
Abstract
Background
The Angolan government recommends three artemisinin-based combinations for the treatment of uncomplicated Plasmodium falciparum malaria: artemether–lumefantrine (AL), artesunate–amodiaquine (ASAQ), and dihydroartemisinin–piperaquine (DP). Due to the threat of emerging anti-malarial drug resistance, it is important to periodically monitor the efficacy of artemisinin-based combination therapy (ACT). This study evaluated these medications’ therapeutic efficacy in Benguela, Lunda Sul, and Zaire Provinces.
Methods
Enrollment occurred between March and July 2017. Study participants were children with P. falciparum monoinfection from each provincial capital. Participants received a 3-day course of a quality-assured artemisinin-based combination and were monitored for 28 (AL and ASAQ arms) or 42 days (DP arm). Each ACT was assessed in two provinces. The primary study endpoints were: (1) follow-up without complications and (2) failure to respond to treatment or development of recurrent P. falciparum infection. Parasites from each patient experiencing recurrent infection were genotyped to differentiate new infection from recrudescence of persistent parasitaemia. These parasites were also analysed for molecular markers associated with ACT resistance.
Results
Of 608 children enrolled in the study, 540 (89%) reached a primary study endpoint. Parasitaemia was cleared within 3 days of medication administration in all participants, and no early treatment failures were observed. After exclusion of reinfections, the corrected efficacy of AL was 96% (91–100%, 95% confidence interval) in Zaire and 97% (93–100%) in Lunda Sul. The corrected efficacy of ASAQ was 100% (97–100%) in Benguela and 93% (88–99%) in Zaire. The corrected efficacy of DP was 100% (96–100%) in Benguela and 100% in Lunda Sul. No mutations associated with artemisinin resistance were identified in the pfk13 gene in the 38 cases of recurrent P. falciparum infection. All 33 treatment failures in the AL and ASAQ arms carried pfmdr1 or pfcrt mutations associated with lumefantrine and amodiaquine resistance, respectively, on day of failure.
Conclusions
AL, ASAQ, and DP continue to be efficacious against P. falciparum malaria in these provinces of Angola. Rapid parasite clearance and the absence of genetic evidence of artemisinin resistance are consistent with full susceptibility to artemisinin derivatives. Periodic monitoring of in vivo drug efficacy remains a priority routine activity for Angola.
Similar content being viewed by others


Background
Malaria is the principal cause of morbidity and mortality in Angola and is endemic throughout the country. The disease led to more than three million cases and 7999 deaths nationwide in 2015. In recent years, the country has made significant gains in reducing malaria burden with aggressive preventive measures, case management, and surveillance [1].
There are currently three medications equally recommended by the Angolan Ministry of Health for treatment of uncomplicated malaria: artemether–lumefantrine (AL), artesunate–amodiaquine (ASAQ), and dihydroartemisinin–piperaquine (DP). These are all artemisinin-based combinations, the drug class recommended as first-line malaria treatment by the World Health Organization (WHO) [2]. Artemisinin-based combination therapy (ACT) comprises an artemisinin derivative plus a partner drug; the artemisinin component has a relatively short half-life and acts quickly to reduce parasite burden, while the partner drugs have longer half-lives and suppress parasitaemia for weeks post-treatment. These pharmacological differences are important when monitoring for drug resistance—inadequate clearance of parasitaemia in the first few days after treatment signals a possible problem with the artemisinin component of an ACT, while recurrent parasitaemia later on points more towards an issue with the partner drug.
WHO recommends biennial surveillance of ACT efficacy to provide an early warning against the emergence and spread of resistance [3]. In Angola, the findings of such surveillance have generated international interest and demonstrate the need for continued monitoring [4]. A study of anti-malarial therapeutic efficacy in Angola in 2002–2003 revealed an efficacy of less than 20% for chloroquine [5] and encouraged the Ministry of Health to transition to ACT for first-line treatment of uncomplicated malaria [1]. Efficacy studies in 2013 and 2015 raised concerns about possible lumefantrine resistance, particularly in Zaire Province where corrected efficacy was below the 90% WHO threshold for changing treatment policy [6, 7]. Another therapeutic efficacy study using data from 2011 to 2013 prompted similar concern in Luanda [8].
Although artemisinin resistance has not yet been documented in Africa, it is widespread in Southeast Asia [9, 10]. In 2013, a Vietnamese man with recent travel to Angola’s Lunda Sul Province was diagnosed with malaria which failed to respond to repeated dosing of artemisinin derivatives [11]. Ultimately, this case was inconclusive for artemisinin failure, largely because of uncertainty around the quality of medications administered and the role of the patient’s functional asplenia in delayed parasite clearance [12, 13]. However, the case does emphasize the importance of continued surveillance, particularly as Africa and Asia forge closer economic links. The extensive use of ACT worldwide also provides a strong selective pressure for artemisinin resistance.
Another important component of anti-malarial resistance monitoring is surveillance for molecular markers of resistance among Plasmodium falciparum parasites. Numerous polymorphisms in the P. falciparum genome have been suggested to provide resistance to ACT, both to the artemisinin component and to various partner drugs. Certain mutations in the propeller domain of pfk13, for instance, have been shown to confer artemisinin resistance [14]. Polymorphisms in the chloroquine resistance transporter gene pfcrt [15], originally identified as a marker of chloroquine resistance, have also been associated with resistance to multiple drugs, including amodiaquine and lumefantrine. Polymorphisms in the gene encoding the multidrug resistance 1 transporter (pfmdr1) have been preliminarily associated with both lumefantrine and amodiaquine resistance, with certain mutations such as N86Y having an opposite effect on lumefantrine and amodiaquine susceptibility [16, 17]. Elevated pfmdr1 copy number has been postulated to confer resistance to lumefantrine [18], and, most recently, elevated copy number of the plasmepsin 2 gene (pfpm2) has been proposed as a marker for piperaquine resistance [19, 20].
This report presents the results of the latest round of therapeutic efficacy monitoring of anti-malarials in Angola’s three fixed anti-malarial surveillance sentinel sites.
Methods
Study design
An in vivo assessment of the therapeutic efficacy of AL, ASAQ, and DP was conducted according to the standard WHO protocol [3]. Study participants were recently febrile children with P. falciparum monoinfection. Participants were followed with physical exams and blood samples for 28–42 days after anti-malarial administration for development of adverse medication effects or recurrent parasitaemia. Molecular analyses were performed on samples from those experiencing treatment failure.
Study population
The study took place at anti-malarial resistance sentinel sites in Benguela, Lunda Sul, and Zaire that were retained from previous therapeutic efficacy studies [6, 7]. Benguela is a southern coastal province with stable mesoendemic malaria transmission. Zaire is a forested province on the northern coast that also has stable mesoendemic transmission. Lunda Sul province, part of Angola’s eastern savannah, features hyperendemic transmission (Fig. 1) [1].

Location of sentinel sites for therapeutic efficacy monitoring in Angola, 2017
Enrollment was divided into six arms, and efficacy of each medication was assessed in two provinces. Target enrollment of one hundred participants per arm provided enough power to estimate efficacy with 95% confidence limits of ± 5%, assuming an expected efficacy of 95% and a maximum loss to follow-up and exclusion rate of 27%. AL and DP were evaluated in Lunda Sul, AL and ASAQ in Zaire, and DP and ASAQ in Benguela (Fig. 1). Two drugs were assessed sequentially in each site. Medication administration was not randomized or blinded; the first hundred participants enrolled at each site received the first ACT to be evaluated there (AL in Zaire and Lunda Sul or DP in Benguela), and the second hundred enrollees received the second drug under evaluation (ASAQ in Benguela and Zaire or DP in Lunda Sul).
Screening for participants took place at two health facilities in each province’s capital from March to July 2017. Study entrants were children with fever (≥ 37.5 °C) or a history of fever in the past 24 h, P. falciparum monoinfection on blood smear, no signs of severe malaria on physical exam, no concomitant illness or severe malnutrition, a haemoglobin greater than 8 g/dL, no anti-malarials within the last 14 days, and caregivers willing to attend all follow-up visits with study staff. Inclusion criteria were broader in Benguela compared to Lunda Sul and Zaire, allowing for older children (< 12 years vs. < 5 years) and lower parasitaemias (between 1000 and 100,000 asexual parasites/μL vs. between 2000 and 200,000 parasites/μL), due to the lower level of malaria transmission in Benguela.
Clinical monitoring
Enrolled children received a 3-day course of one of the anti-malarials under evaluation, dosed according to guidelines from the drug manufacturers. Quality-controlled AL (Ipca Laboratories, Maharashtra, India), ASAQ (Sanofi Aventis, Paris, France), and DP (Eurartesim®; Leadiant Biosciences, Rome, Italy) were provided by WHO. For ASAQ and DP, which have once-daily dosing, all three doses were given under direct observation of study staff. For AL, which requires twice-daily dosing, each morning dose was directly observed. Evening doses were given at home, and as in previous studies, efforts were made to ensure correct administration of each dose. Parents or guardians were given the appropriate weight-based dose, called at home to confirm delivery of the dose, and instructed to bring the empty pill packages back to study staff the following morning. All AL doses were administered with a snack, containing at least 3 g of fat, to facilitate drug absorption. Parents or guardians were given individual packages of yogurt or chocolate milk to take home and were asked to bring the empty package back to study staff the following morning.
After each medication administration, children were observed for 1 h to monitor for vomiting or other side effects, including diarrhea, nausea, or excessive sweating. Children who vomited within half an hour after medication administration were given a repeat dose. Children who vomited between half an hour and 1 h after medication administration were given a repeat dose that was half of the original dose. Those with persistent vomiting were excluded from the study.
Participants made eight or ten visits to study staff over the course of 28 or 42 days. Participants in the AL and ASAQ arms were followed for 28 days, while those in the DP arms required 42-day follow-up due to the longer half-life of piperaquine [3]. All children were monitored daily during the 3 days of drug administration and 1 day following; afterwards, follow-up visits occurred weekly. An interval history, physical exam, thick and thin blood smear, and dried blood spot collection were performed at each visit, except for day 1 of follow-up, in which blood samples were only collected if a child exhibited signs of severe malaria. Participants’ haemoglobin was also measured every 2 weeks. Finally, participants in the AL arm in Zaire underwent an additional dried blood spot collection on day 7 of follow-up to measure blood lumefantrine levels, to provide further insight into the large number of late treatment failures previously observed with AL in this province [6, 7].
Children were excluded from follow-up if they developed a non-falciparum malaria infection, showed signs of severe illness, missed a follow-up visit, or took another anti-malarial. For the preliminary analysis, the primary outcomes were adequate clinical and parasitological response, failure to adequately clear parasitaemia, or recurrent P. falciparum parasitaemia. Participants were categorized as early treatment failures if their parasitaemia increased from day 0 to day 2 of follow-up, failed to decrease by at least 75% by day 3, was associated with fever on day 3, or was associated with signs of severe malaria on days 1–3. Participants who developed any parasitaemia after day 3 were classified as late treatment failures.
Molecular analysis
Microsatellite analysis and Sanger sequencing of P. falciparum parasites were performed on selected blood samples from study participants. Day 0 and day of failure samples from all cases of late treatment failure were genotyped by microsatellite analysis for reclassification as reinfection or recrudescence. Additional day 0 samples from participants not experiencing treatment failure were randomly selected for neutral microsatellite analysis to augment the number of samples for determination of allele frequencies. day 0 and day of failure samples from all cases of late treatment failure were also assessed for pfK13, pfcrt, and pfmdr1 sequence and pfmdr1 and pfpm2 copy number.
Genomic DNA was extracted using the QIAamp blood minikit (Qiagen Inc., California, USA). Seven neutral microsatellite loci spanning six chromosomes (TA1, chromosome 6; poly α, chromosome 4; PfPK2, chromosome 12; TA109, chromosome 6; and 2490, chromosome 10; C2M24, chromosome 2; and C3M69, chromosome 3) were amplified by non-nested or semi-nested PCR and their fragment lengths were measured [21,22,23,24]. Fragments of the pfcrt [25], pfmdr1 [25], and pfK13 [26] genes were amplified using previously published primers. Direct Sanger sequencing of the nested purified PCR products was performed by using a BigDye Terminator v3.1 cycle sequencing kit on an iCycler thermal cycler (Bio-Rad, California, USA). Sequence analysis was performed by using Geneious R7 (Biomatters, Auckland, New Zealand).
Pfmdr1 copy number was assessed using a previously described protocol [27]. The P. falciparum β-tubulin gene was used as the reference gene for relative quantification of pfmdr1 gene copy number. Each run included the 3D7 parasite strain as a single-copy control, the W2Mef strain (two copies), and the Dd2 strain (three to four copies). The assay was performed using the Agilent Mx3005 real-time PCR instrument (Agilent Technologies, California, USA). The copy number was determined by using the relative quantification module in MxPro3005 software (Agilent Technologies, California, USA), using the comparative ΔΔCT method, and rounded to the nearest integer.
For pfpm2 copy number analysis, the P. falciparum β-tubulin gene was again used for reference. The reverse primers of both the pfpm2 and β-tubulin genes were modified with the PET tag and labeled with FAM (pfpm2) and HEX (β-tubulin) fluorophores. The PET-PCR assays were performed using Agilent Mx3005pro thermocyclers (Agilent Technologies, California, USA).
Lumefantrine level measurement
Additional dried blood spots were collected on day 7 of follow-up from participants in the AL arm in Zaire. A volume of 50 μL of whole blood was collected on filter paper pre-treated with 0.75 M tartaric acid and stored at room temperature. Dried blood spots were eluted with an acidified acetonitrile solution followed by solid phase extraction. The extraction procedure was followed by liquid chromatographic separation using a Synergi Hydro-RP, 4 µm, 150 × 2.0 mm analytical column (Phenomenex, California, USA) with an isocratic mobile phase containing a mixture of acetonitrile, water, and formic acid (70:29.8:0.2) at a flow-rate of 300 µL/min. An AB Sciex API 3000 mass spectrometer at unit resolution in the multiple reaction monitoring mode was used to monitor the transition of the protonated precursor ions m/z 530.0 and m/z 539.1 to the product ions m/z 512.1 and m/z 521.3 for lumefantrine and the internal standard, respectively. Electrospray ionization was used for ion production [28, 29].
Day 7 lumefantrine concentrations greater than or equal to 0.2 μg/mL were considered therapeutic [30].
Statistical analysis
Uncorrected efficacy was determined by dividing the number of treatment failures in each study arm by the total number of participants classified as either adequate clinical and parasitological response or treatment failure in that arm. Parasite clearance rates on day 2 and day 3 of follow-up were also examined to evaluate artemisinin delayed response.
Kaplan–Meier estimates of survival curves were calculated using the survival package in R version 3.3.2 (R Foundation for Statistical Computing, Vienna, Austria). Data from participants excluded for reasons other than treatment failure or incorrect enrollment were included in the analysis until the day of study departure.
To determine corrected therapeutic efficacy, microsatellite data were analysed using a previously published algorithm [31] that assigns each late treatment failure a posterior probability of recrudescence. A probability of more than 0.5 was considered a recrudescence, and less than or equal to 0.5 was designated as a reinfection.
The distribution of day 7 lumefantrine drug levels was compared between cases of adequate clinical and parasitological response and cases of recrudescence and reinfection using the Kolmogorov–Smirnov test. The proportion of participants with sub-therapeutic day 7 lumefantrine levels was compared between cases of adequate clinical and parasitological response and cases of recrudescence and reinfection using Fisher’s exact test.
Results
Triage and enrollment
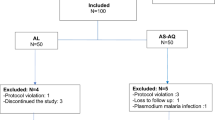
Of 608 children enrolled in the study, 27 (4%) were lost to follow-up (loss to follow-up rates were 7% or less in all study arms), and 41 participants (7%) were excluded (Additional file 1). Participant characteristics at baseline reflected the different inclusion criteria in each site (Table 1). Median age ranged from 2.7 years in the Lunda Sul DP arm to 7.1 years in the Benguela DP arm. Sufficient participants completed follow-up to provide statistical power in all treatment arms, with between 85 and 94 participants reaching a primary study outcome in each arm.
Efficacy
Of 540 children completing follow up, 502 achieved adequate clinical and parasitological response, and 38 were classified as treatment failures (Table 2). Rates of parasite clearance were at least 97% on day 2 in all arms and 100% by day 3 in all arms (Table 3). No children experienced early treatment failure; all 38 treatment failures were late treatment failures.
The uncorrected 28-day efficacy using the Kaplan–Meier estimate of the survival curve was 93% (88–98%) for AL in Zaire, 90% (84–96%) for AL in Lunda Sul, 99% (94–100%, 95% confidence interval) for ASAQ in Benguela, and 82% (75–90%) for ASAQ in Zaire (Table 4). The uncorrected 42-day efficacy was 95% (90–99%) for DP in Benguela and 100% (100–100%) for DP in Lunda Sul.
The majority of the 38 late treatment failures were reinfections (26, 68%) rather than recrudescences (12, 32%) by microsatellite analysis (Table 2). Late treatment failures were categorized with a high degree of confidence, with the posterior probability of recrudescence greater than 90% or less than 10% in all but three instances (Additional file 2). One late treatment failure in the Zaire AL arm had a posterior probability of recrudescence of 29%, and two late treatment failures in the Zaire ASAQ arms had probabilities of 19 and 39%. Four cases of recrudescence were identified in the Zaire AL arm, three in the Lunda Sul AL arm, and five in the Zaire ASAQ arm. No recrudescences were found among participants in the Benguela DP, Lunda Sul DP, or Benguela ASAQ arms.
After exclusion of reinfections, the corrected 28-day efficacy of AL was 96% (91–100%), in Zaire and 97% (93–100%) in Lunda Sul. The corrected 28-day efficacy of ASAQ was 100% (97–100%) in Benguela and 93% (88–99%) in Zaire. The corrected 42-day efficacy of DP was 100% (96–100%) in Benguela and 100% in Lunda Sul.
Safety
Among the 586 participants enrolled within inclusion criteria, 11 participants (2%) reported adverse medication effects: four with vomiting after DP (2% of participants in DP arms), three with nausea after ASAQ (2%), two with excessive sweating after ASAQ (1%), one with vomiting after ASAQ (1%), and one with excessive sweating after DP (1%). No participants reported adverse effects after AL administration, neither under direct observation nor at home. No participants left the study due to adverse medication effects.
Molecular markers of resistance
Almost all (75/76, 99%) of the day 0 and day of failure samples analysed from the 38 treatment failures were wild-type for pfk13 (Table 5). One sample from Zaire exhibited the A504V mutation, which has not been associated with artemisinin resistance. Nearly all day of failure samples in the AL arms carried the pfcrt wildtype K76 genotype (14/16, 88%), and all had the N86 pfmdr1 genotype (16/16, 100%). All ASAQ day of failure samples (17/17, 100%) carried the pfcrt 76T single-nucleotide polymorphism. The only two samples with the pfmdr1 86Y mutation were ASAQ day of failure samples from Benguela.
All 76 analysed samples had only one copy of both the pfmdr1 and pfpm2 genes. However, 75 of 76 samples contained multiple P. falciparum strains.
Lumefantrine concentrations
Median day 7 lumefantrine concentrations in the Zaire AL arm were similar among participants experiencing adequate clinical and parasitological response, reinfection, and recrudescence, ranging from 0.19 to 0.23 μg/mL (Additional files 3, 4). Two (50%) out of the four cases of recrudescence observed in the AL arm had sub-therapeutic day 7 lumefantrine levels, but this did not differ significantly (p value 1.0) from rate of sub-therapeutic day 7 lumefantrine levels in participants with adequate clinical and parasitological response (35/87, 40%). Two children had undetectable lumefantrine levels, one of whom successfully responded to treatment and one of whom had a recrudescent infection. The distribution of day 7 lumefantrine concentrations was not statistically different between cases of adequate clinical and parasitological response and recrudescence (p value 0.94), between cases of adequate clinical and parasitological response and reinfection (p value 0.34), or between cases of adequate clinical and parasitological response and recrudescence/reinfection (0.80).
Discussion
AL, ASAQ, and DP remain efficacious for the treatment of uncomplicated P. falciparum malaria in the sites where they were tested. The WHO recommends changing national malaria treatment policy if efficacy drops below 90% [2], and all corrected efficacies reported here exceed that threshold. All three medications were very well tolerated, with less than 2% of participants experiencing mild adverse effects and no participants experiencing serious adverse effects.
Uncorrected therapeutic efficacy was above 90% in all study arms except ASAQ in Zaire, which had an uncorrected efficacy of 82%. Low uncorrected efficacy has been noted in previous anti-malarial studies in Zaire [6, 7] and may be explained in part by relatively high malaria transmission in this province as compared to the other sites.
There has been concern recently around possible lumefantrine resistance in Angola, particularly in Zaire. The results presented here do not support this concern, with corrected AL efficacy at 96% in Zaire and 97% in Lunda Sul. Greater efforts to administer all AL doses with a fatty meal could explain the higher lumefantrine efficacy observed in this round of monitoring. Nevertheless, a substantial proportion (40%) of cases of adequate clinical and parasitological response had day 7 lumefantrine concentrations below the recommended therapeutic threshold of 0.2 μg/mL. There was no statistically significant difference in day 7 lumefantrine drug levels by treatment outcome in the AL Zaire arm, but this could be due to the small number of treatment failures observed in that arm.
DP demonstrated the highest efficacy of all anti-malarials in this study, with no recrudescences identified throughout 42 days of follow-up among 174 participants taking this medication. This high efficacy, combined with the drug’s relatively long half-life, confirms DP as an appropriate ACT for Angola [32]. Although the utility of this medication is declining in Asia, DP efficacy remains high throughout Africa [33].
Multiple factors provide reassurance that P. falciparum is still highly susceptible to artemisinin derivatives in Angola. No early treatment failures were observed in this study. Parasite clearance was rapid in all study arms, with parasitaemia resolving in at least 97% of participants by day 2 of follow-up and all participants by day 3. Finally, no mutations in pfk13 associated with artemisinin resistance were identified during molecular analysis of treatment failures.
In contrast to the previous rounds of therapeutic efficacy studies [6, 7], the minimum haemoglobin required for inclusion was increased to 8 g/dL from 5 g/dL, possibly explaining why no early treatment failures were observed in the current round. All but one of the early treatment failures observed in both the 2013 and 2015 studies were due to severe anemia. A haemoglobin of less than 5 g/dL in the presence of parasitaemia is a sign of severe malaria and, if observed during follow up, would prompt concern for treatment failure. However, it is common for patients on anti-malarial treatment to experience an initial drop in haemoglobin before improving. Children enrolled with a haemoglobin near 5 g/dL who undergo this customary decrease may be inappropriately classified as early treatment failures, when in fact they were following the expected treatment course.
Analysis of markers of partner drug resistance in AL and ASAQ treatment failures yielded results consistent with previous understanding of these resistance markers. Mutations in pfcrt K76 and pfmdr1 N86 are known to be associated with AL resistance [17], and these were present on day of failure in nearly all AL treatment failures. The pfcrt K76T genotype, which has been associated with decreased ASAQ susceptibility [17], was identified in all ASAQ treatment failures from this study. The only instances of a pfmdr1 86Y mutation, associated with amodiaquine resistance, were identified in ASAQ treatment failures.
As AL and ASAQ exhibit opposite selective pressures on certain loci in the pfmdr1 and pfcrt genes, alternating the use of these drugs might preserve diversity in these genes and minimize the emergence of resistance. Analysis of day 0 samples from the 2015 therapeutic efficacy study has shown that the majority of parasites circulating in the three sentinel provinces in Angola carry the pfmdr1 N86 genotype [34]. A similar analysis of all day 0 samples from this current study could be useful in characterizing how the circulating parasite population is affected by the selective pressures on the pfmdr1 gene.
None of the day 0 or day of failure samples had an elevated copy number of pfmdr1, consistent with the general absence [35, 36] or low rates [37, 38] of pfmdr1 copy number variation in sub-Saharan Africa. As a recently proposed marker, there are few available data on pfpm2 copy number variation in sub-Saharan Africa; absence of elevated copy number in this gene in the samples analysed here is consistent with relatively low rates of DP use in sub-Saharan Africa. However, all but one of the samples analysed for copy number variation contained multiple P. falciparum strains, so this analysis of could have missed an elevated copy number in parasites occurring at low-frequencies in mixed-strain infections.
Although the study was carried out in three provinces of Angola with varied geography and malaria endemicity, it is possible that these results may not be representative of either any one province or the entire country. Furthermore, since assignment of treatment drug was not random, drug efficacies should not be compared across study arms.
Conclusions
All three evaluated artemisinin-based combinations continue to be efficacious against P. falciparum malaria in the three sentinel sites in Angola. Rapid parasite clearance is consistent with full susceptibility to artemisinin derivatives. Periodic monitoring of in vivo drug efficacy remains a critical routine activity for the Angolan NMCP.
References
Angola Ministry of Health. National strategic plan for malaria control in Angola, 2016–2020. Luanda: Angola Ministry of Health; 2015.
WHO. Guidelines for the treatment of malaria. Geneva: World Health Organization; 2015.
WHO. Methods for surveillance of antimalarial drug efficacy. Geneva: World Health Organization; 2009.
Fançony C, Brito M, Gil JP. Plasmodium falciparum drug resistance in Angola. Malar J. 2016;15:72.
Guthmann JP, Ampuero J, Fortes F, van Overmeir C, Gaboulaud V, Tobback S, et al. Antimalarial efficacy of chloroquine, amodiaquine, sulfadoxine–pyrimethamine, and the combinations of amodiaquine + artesunate and sulfadoxine–pyrimethamine + artesunate in Huambo and Bie provinces, central Angola. Trans R Soc Trop Med Hyg. 2005;99:485–92.
Plucinski MM, Talundzic E, Morton L, Dimbu PR, Macaia AP, Fortes F, et al. Efficacy of artemether–lumefantrine and dihydroartemisinin–piperaquine for treatment of uncomplicated malaria in children in Zaire and Uíge provinces, Angola. Antimicrob Agents Chemother. 2015;59:437–43.
Plucinski MM, Dimbu PR, Macaia AP, Ferreira CM, Samutondo C, Quivinja J, et al. Efficacy of artemether–lumefantrine, artesunate–amodiaquine, and dihydroartemisinin–piperaquine for treatment of uncomplicated Plasmodium falciparum malaria in Angola, 2015. Malar J. 2017;16:62.
Kiaco K, Teixeira J, Machado M, do Rosário V, Lopes D. Evaluation of artemether-lumefantrine efficacy in the treatment of uncomplicated malaria and its association with pfmdr1, pfatpase6 and K13-propeller polymorphisms in Luanda, Angola. Malar J. 2015;14:504.
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67.
Amaratunga C, Lim P, Suon S, Sreng S, Mao S, Sopha C, et al. Dihydroartemisinin–piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis. 2016;16:357–65.
Van Hong N, Amambua-Ngwa A, Tuan NQ, Cuong DD, Giang NTH, Van Dung N, et al. Severe malaria not responsive to artemisinin derivatives in man returning from Angola to Vietnam. Emerg Infect Dis. 2014;20:1207.
Ringwald P, Dondorp A. Severe malaria not responsive to artemisinin derivatives in man returning from Angola to Vietnam. Emerg Infect Dis. 2015;21:1264–5.
Van Hong N, Amambua-Ngwa A, Tuan NQ, Cuong DD, Giang NTH, Van Dung N, et al. Severe malaria not responsive to artemisinin derivatives in man returning from Angola to Vietnam. Emerg Infect Dis. 2015;20:1207.
Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, et al. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science. 2015;347:428–31.
Ecker A, Lehane AM, Clain J, Fidock DA. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012;28:504–14.
Malmberg M, Ferreira PE, Tarning J, Ursing J, Ngasala B, Bjorkman A, et al. Plasmodium falciparum drug resistance phenotype as assessed by patient antimalarial drug levels and its association with pfmdr1 polymorphisms. J Infect Dis. 2013;207:842–7.
Venkatesan M, Gadalla NB, Stepniewska K, Dahal P, Nsanzabana C, Moriera C, et al. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P. falciparum malaria after artemether–lumefantrine and artesunate–amodiaquine. Am J Trop Med Hyg. 2014;91:833–43.
Price RN, Uhlemann AC, van Vugt M, Brockman A, Hutagalung R, Nair S, et al. Molecular and pharmacological determinants of the therapeutic response to artemether–lumefantrine in multidrug-resistant Plasmodium falciparum malaria. Clin Infect Dis. 2006;42:1570–7.
Witkowski B, Duru V, Khim N, Ross LS, Saintpierre B, Beghain J, et al. A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: a phenotype-genotype association study. Lancet Infect Dis. 2017;17:174–83.
Amato R, Lim P, Miotto O, Amaratunga C, Dek D, Pearson RD, et al. Genetic markers associated with dihydroartemisinin–piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype-phenotype association study. Lancet Infect Dis. 2017;17:164–73.
Snounou G, Viriyakosol S, Zhu XP, Jarra W, Pinheiro L, do Rosario VE, et al. High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol. 1993;61:315–20.
Anderson TJ, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, et al. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000;17:1467–82.
Anderson TJ, Su XZ, Bockarie M, Lagog M, Day KP. Twelve microsatellite markers for characterization of Plasmodium falciparum from finger-prick blood samples. Parasitology. 1999;119(Pt 2):113–25.
McCollum AM, Mueller K, Villegas L, Udhayakumar V, Escalante AA. Common origin and fixation of Plasmodium falciparum dhfr and dhps mutations associated with sulfadoxine–pyrimethamine resistance in a low-transmission area in South America. Antimicrob Agents Chemother. 2007;51:2085–91.
Alam MT, de Souza DK, Vinayak S, Griffing SM, Poe AC, Duah NO, et al. Selective sweeps and genetic lineages of Plasmodium falciparum drug-resistant alleles in Ghana. J Infect Dis. 2011;203:220–7.
Isozumi R, Uemura H, Kimata I, Ichinose Y, Logedi J, Omar AH, et al. Novel mutations in K13 propeller gene of artemisinin-resistant Plasmodium falciparum. Emerg Infect Dis. 2015;21:490–2.
Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004;364:438–47.
Blessborn D, Romsing S, Annerberg A, Sundquist D, Bjorkman A, Lindegardh N, et al. Development and validation of an automated solid-phase extraction and liquid chromatographic method for determination of lumefantrine in capillary blood on sampling paper. J Pharm Biomed Anal. 2007;45:282–7.
Kredo T, Mauff K, Van der Walt JS, Wiesner L, Maartens G, Cohen K, et al. Interaction between artemether–lumefantrine and nevirapine-based antiretroviral therapy in HIV-1-infected patients. Antimicrob Agents Chemother. 2011;55:5616–23.
WorldWide Antimalarial Resistance Network (WWARN), Lumefantrine PK/PD Study Group. Artemether–lumefantrine treatment of uncomplicated Plasmodium falciparum malaria: a systematic review and meta-analysis of day 7 lumefantrine concentrations and therapeutic response using individual patient data. BMC Med. 2015;13:227.
Plucinski MM, Morton L, Bushman M, Dimbu PR, Udhayakumar V. Robust algorithm for systematic classification of malaria late treatment failures as recrudescence or reinfection using microsatellite genotyping. Antimicrob Agents Chemother. 2015;59:6096–100.
Okell LC, Cairns M, Griffin JT, Ferguson NM, Tarning J, Jagoe G, et al. Nat Commun. 2014;5:5606.
Sinclair D, Zani B, Donegan S, Olliaro P, Garner P. Artemisinin-based combination therapy for treating uncomplicated malaria. Cochrane Database Syst Rev. 2009:Cd007483.
Ljolje D, Dimbu PR, Kelley J, Goldman I, Nace D, Macaia A, et al. Prevalence of molecular markers of artemisinin and lumefantrine resistance in three provinces in Angola, 2015. Malar J. 2018;17:84.
Ursing J, Kofoed PE, Rombo L, Gil JP. No pfmdr1 amplifications in samples from Guinea-Bissau and Liberia collected between 1981 and 2004. J Infect Dis. 2006;194:716–8 (author reply 718–719).
Happi CT, Gbotosho GO, Folarin OA, Sowunmi A, Hudson T, O’Neil M, et al. Selection of Plasmodium falciparum multidrug resistance gene 1 alleles in asexual stages and gametocytes by artemether–lumefantrine in Nigerian children with uncomplicated falciparum malaria. Antimicrob Agents Chemother. 2009;53:888–95.
Witkowski B, Nicolau ML, Soh PN, Iriart X, Menard S, Alvarez M, et al. Plasmodium falciparum isolates with increased pfmdr1 copy number circulate in West Africa. Antimicrob Agents Chemother. 2010;54:3049–51.
Nguetse CN, Adegnika AA, Agbenyega T, Ogutu BR, Krishna S, Kremsner PG, et al. Molecular markers of anti-malarial drug resistance in Central, West and East African children with severe malaria. Malar J. 2017;16:217.
Authors’ contributions
PRD, ESH, FF, and MMP planned and designed the study. ED, PRD, MF, CMF, ED, BNA, EM, CO, DP, JF, ES, and MMP supervised the study teams collecting the data. SSS, VK, and ET performed the molecular analyses. LW performed lumefantrine level analysis. ED, PRD, SSS, JFM, LW and MMP prepared the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Local support was provided by the Provincial health directorates of Benguela, Lunda Sul, and Zaire; the Mentor Initiative; and PSI Angola. The authors would like to thank all the study team members in each of the six sentinel health facilities and all study participants and their guardians.
Competing interests
The authors declare that they have no competing interests. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Availability of data and materials
Data are available through the Worldwide Antimalarial Resistance Network, http://www.wwarn.org/.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study received human subjects approval from both the Angolan Ministry of Health and the United States Centers for Disease Control and Prevention. Parents or guardians gave written informed consent for each participant before enrollment.
Funding
This study was funded by the United States President’s Malaria Initiative (PMI). Molecular marker testing was funded as part of the PMI Artemisinin Resistance Monitoring in Africa (PARMA) network.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1.
Consort flow diagrams for follow-up outcomes in all six study arms, 2017.
Additional file 2.
Microsatellite marker results, Angola therapeutic efficacy monitoring, 2017.
Additional file 3.
Day 7 lumefantrine drug levels in patients treated with artemether-lumefantrine, by treatment outcome, Zaire, Angola.
Additional file 4.
Day 7 lumefantrine levels in patients treated with artemether-lumefantrine, by treatment outcome, Zaire, Angola.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Davlantes, E., Dimbu, P.R., Ferreira, C.M. et al. Efficacy and safety of artemether–lumefantrine, artesunate–amodiaquine, and dihydroartemisinin–piperaquine for the treatment of uncomplicated Plasmodium falciparum malaria in three provinces in Angola, 2017. Malar J 17, 144 (2018). https://doi.org/10.1186/s12936-018-2290-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-018-2290-9