
Abstract
Background
The widespread presence of low-density asymptomatic infections with concurrent gametocytes may be a stumbling block for malaria elimination. This study investigated the asymptomatic reservoir of Plasmodium falciparum and Plasmodium vivax infections in schoolchildren from five settings in northwest Ethiopia.
Methods
Two cross-sectional surveys were conducted in June and November 2015, enrolling 551 students from five schools and 294 students from three schools, respectively. Finger prick whole blood and plasma samples were collected. The prevalence and density of P. falciparum and P. vivax parasitaemia and gametocytaemia were determined by 18S rRNA quantitative PCR (qPCR) and pfs25 and pvs25 reverse transcriptase qPCR. Antibodies against blood stage antigens apical membrane antigen-1 (AMA-1) and merozoite surface protein-1 (MSP-119) were measured for both species.
Results
Whilst only 6 infections were detected by microscopy in 881 slides (0.7%), 107 of 845 blood samples (12.7%) were parasite positive by (DNA-based) qPCR. qPCR parasite prevalence between sites and surveys ranged from 3.8 to 19.0% for P. falciparum and 0.0 to 9.0% for P. vivax. The median density of P. falciparum infections (n = 85) was 24.4 parasites/µL (IQR 18.0–34.0) and the median density of P. vivax infections (n = 28) was 16.4 parasites/µL (IQR 8.8–55.1). Gametocyte densities by (mRNA-based) qRT-PCR were strongly associated with total parasite densities for both P. falciparum (correlation coefficient = 0.83, p = 0.010) and P. vivax (correlation coefficient = 0.58, p = 0.010). Antibody titers against P. falciparum AMA-1 and MSP-119 were higher in individuals who were P. falciparum parasite positive in both surveys (p < 0.001 for both comparisons).
Discussion
This study adds to the available evidence on the wide-scale presence of submicroscopic parasitaemia by quantifying submicroscopic parasite densities and concurrent gametocyte densities. There was considerable heterogeneity in the occurrence of P. falciparum and P. vivax infections and serological markers of parasite exposure between the examined low endemic settings in Ethiopia.
Similar content being viewed by others

Background
The remarkable success that was documented in the control of malaria in the last decade and half has intensified efforts to achieve malaria elimination and brought eradication back on the table [1]. Detailed assessments of parasite carriage by conventional diagnostics alongside molecular investigation have uncovered a considerable proportion of malaria infections is undetected by microscopy and rapid diagnostic tests (RDT) [2, 3]. In settings where recent malaria control efforts have been successful, submicroscopic infections frequently outnumber microscopically detectable infections [3–5]. Whilst the clinical consequences of apparently asymptomatic submicroscopic infections are unknown [6, 7], infections may last for several months [8] and are associated with gametocyte production [9–11]. Gametocytes are essential for onward malaria transmission to mosquitoes. Although non-linear, the likelihood of mosquito infections increases with increasing gametocyte density [12] and microscopically detectable gametocytes are thus more likely to result in mosquito infections than submicroscopic gametocyte densities [4, 13]. Submicroscopic infections may nevertheless contribute considerably to malaria transmission because of their relative abundance in populations [10, 14].
Ethiopia has experienced a 66% decline in confirmed malaria cases between 2001 and 2011 [15]. Motivated by this decline, mainly attributed to wide-scale deployment of long-lasting insecticide-treated nets and implementation of artemisinin-based combination therapy (ACT) [15], the country set a plan to eliminate malaria in selected low-transmission settings by the end of 2020 [16]. Microscopy and RDT may be insufficiently sensitive to guide or evaluate these elimination efforts. Molecular assays to sensitively detect Plasmodium falciparum and Plasmodium vivax infections and serological assays of malaria exposure may be of great value in low-endemic settings [17, 18].
Previous studies in Ethiopia reported a high degree of submicroscopic parasite carriage [19–24] and underlined the relevance of (school) surveys using serological markers of malaria exposure to determine spatial and temporal variations [25]. However, none of these studies used serological and molecular assays together or quantified the low-density infections or circulating gametocytes. A better understanding of the distribution and contribution of submicroscopic infections to the overall parasite reservoir in low-endemic settings is a prerequisite for elimination efforts in order to shape the measures to be taken. The present study evaluated spatial and temporal variation in submicroscopic parasite and gametocyte carriage along with serological markers of malaria exposure in asymptomatic schoolchildren at five different sites in northwest Ethiopia.
Methods
Ethics statement
The study was reviewed and approved by the Institutional Ethics Review Board of the College of Natural Sciences at Addis Ababa University (ref. CNSDO/1/07/14), AHRI/ALERT Ethical Review Committee (ref. PO52/14) at Armauer Hansen Research Institute (AHRI), the Observational/Interventions Research Ethics Committee (ref. 8626) at London School of Hygiene and Tropical Medicine (LSHTM) and the National Research Ethics Review Committee (3.10|016\20) at the Ministry of Science and Technology of the Federal Democratic Republic of Ethiopia. Community sensitization was conducted using a cascade approach [26]. All parents of children who met the basic recruitment criteria were informed about the study and offered the choice to participate through an oral informed consent process. Written consent for the study was provided by a committee that comprised the school principal, deputy principal, classroom leaders, woreda (district) officials, elders, religious leaders and representatives of the family school association while parents maintained the right to withdraw their child from the survey.
Study area and population
The study was conducted in five elementary schools located in the kebeles of Andassa (1730masl, N 11°30′14.5″ and E°37°29′07.9″); Yinessa (1853masl, N 11°31′42.0″ and E°37°18′26.7″), Ahuri (2010masl, N 11°24′00.7″ and E°36°56′53.0″); Yeboden (1997masl, N 11°18′50.8″ and E°36°57′49.3″) and Fendika (1218masl, N 11°34′00.3″ and E °36°29′22.9″) in the Amhara Regional State of Ethiopia (Fig. 1). Andassa and Yinessa are located in Bahir Dar Zuria woreda; Ahuri and Yeboden in Debub Achefer woreda and Fendika is the administrative town of Jawi woreda. Two seasonal peaks of malaria transmission occur in the study areas; the main peak follows the heaviest rainfall that lasts from June to September, with a smaller peak in transmission in April/May following the short rains. The study sites, except Jawi, are located in the highlands of the North-Western part of Ethiopia, around the Lake Tana and Blue Nile river basin. Most of the study sites are characterized by swampy areas or irrigation lines that are associated with small water bodies conducive for anopheline breeding.

Map of study districts. The study was conducted in two surveys in the northwestern part of Ethiopia around the Lake Tana and Blue Nile river basin. Children from five elementary schools (Andassa, Yinessa, Ahuri, Yeboden and Jawi) were studied in the first survey in June 2015. During the second survey, in November 2015, students from three of the same schools (Andassa, Ahuri, Jawi) were studied
Two cross-sectional surveys were undertaken in June and November 2015, representing the beginning of the rainy season and the peak malaria transmission season, respectively. However, due to the recent El Ǹino phenomenon that occurred during the study period the rain was intermittent and most of the areas were dry. Study participants were selected randomly among students attending the elementary schools stratified by age following protocols developed by Brooker et al. [26]. One hundred and ten students were targeted for each school from five class levels (2–6); 22 students from each class comprising 11 boys and 11 girls. The same students were invited for both surveys.
Sample collection and microscopic parasite detection
Detailed clinical and demographic data and utilization of intervention services was recorded with a pre-tested semi-structured interview based questionnaire. Axillary body temperature was first checked for all students. If the student was found febrile (axillary temperature ≥37.5 °C) or reported a history of fever in the past 48 h, an RDT (First Response Malaria Ag (pLDH/HRP2) Combo RDT, Premier Medical Corporation Ltd., India) was immediately used to diagnose the presence of malaria parasites. If the child was not febrile, a finger-prick sample (0.3–0.4 mL blood) was collected for microscopy examination of infection with thick and thin blood smears, dried blood spots (DBS) on 3MM Whatman filter paper (Whatman, Maidstone, UK) and in an EDTA-coated microtainer tube (BD). EDTA blood samples were centrifuged immediately using portable centrifuge powered from a car battery for 3 min (at 3000 rpm) and the plasma was put into an equal volume of 0.05% sodium azide (SIGMA-ALDRICH) solution. The cell pellet was put into RNAprotect buffer (QIAGEN) in a 1:5 ratio and was used for extraction of nucleic acids to determine parasite density by qPCR and RNA-based gametocyte detection. DBS was stored in self-indicating silica gel desiccant beads (Geejay Chemicals Ltd). Plasma, cell pellet and DBS samples were shipped on ice packs from the field to the Amhara Regional Laboratory in Bahir Dar. There, samples were stored at −20 °C, before being shipped in liquid nitrogen containers to Addis Ababa and from there shipped on dry-ice to laboratories in London and Nijmegen where they were stored at −80 °C until further investigation.
Blood smears were stained at the sites for 10 min with 10% Giemsa (VWR) and then screened for malaria asexual parasites and gametocyte stages. Slides were declared negative if no parasites were observed in 100 microscopic fields. Asexual parasites and gametocytes were counted against 200 and 500 white blood cells, respectively. Slides were read independently by two microscopists in the field and at AHRI’s facility. A third, WHO certified, professional microscopist read the slides to verify the disagreement in the reading results from the two readers.
Nucleic acid extraction, parasite and gametocyte detection by qPCR and qRT-PCR
Total nucleic acids were extracted from 600 μL aliquots containing 100 μL cell pellet plus 500 μL RNAprotect buffer (QIAGEN) using a MagNAPure LC automatic extractor and total Nucleic Acid Isolation Kit—High Performance (Roche Applied Science) and eluted in 50 μL elution buffer. qPCR for parasite detection was performed by targeting the 18S rRNA small subunit gene on DNA for P. falciparum and P. vivax using primer and probe sequences described in Hermsen et al. [27] and Wampfler et al. [28], respectively. Genomic human and parasite DNA was digested with the RQ1 DNaseI Digest Kit (Promega), then cDNA was synthesized with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) for evaluation of transcription level of Pfs25 and Pvs25 to quantify P. falciparum and P. vivax gametocytes, respectively. Parasite and gametocyte quantification was done based on models developed by Wampfler et al. [28] and Koepfli et al. [11]. Briefly, plasmid constructs from Wampfler et al. [28], except the P. falciparum 18S rRNA target on DNA, were used to infer copy numbers, then the model was applied to calculate the corresponding parasite and gametocyte counts for P. falciparum and P. vivax parasitaemia and P. falciparum gametocytaemia [28] and for the P. vivax gametocytaemia [11]. The P. falciparum 18S rRNA gene was cloned into TOPO®-TA Cloning vector (Thermo Fisher Scientific) following the company’s protocol. Serial dilutions (106, 105 and 104) of plasmids containing the respective amplicons in triplicates were run in parallel on every plate to generate standard curves for quantification of copy numbers. Ten percent of positive samples were retested as quality control. All cDNA samples that passed through DNA digestion were further checked for genomic DNA contamination.
The Pfs25 sequence was amplified using GoTaq® qPCR Master Mix (Promega) that contains a proprietary dye and the rest (the Pvs25 and the P. falciparum and P. vivax 18S rRNA targets on DNA) were amplified using probe based detection using TaqMan Fast Advanced Master Mix (Applied Biosystems). Melt curves were run to evaluate detection of the specific product when the detection was not probe based. In all assays, both qPCR and qRT-PCR, 104 nM probe and 833 nM primer concentrations were used. All probes were from Life technologies (Applied Biosystems), primers were from SIGMA ALDRICH, and CFX96™ Real-Time PCR Detection System (BIO-RAD) was used.
Serological detection of immune response to malaria antigens
Antibodies to P. falciparum and P. vivax Apical Membrane Antigen-1 (AMA-1) and Merozoite Surface Protein-119 (MSP-119) were detected using plasma samples that were collected and stored in equal volume of 0.05% sodium azide by Enzyme Linked Immunosorbent Assay (ELISA), as previously described [29].
Data analysis
All analyses were performed with STATA 12 (StataCorp., TX, USA) and Graph Pad Prism 5.0 (Graph Pad Software Inc., CA, USA). Asymptomatic malaria was defined as malaria infections that lack typical clinical symptoms (absence of fever at the time of sampling or absence of history of fever 48 h before sampling) but are detectable by microscopy, rapid diagnostic test or molecular methods [30]. Antibody prevalence was determined per antigen after defining a cut-off optical density (OD) using the mixture model [31]. Prevalence was then used for the separate antigens and for the combined P. falciparum and the combined P. vivax antigens [17]. Chi square or Fisher’s Exact tests were used for dichotomous analyses; logistic or linear regression models were used for multivariate analyses. The correlation between continuous variables was assessed by Spearman correlation coefficient; continuous variables were compared between groups using the non-parametric Wilcoxon Rank Sum test.
Results
Parasite and gametocyte prevalence by microscopy and qPCR
During the first cross-sectional survey (June 2015), 551 students were enrolled from 5 elementary schools. The median age of the students was 12 years (IQR 11–14) (Table 1). During the second survey (November 2015), three of the five schools were revisited and 87.8% (294/335) students were successfully sampled for a second time. Microscopy detected only 6 infections (3 in each survey), which were all from Jawi. There were seven febrile students (axillary temperature ≥37.5 °C) who were all RDT negative but 1 was positive for P. vivax by microscopy with 6267 parasites/μL. During the second survey, 14.6% (43/294) of the children reported that they had been treated for malaria in the time-period between the two surveys (Table 1).
Parasite prevalence by (DNA-based) qPCR during the first survey was 7.8% (43/551) for P. falciparum and 2.4% (13/551) for P. vivax (Table 2). During the second survey parasite prevalence was 12.9% (38/294) for P. falciparum and 4.8% (14/294) for P. vivax. Of all parasite-positive individuals, 7.1% (4/56) and 5.4% (3/52) had mixed P. falciparum and P. vivax infections in the first and second survey, respectively. Nine individuals (29%, 9/31) who were P. falciparum positive in the first survey and sampled again in the second survey were again P. falciparum positive; 28 (10.7%, 28/263) acquired a P. falciparum infection after being negative in the first survey. Two individuals (22%, 2/9) who were P. vivax positive in the first survey and sampled again in the second survey remained P. vivax positive; 12 (4.2%, 12/285) acquired a P. vivax infection.
Jawi, the study site at the lowest altitude (1218 metres above sea level), had the highest prevalence of Plasmodium infections; 16.8% (18/107) and 25.0% (25/100) during the first and second surveys, respectively (Fig. 2). While P. falciparum and P. vivax infections were detected in Andassa, Jawi, Yeboden and Yinessa, only P. falciparum infections were found in Ahuri (2010 metres above sea level), where the prevalence of P. falciparum infection was 9.9% (11/111) during the first and 8.2% (8/98) during the second survey.

Prevalence of submicroscopic P. falciparum and P. vivax malaria carriage by quantitative PCR. Prevalence of submicroscopic carriage of P. falciparum (gray bars), P. vivax (colorless bars) and mixed infections (hatched bars) among children from the five schools in the first survey and three schools from the second survey is presented. Error bars indicate the upper limit of the 95% confidence interval
Gametocyte carriage was tested for all qPCR parasite positive and negative individuals. Of all samples from the first and second survey, 1.3% (7/551) and 2.0% (6/294) were P. falciparum gametocyte positive. Among P. falciparum qPCR positive individuals, P. falciparum gametocyte prevalence by (RNA-based) qRT-PCR was 9.3% (4/43) in the first and 10.8% (4/37) in the second survey (Table 2). Of all samples from the first and second survey, 2.0% (11/551) and 4.1% (12/294) were P. vivax gametocyte positive. Among P. vivax qPCR positive individuals, P. vivax gametocyte prevalence was 69.2% (9/13) in the first and 57.1% (8/14) in the second survey (Table 2).
Parasite densities in relation to detectability by microscopy and gametocyte densities
Densities of P. falciparum and P. vivax infections were commonly low (Fig. 3). The 4 samples, that had microscopically detectable P. falciparum parasites showed higher median parasite density by qPCR (median density 326.1 parasites/μL; IQR 227.3–759.6,) compared to the 81 samples that were microscopy negative (median density 24.1 parasites/μL; IQR 17.7–32.4, p = 0.001). Similarly, the two samples, that had microscopically detectable P. vivax parasites also had higher parasite density by qPCR (density 56.0 and 8627.4 parasites/μL) compared to the 26 samples that were microscopy negative (median density 15.6 parasites/μL; IQR 8.5–30.5, p = 0.03). Estimated gametocyte densities were low for both P. falciparum (median 10.4 gametocytes/μL; IQR 3.1–51.2) and especially P. vivax (median 2.3 gametocytes/μL; IQR 0.7–32.0). P. falciparum qRT-PCR gametocyte density was positively associated with P. falciparum qPCR parasite density (Fig. 4; spearman correlation coefficient 0.83, p = 0.010). Similarly, P. vivax qRT-PCR gametocyte density was positively associated with P. vivax qPCR parasite density (Fig. 4; spearman correlation coefficient 0.58, p = 0.010).

Parasite density of asymptomatic infections. Submicroscopic parasite densities as measured by 18S based qPCR for P. falciparum (a) and P. vivax (b) in microscopically positive (black circles) and only submicroscopic (colorless) asymptomatic malaria parasite carriers. Presented in the Y-axis is the Log10 transformed parasite density per µL of blood from the two surveys in school children from five schools (X-axis). The lines refer to the median parasite density from children in the specific school. Numbers below the figure indicate the qPCR parasite prevalence during the two surveys in the respective school. ND not determined

Association between parasite density and gametocyte density. The association between P. falciparum total parasite density versus gametocyte density (a) and P. vivax total parasite density versus gametocyte density (b). P. falciparum qRT-PCR gametocyte density was positively associated with P. falciparum qPCR parasite density (Spearman correlation coefficient 0.83, p = 0.010). P. vivax qRT-PCR gametocyte density was positively associated with P. vivax qPCR parasite density (Spearman correlation coefficient 0.58, p = 0.010)
Seroprevalence and dynamics of antibody titres to malaria antigens
The prevalence of antibody responses to P. falciparum and P. vivax antigens was assessed as a more sensitive indicator of malaria exposure than current parasite carriage by qPCR. Seroprevalence to P. falciparum and P. vivax AMA-1 and MSP-119 was strongly dependent on site (Fig. 5). The prevalence of antibodies to P. falciparum AMA-1 and/or MSP-119 in the first survey was 11.0% (11/100) in Ahuri, 45.1% (41/91) in Andassa and 44.0% (44/100) in Jawi. In the second survey, 3.2% (3/95), 24.4% (22/90), and 34.7% (33/95) prevalence was detected, respectively. The prevalence of antibodies to P. vivax AMA-1 and/or MSP-119 in the first survey was 9.0% (9/100) in Ahuri, 36.3% (33/91) in Andassa and 27.0% (27/100) in Jawi. In the second survey, the prevalence of any P. vivax antibodies was 8.5% (8/94), 24.4% (22/90), and 26.3% (25/95), respectively. After adjustment for study site, the prevalence of antibodies against either P. falciparum antigen was strongly associated with age in both the first (odds ratio for each year increase in age 1.29, 95% CI 1.14–1.46, p < 0.001) and second survey (OR 1.50, 95% CI 1.26–1.79, p < 0.001). For P. vivax antibodies, there was no statistically significant association with age (p ≥ 0.19). The prevalence and titer of P. falciparum antibodies was associated with parasite carriage by qPCR. Individuals who were parasite positive by qPCR in both surveys (n = 9) had a borderline significantly higher odds of being positive to either P. falciparum antigen in the second survey (OR 4.36, 95% CI 0.86–22.08, p = 0.076) compared to children who were parasite-negative in both surveys (n = 235), after adjustment for age and site. Individuals who were parasite positive in one but not both surveys (n = 50) had no significantly higher odds of being P. falciparum antibody positive in the second survey (OR 1.03, 95% CI 0.44–2.45, p = 0.94) compared to children who were parasite-negative in both surveys, after adjustment for age and site (Fig. 6). The titer of antibodies to P. falciparum AMA-1 (p < 0.001) and MSP-119 (p < 0.001) in the second survey was also higher in individuals who were parasite positive by qPCR in both surveys compared to those remaining parasite-free, after adjustment for age and study site (Fig. 6). Individuals who were parasite positive in one but not both surveys did not have statistically significant higher antibody titers to P. falciparum MSP-119 (p = 0.35) or AMA-1 (p = 0.93) compared to individuals who remained parasite-free. No associations between qPCR parasite carriage and antibody responses to P. vivax antigens were observed (Fig. 6).

Prevalence of antibodies generated to P. falciparum and P. vivax AMA-1 and MSP-119 in the study sites. The seroprevalence, positivity to antibodies generated against the P. falciparum AMA-1 (a) and MSP-119 (b), the P. vivax AMA-1 (d) and MSP-119 (e) during the first survey in June 2015 (black bars) and the second survey in November 2015 (colorless bars). Any Pf (c) and Any Pv (d) refers to positivity to antibodies to either of the two P. falciparum (c) or P. vivax (d) antigens. The three schools that were surveyed for the two rounds are presented in the X axis

Dynamics of antibody responses. Antibody prevalence (top) and titre (bottom) for P. falciparum and P. vivax antigens in relation to qPCR detected parasite prevalence in neither of the surveys (parasite-free), in just one of the surveys (once positive) or in both surveys (twice positive). Antibody titre is presented for all individuals, including individuals whose ELISA optical density was below the threshold for positivity. The two individuals who were P. vivax positive in both surveys had optical density values for P. vivax AMA-1 and MSP-119 that were below the threshold for positivity and are therefore not presented in the antibody prevalence plot
Gametocyte carriage is essential for onward transmission to mosquitoes and associated with total parasite biomass [11]. At the end of follow-up (second survey), the prevalence of antibody responses to either of the P. falciparum antigens was 75.0% (9/12) for individuals who had P. falciparum gametocytes detected by qRT-PCR during the study period compared to 18.3% (49/268) for non-carriers (p < 0.001). The prevalence of antibody responses to either of the P. vivax antigens was 31.3% (5/16) for individuals who had P. vivax gametocytes detected by qRT-PCR during the study period compared to 19.0% (50/263) for non-carriers (p = 0.23).
Discussion
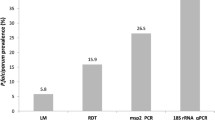
The current study adds to the available evidence on the wide-scale presence of submicroscopic parasitaemia. This study quantified submicroscopic parasites and concurrent gametocytes using highly sensitive molecular assays in asymptomatic schoolchildren. Whilst only 6 infections were detected by microscopy in 881 slides, 107 of 845 blood samples were parasite positive by qPCR. A higher prevalence of malaria infections by molecular methods is commonly reported and the proportion of infections that is submicroscopic is highest in low-endemic settings [2, 3]. Nevertheless, the 18-fold higher parasite prevalence observed by qPCR compared to microscopy is remarkable [3]. Median densities were <10 parasites/μL for both P. falciparum and P. vivax, below the density of 50–100 parasites/μL that is detectable by routine microscopy [32]. Expert microscopy may detect lower parasite densities but even expert microscopy may only have a sensitivity of ~29% for detecting parasite densities in the range of 1–10 parasites/μL [33].
The findings further demonstrated that approximately 10% of all P. falciparum infections and 60% of all P. vivax infections had concurrent detectable low-density gametocytaemia [9, 11]. For both Plasmodium species gametocyte densities were positively associated with total parasite densities [11]. In P. falciparum gametocytes typically comprise <5% of the total parasite biomass. With the very low parasite densities that were detected in this study, it is conceivable that concurrent gametocyte densities were too low to be detected by qRT-PCR. On the other hand, the finding that some individuals were gametocyte positive but negative for parasites by qPCR is likely to reflect the difference in sensitivity between DNA- and RNA-based assays [4] and illustrates that some infections could have been missed by qPCR [5]. This might partly explain infection of mosquitoes even when no parasites were detected by molecular methods [34]. Higher blood volume PCR [5] or more sensitive PCR targets [35] may reduce the proportion of false-negative PCR samples although it is likely that a fraction of infections will remain undetected with currently available diagnostics [5].
The current study identified heterogeneous transmission of Plasmodium species in the five study sites. At four sites both P. falciparum and P. vivax were prevalent; the former was the dominant species. In one highland site, Ahuri at 2010 metres above sea level, only P. falciparum infections were detected. Although parasite prevalence by qPCR was relatively low in Ahuri, it remains to be demonstrated whether infections were acquired locally. The finding may reflect a shift in altitude in P. falciparum distribution. In warmer years, an increase in the altitude at which malaria is observed has been reported for Ethiopia and Colombia [36]. The current findings may also be influenced by climatic changes, including the El Ǹino phenomenon that occurred during the study year and resulted in intermittent rain and increased temperature across the region. Intriguingly, antibodies to P. vivax AMA-1 and MSP-119 were detected in approximately 9% of children in Ahuri. This may reflect past exposure to P. vivax [31], exposure outside the area of residence, P. vivax exposure that is too infrequent to be detected in parasitological surveys [17], or cross-reactivity of antibodies acquired following P. falciparum exposure. For P. falciparum, antibody responses were associated with P. falciparum infections measured by qPCR. Individuals who were parasite positive in both surveys had higher prevalence and density of P. falciparum antibodies [37]. It is unclear whether this reflects boosting of antibody responses by circulating low densities of parasite antigens [38] or the fact that current infections are strongly associated with cumulative past malaria exposure [39]. Parasite prevalence and the incidence of new infections in the study populations was too low for meaningful assessments of serological markers of malaria exposure as reliable indicator of recent malaria exposure or explore the longevity of antibody responses in relation to time since last infection. However, the current findings of an association between antibody prevalence and qPCR-detected infections and qRT-PCR detected gametocyte carriage, suggest that serological markers might be useful to identify P. falciparum exposed individuals or populations whose infections are undetectable by microscopy but may contribute to onward transmission. It is currently unclear why this association was not observed for P. vivax and more detailed longitudinal studies are required to robustly demonstrate the ability of serological markers of malaria exposure to detect (recent or current) malaria infections. Recently described serological markers of recent malaria infection [40] have the potential to perform considerably better in this respect since antibodies to AMA-1 and MSP-119 have a long half-life [29] and may be less informative to indicate ongoing malaria transmission in settings where transmission has declined. The age-range of the current study population was too narrow to generate a full age-seroprevalence curve that may demonstrate whether recent changes in malaria exposure have occurred [31, 41]. The association between antibody responses to P. falciparum blood stage antigens and P. falciparum gametocyte carriage is plausibly a consequence of a larger total parasite biomass in gametocyte carriers [11] that forms a stimulus for antibody production.
The relevance of the low-density parasite reservoir that was described in the current study remains to be established. Whilst chronic low-density infections may have clinical consequences [6], most attention goes to their possible role in the human infectious reservoir for malaria [10, 13]. The current study demonstrates that a proportion of the low-density infections had detectable gametocyte densities. For P. falciparum this proportion was only 10% which is lower than commonly reported in parasite carriers detected in cross-sectional or clinical surveys [9]. It may be argued that these infections may produce detectable gametocytes at a later stage, but the dynamics of gametocyte production and infectivity of submicroscopic infections is currently unknown. By comparison, the prevalence of gametocytes in P. vivax infections was higher, as has been described before, and may reflect the more rapid production and maturation of P. vivax gametocytes [9, 11, 42]. Efforts are currently ongoing to establish mosquito feeding facilities in the study area to determine the contribution of symptomatic, asymptomatic and submicroscopic infections to onward transmission to mosquitoes.
Conclusions
The current findings demonstrate a considerable pool of low density infections in schoolchildren in five low endemic sites in Ethiopia that are potentially to be included in malaria elimination initiatives by the Ethiopian Ministry of Health. Serological markers may play a role in identifying individuals or populations who are disproportionately exposed to malaria and may be preferentially targeted with interventions.
References
WHO. World Malaria Report 2015. Geneva, World Health Organization, 2015. http://apps.who.int/iris/bitstream/10665/200018/1/9789241565158_eng.pdf. Accessed 08 May 2016.
Wu L, van den Hoogen LL, Slater H, Walker PG, Ghani AC, Drakeley CJ, et al. Comparison of diagnostics for the detection of asymptomatic Plasmodium falciparum infections to inform control and elimination strategies. Nature. 2015;528:S86–93.
Okell LC, Bousema T, Griffin JT, Ouedraogo AL, Ghani AC, Drakeley CJ. Factors determining the occurrence of submicroscopic malaria infections and their relevance for control. Nat Commun. 2012;3:1237.
Bousema T, Okell L, Felger I, Drakeley C. Asymptomatic malaria infections: detectability, transmissibility and public health relevance. Nat Rev Microbiol. 2014;12:833–40.
Imwong M, Stepniewska K, Tripura R, Peto TJ, Lwin KM, Vihokhern B, et al. Numerical distributions of parasite densities during asymptomatic malaria. J Infect Dis. 2016;213:1322–9.
Chen I, Clarke SE, Gosling R, Hamainza B, Killeen G, Magill A, et al. “Asymptomatic” malaria: a chronic and debilitating infection that should be treated. PLoS Med. 2016;13:e1001942.
de Mast Q, Brouwers J, Syafruddin D, Bousema T, Baidjoe AY, de Groot PG, et al. Is asymptomatic malaria really asymptomatic? Hematological, vascular and inflammatory effects of asymptomatic malaria parasitaemia. J Infect. 2015;71:587–96.
Tripura R, Peto TJ, Chalk J, Lee SJ, Sirithiranont P, Nguon C, et al. Persistent Plasmodium falciparum and Plasmodium vivax infections in a western Cambodian population: implications for prevention, treatment and elimination strategies. Malar J. 2016;15:181.
Bousema T, Drakeley C. Epidemiology and infectivity of Plasmodium falciparum and Plasmodium vivax gametocytes in relation to malaria control and elimination. Clin Microbiol Rev. 2011;24:377–410.
Slater HC, Ross A, Ouedraogo AL, White LJ, Nguon C, Walker PG, et al. Assessing the impact of next-generation rapid diagnostic tests on Plasmodium falciparum malaria elimination strategies. Nature. 2015;528:S94–101.
Koepfli C, Robinson LJ, Rarau P, Salib M, Sambale N, Wampfler R, et al. Blood-stage parasitaemia and age determine Plasmodium falciparum and Plasmodium vivax gametocytaemia in Papua New Guinea. PLoS ONE. 2015;10:e0126747.
Churcher TS, Bousema T, Walker M, Drakeley C, Schneider P, Ouédraogo AL, et al. Predicting mosquito infection from Plasmodium falciparum gametocyte density and estimating the reservoir of infection. Elife. 2013;2:e00626.
Lin JT, Saunders DL, Meshnick SR. The role of submicroscopic parasitaemia in malaria transmission: what is the evidence? Trends Parasitol. 2014;30:183–90.
Gaye A, Bousema T, Libasse G, Ndiath MO, Konate L, Jawara M, et al. Infectiousness of the human population to Anopheles arabiensis by direct skin feeding in an area hypoendemic for malaria in Senegal. Am J Trop Med Hyg. 2015;92:648–52.
Aregawi M, Lynch M, Bekele W, Kebede H, Jima D, Taffese HS, et al. Time series analysis of trends in malaria cases and deaths at hospitals and the effect of antimalarial interventions, 2001–2011, Ethiopia. PLoS ONE. 2014;9:e106359.
Health Federal Democratic Republic of Ethiopia Ministry of Health. National Malaria Programme Monitoring and Evaluation Plan 2014–2020. http://www.moh.gov.et/documents/26765/0/National+Malaria+Program+Plan+2014-2020/d1189778-9dd0-4523-ba79-509aae69c4b4?version=1.1. Accessed 10 March 2016.
Bousema T, Youssef RM, Cook J, Cox J, Alegana VA, Amran J, et al. Serologic markers for detecting malaria in areas of low endemicity, Somalia, 2008. Emerg Infect Dis. 2010;16:392–9.
Drakeley C, Cook J. Potential contribution of sero-epidemiological analysis for monitoring malaria control and elimination: historical and current perspectives. Adv Parasitol. 2009;69:299–352.
Santana-Morales MA, Afonso-Lehmann RN, Quispe MA, Reyes F, Berzosa P, Benito A, et al. Microscopy and molecular biology for the diagnosis and evaluation of malaria in a hospital in a rural area of Ethiopia. Malar J. 2012;11:199.
Golassa L, Enweji N, Erko B, Aseffa A, Swedberg G. Detection of a substantial number of sub-microscopic Plasmodium falciparum infections by polymerase chain reaction: a potential threat to malaria control and diagnosis in Ethiopia. Malar J. 2013;12:352.
Alemu A, Fuehrer HP, Getnet G, Kassu A, Getie S, Noedl H. Comparison of Giemsa microscopy with nested PCR for the diagnosis of malaria in North Gondar, north-west Ethiopia. Malar J. 2014;13:174.
Mekonnen SK, Aseffa A, Medhin G, Berhe N, Velavan TP. Re-evaluation of microscopy confirmed Plasmodium falciparum and Plasmodium vivax malaria by nested PCR detection in southern Ethiopia. Malar J. 2014;13:48.
Golassa L, Baliraine FN, Enweji N, Erko B, Swedberg G, Aseffa A. Microscopic and molecular evidence of the presence of asymptomatic Plasmodium falciparum and Plasmodium vivax infections in an area with low, seasonal and unstable malaria transmission in Ethiopia. BMC Infect Dis. 2015;15:310.
Tadesse FG, Pett H, Baidjoe A, Lanke K, Grignard L, Sutherland C, et al. Submicroscopic carriage of Plasmodium falciparum and Plasmodium vivax in a low endemic area in Ethiopia where no parasitaemia was detected by microscopy or rapid diagnostic test. Malar J. 2015;14:303.
Ashton RA, Kefyalew T, Rand A, Sime H, Assefa A, Mekasha A, et al. Geostatistical modeling of malaria endemicity using serological indicators of exposure collected through school surveys. Am J Trop Med Hyg. 2015;93:168–77.
Brooker S, Kolaczinski JH, Gitonga CW, Noor AM, Snow RW. The use of schools for malaria surveillance and programme evaluation in Africa. Malar J. 2009;8:231.
Hermsen CC, Telgt DS, Linders EH, van de Locht LA, Eling W, Mensink EJ, et al. Detection of Plasmodium falciparum malaria parasites in vivo by real-time quantitative PCR. Mol Biochem Parasitol. 2001;118:247–51.
Wampfler R, Mwingira F, Javati S, Robinson L, Betuela I, Siba P, et al. Strategies for detection of Plasmodium species gametocytes. PLoS ONE. 2013;8:e76316.
Drakeley CJ, Corran PH, Coleman PG, Tongren JE, McDonald SL, Carneiro I, et al. Estimating medium- and long-term trends in malaria transmission by using serological markers of malaria exposure. Proc Natl Acad Sci USA. 2005;102:5108–13.
Bousema T, Okell L, Felger I, Drakeley C. Asymptomatic malaria infections: detectability, transmissibility and public health relevance. Nat Rev Microbiol. 2014;12:833–40.
Cook J, Reid H, Iavro J, Kuwahata M, Taleo G, Clements A, et al. Using serological measures to monitor changes in malaria transmission in Vanuatu. Malar J. 2010;9:169.
Wongsrichanalai C, Barcus MJ, Muth S, Sutamihardja A, Wernsdorfer WH. A review of malaria diagnostic tools: microscopy and rapid diagnostic test (RDT). Am J Trop Med Hyg. 2007;77:119–27.
Bejon P, Andrews L, Hunt-Cooke A, Sanderson F, Gilbert SC, Hill AV. Thick blood film examination for Plasmodium falciparum malaria has reduced sensitivity and underestimates parasite density. Malar J. 2006;5:104.
Kiattibutr K, Roobsoong W, Sriwichai P, Saeseu T, Rachaphaew N, Suansomjit C, et al. Infectivity of symptomatic and asymptomatic Plasmodium vivax infections to a Southeast Asian vector, Anopheles dirus. Int J Parasitol. 2017;47:163–70.
Hofmann N, Mwingira F, Shekalaghe S, Robinson LJ, Mueller I, Felger I. Ultra-sensitive detection of Plasmodium falciparum by amplification of multi-copy subtelomeric targets. PLoS Med. 2015;12:e1001788.
Siraj AS, Santos-Vega M, Bouma MJ, Yadeta D, Ruiz Carrascal D, Pascual M. Altitudinal changes in malaria incidence in highlands of Ethiopia and Colombia. Science. 2014;343:1154–8.
Proietti C, Verra F, Bretscher MT, Stone W, Kanoi BN, Balikagala B, et al. Influence of infection on malaria-specific antibody dynamics in a cohort exposed to intense malaria transmission in northern Uganda. Parasite Immunol. 2013;35:164–73.
Shekalaghe S, Alifrangis M, Mwanziva C, Enevold A, Mwakalinga S, Mkali H, et al. Low density parasitaemia, red blood cell polymorphisms and Plasmodium falciparum specific immune responses in a low endemic area in northern Tanzania. BMC Infect Dis. 2009;9:69.
Kinyanjui SM, Bejon P, Osier FH, Bull PC, Marsh K. What you see is not what you get: implications of the brevity of antibody responses to malaria antigens and transmission heterogeneity in longitudinal studies of malaria immunity. Malar J. 2009;8:242.
Helb DA, Tetteh KK, Felgner PL, Skinner J, Hubbard A, Arinaitwe E, et al. Novel serologic biomarkers provide accurate estimates of recent Plasmodium falciparum exposure for individuals and communities. Proc Natl Acad Sci USA. 2015;112:E4438–47.
Stewart L, Gosling R, Griffin J, Gesase S, Campo J, Hashim R, et al. Rapid assessment of malaria transmission using age-specific sero-conversion rates. PLoS ONE. 2009;4:e6083.
McKenzie FE, Jeffery GM, Collins WE. Gametocytaemia and fever in human malaria infections. J Parasitol. 2007;93:627–33.
Authors’ contributions
FGT planned and executed the field and laboratory works and analyzed results and drafted the manuscript; TB conceived the project, advised the field and lab works and critically commented on the draft. EG participated in the planning, execution of the field work and discussion and commented on the draft manuscript. LvH and JS participated in the ELISA assays and commented on the manuscript. KL and IF participated in the molecular analyses and troubleshooting and commented on the manuscript. CD and KT advised the execution of the ELISA assays and guided data analysis and commented on the manuscript, HA, HM, RS, and AA participated in the study and commented on the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We are indebted to the schoolchildren and their parents for their willingness to take part in this study. We thank Nuffic for partially sponsoring this study via its NFP scheme. Thanks also goes to the Armauer Hansen Research Institute and Institute of Biotechnology at Addis Ababa University for administrative and technical support during the field work in Ethiopia. We thank the school principals, teachers, Amhara Regional Laboratory, district and regional health bureau officers and lab technicians and nurses for their assistance during sample collection. We thank Elias F Mekuriaw for constructing the map.
Competing interests
The authors declare that they have no competing interests.
Project funding statement
This project was partly funded by Grants from the Netherlands Organization for Scientific Research (Vidi fellowship; NWO Project Number 016.158.306 and fellowship from the European Research Council (ERC-2014-StG 639776) to TB and the Netherlands organization for international cooperation in higher education (Nuffic; grant number NFP-PhD.14/150 to FGT) and the Armauer Hansen Research Institute (via its core funding from NORAD and SIDA).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tadesse, F.G., van den Hoogen, L., Lanke, K. et al. The shape of the iceberg: quantification of submicroscopic Plasmodium falciparum and Plasmodium vivax parasitaemia and gametocytaemia in five low endemic settings in Ethiopia. Malar J 16, 99 (2017). https://doi.org/10.1186/s12936-017-1749-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-017-1749-4