, Opoka RO, Lavstsen T, John CC. Plasmodium falciparum EPCR-binding PfEMP1 expression increases with malaria disease severity and is elevated in retinopathy negative cerebral malaria. BMC Med. 2017;15:183.
Wassmer SC, Moxon CA, Taylor T, Grau GE, Molyneux ME, Craig AG. Vascular endothelial cells cultured from patients with cerebral or uncomplicated malaria exhibit differential reactivity to TNF. Cell Microbiol. 2011;13:198–209.
Armah H, Dodoo AK, Wiredu EK, Stiles JK, Adjei AA, Gyasi RK, et al. High-level cerebellar expression of cytokines and adhesion molecules in fatal, paediatric, cerebral malaria. Ann Trop Med Parasitol. 2005;99:629–47.
Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J Infect Dis. 2007;195:942–50.
Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006;74:3262–70.
Feintuch CM, Saidi A, Seydel K, Chen G, Goldman-Yassen A, Mita-Mendoza NK, et al. Activated neutrophils are associated with pediatric cerebral malaria vasculopathy in malawian children. MBio. 2016;7:e01300–15.
Brown H, Rogerson S, Taylor T, Tembo M, Mwenechanya J, Molyneux M, et al. Blood-brain barrier function in cerebral malaria in Malawian children. Am J Trop Med Hyg. 2001;64:207–13.
Turner G. Cerebral malaria. Brain Pathol. 1997;7:569–82.
Introini V, Carciati A, Tomaiuolo G, Cicuta P, Guido S. Endothelial glycocalyx regulates cytoadherence in Plasmodium falciparum malaria. J R Soc Interface. 2018;15:20180773.
Yeo TW, Bush PA, Chen Y, et al. Glycocalyx breakdown is increased in African children with cerebral and uncomplicated falciparum malaria. FASEB J. 2019;33:14185–93.
Hempel C, Sporring J, Kurtzhals JAL. Experimental cerebral malaria is associated with profound loss of both glycan and protein components of the endothelial glycocalyx. FASEB J. 2019;33:2058–71.
Sierro F, Grau GER. The ins and outs of cerebral malaria pathogenesis: immunopathology, extracellular vesicles, immunometabolism, and trained immunity. Front Immunol. 2019;10:830.
Pongponratn E, Riganti M, Punpoowong B, Aikawa M. Microvascular sequestration of parasitized erythrocytes in human falciparum malaria: a pathological study. Am J Trop Med Hyg. 1991;44:168–75.
Ponsford MJ, Medana IM, Prapansilp P, et al. Sequestration and microvascular congestion are associated with coma in human cerebral malaria. J Infect Dis. 2012;205:663–71.
Silamut K, Phu NH, Whitty C, Turner GD, Louwrier K, Mai NT, et al. A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am J Pathol. 1999;155:395–410.
Mandala WL, Msefula CL, Gondwe EN, Drayson MT, Molyneux ME, MacLennan CA. Cytokine profiles in Malawian children presenting with uncomplicated malaria, severe malarial anemia, and cerebral malaria. Clin Vaccine Immunol. 2017;24:e00533–616.
Eeka P, Phanithi PB. Cytotoxic T lymphocyte granzyme-b mediates neuronal cell death during Plasmodium berghei ANKA induced experimental cerebral malaria. Neurosci Lett. 2018;664:58–65.
Huggins MA, Johnson HL, Jin F, et al. Perforin expression by CD8 T cells is sufficient to cause fatal brain edema during experimental cerebral malaria. Infect Immun. 2017;85:e00985–1016.
Medana IM, Mai NT, Day NP, Hien TT, Bethell D, Phu NH, et al. Cellular stress and injury responses in the brains of adult Vietnamese patients with fatal Plasmodium falciparum malaria. Neuropathol Appl Neurobiol. 2001;27:421–33.
Finney CA, Hawkes CA, Kain DC, Dhabangi A, Musoke C, Cserti-Gazdewich C, et al. S1P is associated with protection in human and experimental cerebral malaria. Mol Med. 2011;17:717–25.
Harawa V, Njie M, Kessler A, Choko A, Kumwenda B, Kampondeni S, et al. Brain swelling is independent of peripheral plasma cytokine levels in Malawian children with cerebral malaria. Malar J. 2018;17:435.
Gupta S, Seydel K, Miranda-Roman MA, Feintuch CM, Saidi A, Kim RS, et al. Extensive alterations of blood metabolites in pediatric cerebral malaria. PLoS ONE. 2017;12:e0175686.
McDonald CR, Conroy AL, Hawkes M, Elphinstone RE, Gamble JL, Hayford K, et al. Brain-derived neurotrophic factor is associated with disease severity and clinical outcome in Ugandan children admitted to hospital with severe malaria. Pediatr Infect Dis J. 2017;36:146–50.
Molyneux ME, Engelmann H, Taylor TE, Wirima JJ, Aderka D, Wallach D, et al. Circulating plasma receptors for tumour necrosis factor in Malawian children with severe falciparum malaria. Cytokine. 1993;5:604–9.
Moussa EM, Huang H, Thezenas ML, Fischer R, Ramaprasad A, Sisay-Joof F, et al. Proteomic profiling of the plasma of Gambian children with cerebral malaria. Malar J. 2018;17:337.
Punsawad C, Viriyavejakul P. Reduction in serum sphingosine 1-phosphate concentration in malaria. PLoS ONE. 2017;12:e0180631.
Esamai F, Ernerudh J, Janols H, Welin S, Ekerfelt C, Mining S, et al. Cerebral malaria in children: serum and cerebrospinal fluid TNF-alpha and TGF-beta levels and their relationship to clinical outcome. J Trop Pediatr. 2003;49:216–23.
Park GS, Ireland KF, Opoka RO, John CC. Evidence of endothelial activation in asymptomatic Plasmodium falciparum parasitemia and effect of blood group on levels of von Willebrand factor in malaria. J Pediatric Infect Dis Soc. 2012;1:16–25.
Phiri HT, Bridges DJ, Glover SJ, van Mourik JA, de Laat B, M’baya B, et al. Elevated plasma von Willebrand factor and propeptide levels in Malawian children with malaria. PLoS ONE. 2011;6:e25626.
Carter JA, Ross AJ, Neville BG, Obiero E, Katana K, Mung’ala-Odera V, et al. Developmental impairments following severe falciparum malaria in children. Trop Med Int Health. 2005;10:3–10.
Carter JA, Mung’ala-Odera V, Neville BG, Murira G, Mturi N, Musumba C, et al. Persistent neurocognitive impairments associated with severe falciparum malaria in Kenyan children. J Neurol Neurosurg Psychiatry. 2005;76:476–81.
Oluwayemi IO, Brown BJ, Oyedeji OA, Oluwayemi MA. Neurological sequelae in survivors of cerebral malaria. Pan Afr Med J. 2013;15:88.
Brim R, Mboma S, Semrud-Clikeman M, Kampondeni S, Magen J, Taylor T, et al. Cognitive outcomes and psychiatric symptoms of retinopathy-positive cerebral malaria: cohort description and baseline results. Am J Trop Med Hyg. 2017;97:225–31.
Holding PA, Stevenson J, Peshu N, Marsh K. Cognitive sequelae of severe malaria with impaired consciousness. Trans R Soc Trop Med Hyg. 1999;93:529–34.
Boivin MJ, Gladstone MJ, Vokhiwa M, Birbeck GL, Magen JG, Page C, et al. Developmental outcomes in Malawian children with retinopathy-positive cerebral malaria. Trop Med Int Health. 2011;16:263–71.
Idro R, Kakooza-Mwesige A, Asea B, Ssebyala K, Bangirana P, Opoka RO, et al. Cerebral malaria is associated with long-term mental health disorders: a cross sectional survey of a long-term cohort. Malar J. 2016;15:184.
Ssenkusu JM, Hodges JS, Opoka RO, Idro R, Shapiro E, John CC, et al. Long-term behavioral problems in children with severe malaria. Pediatrics. 2016;138:e20161965.
Idro R, Kakooza-Mwesige A, Balyejjussa S, Mirembe G, Mugasha C, Tugumisirize J, et al. Severe neurological sequelae and behaviour problems after cerebral malaria in Ugandan children. BMC Res Notes. 2010;3:104.
Postels DG, Birbeck GL. Children with retinopathy-negative cerebral malaria: a pathophysiologic puzzle. Pediatr Infect Dis J. 2011;30:953–6.
Ngoungou EB, Koko J, Druet-Cabanac M, et al. Cerebral malaria and sequelar epilepsy: first matched case-control study in Gabon. Epilepsia. 2006;47:2147–53.
Christensen SS, Eslick GD. Cerebral malaria as a risk factor for the development of epilepsy and other long-term neurological conditions: a meta-analysis. Trans R Soc Trop Med Hyg. 2015;109:233–8.
Opoka RO, Bangirana P, Boivin MJ, John CC, Byarugaba J. Seizure activity and neurological sequelae in Ugandan children who have survived an episode of cerebral malaria. Afr Health Sci. 2009;9:75–81.
Ibiwoye MO, Howard CV, Sibbons P, Hasan M, van Velzen D. Cerebral malaria in the rhesus monkey (Macaca mulatta): observations on host pathology. J Comp Pathol. 1993;108:303–10.
Pasini EM, Zeeman AM, van der Wel A, Kocken CHM. Plasmodium knowlesi: a relevant, versatile experimental malaria model. Parasitology. 2018;145:56–70.
Rest JR. Cerebral malaria in inbred mice I A new model and its pathology. Trans R Soc Trop Med Hyg. 1982;76:410–5.
Hunt NH, Golenser J, Chan-Ling T, Parekh S, Rae C, Potter S, et al. Immunopathogenesis of cerebral malaria. Int J Parasitol. 2006;36:569–82.
Desruisseaux MS, Gulinello M, Smith DN, Lee SC, Tsuji M, Weiss LM, et al. Cognitive dysfunction in mice infected with Plasmodium berghei strain ANKA. J Infect Dis. 2008;197:1621–7.
White NJ, Turner GD, Medana IM, Dondorp AM, Day NP. The murine cerebral malaria phenomenon. Trends Parasitol. 2010;26:11–5.
Riley EM, Couper KN, Helmby H, Hafalla JC, de Souza JB, Langhorne J, et al. Neuropathogenesis of human and murine malaria. Trends Parasitol. 2010;26:277–8.
Craig AG, Grau GE, Janse C, Kazura JW, Milner D, Barnwell JW, et al. The role of animal models for research on severe malaria. PLoS Pathog. 2012;8:e1002401.
de Souza JB, Hafalla JC, Riley EM, Couper KN. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology. 2010;137:755–72.
Langhorne J, Buffet P, Galinski M, Good M, Harty J, Leroy D, et al. The relevance of non-human primate and rodent malaria models for humans. Malar J. 2011;10:23.
Ghazanfari N, Mueller SN, Heath WR. Cerebral malaria in mouse and man. Front Immunol. 2018;9:2016.
Renia L, Grau GE, Wassmer SC. CD8+T cells and human cerebral malaria: a shifting episteme. J Clin Invest. 2020;130:1109–11.
de Miranda AS, Lacerda-Queiroz N, de Carvalho Vilela M, Rodrigues DH, Rachid MA, Quevedo J, et al. Anxiety-like behavior and proinflammatory cytokine levels in the brain of C57BL/6 mice infected with Plasmodium berghei (strain ANKA). Neurosci Lett. 2011;491:202–6.
Brant F, Miranda AS, Esper L, Gualdron-Lopez M, Cisalpino D, de Souza DDG, et al. Suppressor of cytokine signaling 2 modulates the immune response profile and development of experimental cerebral malaria. Brain Behav Immun. 2016;54:73–85.
Campos AC, Brant F, Miranda AS, Machado FS, Teixeira AL. Cannabidiol increases survival and promotes rescue of cognitive function in a murine model of cerebral malaria. Neuroscience. 2015;289:166–80.
Liu M, Solomon W, Cespedes JC, Wilson NO, Ford B, Stiles JK. Neuregulin-1 attenuates experimental cerebral malaria (ECM) pathogenesis by regulating ErbB4/AKT/STAT3 signaling. J Neuroinflammation. 2018;15:104.
D’Orleans-Juste P, Akide Ndunge OB, Desbiens L, Tanowitz HB, Desruisseaux MS. Endothelins in inflammatory neurological diseases. Pharmacol Ther. 2019;194:145–60.
Dai M, Freeman B, Bruno FP, Shikani HJ, Tanowitz HB, Weiss LM, et al. The novel ETA receptor antagonist HJP-272 prevents cerebral microvascular hemorrhage in cerebral malaria and synergistically improves survival in combination with an artemisinin derivative. Life Sci. 2012;91:687–92.
Freeman BD, Martins YC, Akide-Ndunge OB, Bruno FP, Wang H, Tanowitz HB, et al. Endothelin-1 mediates brain microvascular dysfunction leading to long-term cognitive impairment in a model of experimental cerebral malaria. PLoS Pathog. 2016;12:e1005477.
Darling TK, Mimche PN, Bray C, Umaru B, Brady LM, Stone C, et al. EphA2 contributes to disruption of the blood-brain barrier in cerebral malaria. PLoS Pathog. 2020;16:e1008261.
Varo R, Crowley VM, Sitoe A, Madrid L, Serghides L, Kain KC, et al. Adjunctive therapy for severe malaria: a review and critical appraisal. Malar J. 2018;17:47.
Hemmer CJ, Kern P, Holst FG, Nawroth PP, Dietrich M. Neither heparin nor acetylsalicylic acid influence the clinical course in human Plasmodium falciparum malaria: a prospective randomized study. Am J Trop Med Hyg. 1991;45:608–12.
Charunwatthana P, Abul Faiz M, Ruangveerayut R, Maude RJ, Rahman MR, Roberts LJ, et al. N-acetylcysteine as adjunctive treatment in severe malaria: a randomized, double-blinded placebo-controlled clinical trial. Crit Care Med. 2009;37:516–22.
Maude RJ, Silamut K, Plewes K, Charunwatthana P, Ho M, Abul Faiz M, et al. Randomized controlled trial of levamisole hydrochloride as adjunctive therapy in severe falciparum malaria with high parasitemia. J Infect Dis. 2014;209:120–9.
Watt G, Jongsakul K, Ruangvirayuth R. A pilot study of N-acetylcysteine as adjunctive therapy for severe malaria. QJM. 2002;95:285–90.
Treeprasertsuk S, Krudsood S, Tosukhowong T, Maek-A-Nantawat W, Vannaphan S, Saengnetswang T, et al. N-acetylcysteine in severe falciparum malaria in Thailand. Southeast Asian J Trop Med Public Health. 2003;34:37–42.
Warrell DA, Looareesuwan S, Warrell MJ, Kasemsarn P, Intaraprasert R, Bunnag D, et al. Dexamethasone proves deleterious in cerebral malaria A double-blind trial in 100 comatose patients. N Engl J Med. 1982;306:313–9.
Namutangula B, Ndeezi G, Byarugaba JS, Tumwine JK. Mannitol as adjunct therapy for childhood cerebral malaria in Uganda: a randomized clinical trial. Malar J. 2007;6:138.
van Hensbroek MB, Palmer A, Onyiorah E, Schneider G, Jaffar S, Dolan G, et al. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis. 1996;174:1091–7.
Das BK, Mishra S, Padhi PK, Manish R, Tripathy R, Sahoo PK, et al. Pentoxifylline adjunct improves prognosis of human cerebral malaria in adults. Trop Med Int Health. 2003;8:680–4.
Di Perri G, Di Perri IG, Monteiro GB, Bonora S, Hennig C, Cassatella M, et al. Pentoxifylline as a supportive agent in the treatment of cerebral malaria in children. J Infect Dis. 1995;171:1317–22.
Hemmer CJ, Hort G, Chiwakata CB, Seitz R, Egbring R, Gaus W, et al. Supportive pentoxifylline in falciparum malaria: no effect on tumor necrosis factor alpha levels or clinical outcome: A prospective, randomized, placebo-controlled study. Am J Trop Med Hyg. 1997;56:397–403.
Wenisch C, Looareesuwan S, Wilairatana P, Parschalk B, Vannapann S, Wanaratana V, et al. Effect of pentoxifylline on cytokine patterns in the therapy of complicated plasmodium falciparum malaria. Am J Trop Med Hyg. 1998;58:343–7.
Lell B, Kohler C, Wamola B, Olola CH, Kivaya E, Kokwaro G, et al. Pentoxifylline as an adjunct therapy in children with cerebral malaria. Malar J. 2010;9:368.
Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, et al. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26:9703–12.
Clelland CD, Choi M, Romberg C, Clemenson GD Jr, Fragniere A, Tyers P, et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325:210–3.
Deng W, Saxe MD, Gallina IS, Gage FH. Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci. 2009;29:13532–42.
Gimenez F, de Lagerie S, Fernandez C, Pino P, Mazier D. Tumor necrosis factor alpha in the pathogenesis of cerebral malaria. Cell Mol Life Sci. 2003;60:1623–35.
Lou J, Gasche Y, Zheng L, Critico B, Monso-Hinard C, Juillard P, et al. Differential reactivity of brain microvascular endothelial cells to TNF reflects the genetic susceptibility to cerebral malaria. Eur J Immunol. 1998;28:3989–4000.
Armah HB, Wilson NO, Sarfo BY, Powell MD, Bond VC, Anderson W, et al. Cerebrospinal fluid and serum biomarkers of cerebral malaria mortality in Ghanaian children. Malar J. 2007;6:147.
Solomon W, Wilson NO, Anderson L, Pitts S, Patrickson J, Liu M, et al. Neuregulin-1 attenuates mortality associated with experimental cerebral malaria. J Neuroinflammation. 2014;11:9.
Patel SN, Berghout J, Lovegrove FE, Ayi K, Conroy A, Serghides L, et al. C5 deficiency and C5a or C5aR blockade protects against cerebral malaria. J Exp Med. 2008;205:1133–43.
Serghides L, Kim H, Lu Z, Kain DC, Miller C, Francis RC, et al. Inhaled nitric oxide reduces endothelial activation and parasite accumulation in the brain, and enhances survival in experimental cerebral malaria. PLoS ONE. 2011;6:e27714.
Gramaglia I, Sobolewski P, Meays D, Contreras R, Nolan JP, Frangos JA, et al. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006;12:1417–22.
Yeo TW, Lampah DA, Gitawati R, Tjitra E, Kenangalem E, McNeil YR, et al. Impaired nitric oxide bioavailability and l-arginine reversible endothelial dysfunction in adults with falciparum malaria. J Exp Med. 2007;204:2693–704.
Gramaglia I, Velez J, Chang YS, Caparros-Wanderley W, Combes V, Grau G, et al. Citrulline protects mice from experimental cerebral malaria by ameliorating hypoargininemia, urea cycle changes and vascular leak. PLoS ONE. 2019;14:e0213428.
Serghides L, Patel SN, Ayi K, Lu Z, Gowda DC, Liles WC, et al. Rosiglitazone modulates the innate immune response to Plasmodium falciparum infection and improves outcome in experimental cerebral malaria. J Infect Dis. 2009;199:1536–45.
Serghides L, McDonald CR, Lu Z, Friedel M, Cui C, Ho KT, et al. PPARgamma agonists improve survival and neurocognitive outcomes in experimental cerebral malaria and induce neuroprotective pathways in human malaria. PLoS Pathog. 2014;10:e1003980.
Wilson NO, Solomon W, Anderson L, Patrickson J, Pitts S, Bond V, et al. Pharmacologic inhibition of CXCL10 in combination with anti-malarial therapy eliminates mortality associated with murine model of cerebral malaria. PLoS ONE. 2013;8:e60898.
Taoufiq Z, Pino P, Ndilimabaka N, Arrouss I, Assi S, Soubrier F, et al. Atorvastatin prevents Plasmodium falciparum cytoadherence and endothelial damage. Malar J. 2011;10:52.
Wiese L, Hempel C, Penkowa M, Kirkby N, Kurtzhals JA. Recombinant human erythropoietin increases survival and reduces neuronal apoptosis in a murine model of cerebral malaria. Malar J. 2008;7:3.
Du Y, Chen G, Zhang X, Yu C, Cao Y, Cui L. Artesunate and erythropoietin synergistically improve the outcome of experimental cerebral malaria. Int Immunopharmacol. 2017;48:219–30.
Shabani E, Opoka RO, Idro R, Schmidt R, Park GS, Bangirana P, et al. High plasma erythropoietin levels are associated with prolonged coma duration and increased mortality in children with cerebral malaria. Clin Infect Dis. 2015;60:27–35.
Acknowledgements
Not applicable.
Funding
NIH RO3 NS087303-02, RO1 HL130649, Johns Hopkins Malaria Research Institute and Bloomberg Philanthropies to MFS
Author information
Authors and Affiliations
Contributions
MFS, AVR, KS, KC and NS: drafted and contributed to the writing of this manuscript. MFS edited the manuscript, JC edited the manuscript and provided valuable comments. AVR, with MFS also generated the graphical abstract. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors read and approved consent to publication.
Competing interests
All authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Schiess, N., Villabona-Rueda, A., Cottier, K.E. et al. Pathophysiology and neurologic sequelae of cerebral malaria. Malar J 19, 266 (2020). https://doi.org/10.1186/s12936-020-03336-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-020-03336-z