
Abstract
Background
Plasmodium falciparum gametocytes are vital to sustaining malaria transmission. Parasite densities, multiplicity of infection as well as asexual genotype are features that have been found to influence gametocyte production. Measurements of the prevalence of Plasmodium sp. gametocytes may serve as a tool to monitor the success of malaria eradication efforts.
Methods
Whole blood was collected from 112 children aged between 6 months and 13 years with uncomplicated P. falciparum malaria attending three health facilities in southern Ghana from June to August, 2014 before (day 0) and 4 days after completion of anti-malaria drug treatment (day 7). Malaria parasites were observed by microscopy and polymerase chain reaction (PCR); submicroscopic gametocyte carriage was measured by a Pfs25 (PF3D7_1031000) mRNA real time reverse transcriptase polymerase chain reaction (RT-PCR). Parasite genotyping was performed on gDNA extracted from dried filter paper blood blots by amplification of the polymorphic regions of msp1 (PF3D7_0930300) and msp2 (PF3D7_0206800) using PCR.
Results
Microscopy estimated 3.1% (3/96) of the total population to carry gametocytes on day 0, which decreased to 2.1% (2/96) on day 7. In contrast, reverse transcriptase-real time PCR (RT-PCR) analysis of a subset of 35 samples estimated submicroscopic gametocyte carriage to be as high as 77% (27/35) using primers specific for Pfs25 (CT < 35) on day 0 and by day 7 this only declined to 60% (21/35). Genotyping the msp2 gene identified higher levels of MOI than the msp1 gene.
Conclusions
Although below detection by microscopy, gametocyte prevalence at submicroscopic levels are high in this region and emphasize the need for more effective elimination approaches like the development of transmission-blocking vaccines and safer gametocytocidal drugs.
Similar content being viewed by others


Background
In Ghana, malaria is still one of the leading causes of outpatient attendance and mortality in children under the age of 5 years [1], despite enhanced control efforts. Plasmodium falciparum, the most lethal of the five species that cause human malaria, is responsible for about 90% of all malaria cases in Ghana [2]. Malaria transmission requires the production of sexual stage parasites that are stimulated to fertilize after being taken up during a blood meal by a mosquito [3]. The zygote continues development in the mosquito producing an oocyst containing sporozoites that can initiate an infection in humans during a subsequent blood meal. Sexual reproduction coupled with high genetic diversity in the local parasite population and concurrent infections with polymorphic parasite lines provides genetic flexibility that allow adaptation to immune and drug pressure [4] and also influences malaria transmission success [5]. For example, an increase in the rate of sexual recombination has been found to give rise to parasites with different drug resistant profiles [6–9]. Low haematocrit and history of prolonged illness have been associated with gametocyte prevalence detected using microscopy [10]. Genetic factors are also likely to play a role since gametocyte production and mosquito infectivity have been shown to vary between parasite lines [11–14]. Together the dynamics of parasite diversity and gametocyte production have important implications for the acquisition of immunity by the host and the spread of drug resistant parasites. However, monitoring gametocyte production in the human host is complicated by low production levels and sequestration of immature gametocytes during the 10–12 days required for the development of stage V P. falciparum gametocytes. Only mature stage V gametocytes circulate and can be detected in peripheral blood. Previous work in East Africa and Asia demonstrated that gametocytes are resistant to artemisinin-based combination therapy (ACT) and, consequently, patients remain infectious for over a week after asexual parasite clearance [15, 16]. The role of the immune response in controlling gametocyte levels in the human host has not been well established [17]. However, Pfs230 and Pfs48/45 are expressed on the gametocyte surface during development in the RBC in the human host [18–20] and anti-Pfs230 and Pfs48/45 antibodies are generated during a natural infection [19–24] and thus can serve as a marker for recent gametocyte exposure.
This study assessed the prevalence of submicroscopic gametocytes levels and asexual parasite diversity in patients aged between 6 months and 13 years with uncomplicated P. falciparum infections. Understanding these patterns is critical to the development of intervention strategies in high transmission areas. The persistence of gametocytes in children with uncomplicated malaria 4 days after a 3-day anti-malarial drug course (day 7) was also analysed.
Methods
Ethical considerations
The study was approved by the Institutional Review Board of the Noguchi Memorial Institute for Medical Research (NMIMR) and Ghana Health Services. Before recruitment each parent/guardian was informed of the objectives, methods, anticipated benefits and potential hazards of the study. The parents/guardians were encouraged to ask questions about any aspect of the study that was unclear to them and informed about their liberty to withdraw their children at any time without penalty. Children were enrolled only after written parental consent had been obtained. All patient information is treated as confidential.
Study site and population
The study was conducted in three health facilities, the Ghana Atomic Energy Commission (GAEC) Clinic in Accra, Ewim Health Centre and Elmina Health Centre, both in Cape Coast. Cape Coast (05°05′ N, 01°15′ W), an urban setting, has an estimated population of 227,269 and lies in the Coastal savannah region (Fig. 1). Cape Coast, which is the capital of the Central Region, is about 165 km from Accra. Malaria transmission in this area is perennial with most of the disease occurring during the major rainy season in June/July. Accra (05°35′ N, 00°06′ W), an urban setting has an estimated population of 2,291,352. Accra is the capital city of Ghana with a total land area of 201 sq km. Accra is one of the most populated and fast growing metropolis in Africa with an annual growth rate of 4.3% [25] and lies in the coastal savannah region. Malaria transmission in this area is also perennial with most of the disease occurring during the major rainy season in June/July. The study group comprised of 112 children between the ages of 4 months–13 years, where 55 participants where between 8 and 60 months and the rest between 72 and 156 months (Table 1). The malaria patients enrolled in this study were prescribed ACT (artemether-lumefantrine) at the health centre, BUT there was no evaluation/monitoring of ACT intake.

A geographic map showing location of the study sites in Southern Ghana
Sampling
A total of 112 samples were collected from patients aged 6 months–13 years, who visited the health centre with uncomplicated malaria from June to August 2014. Children who were found to be P. falciparum positive by microscopy were enrolled after parental consent was obtained and in accordance with the study inclusion criteria [26]. Sixteen of the children did not return for the 1 week follow up visit. Prior to treatment (day 0) and during the 7 day follow up visit (day 7), 2 ml of venous blood was collected into EDTA vacutainer tubes and an aliquot spotted onto filter paper (Whatman® 3 mm). The filter paper was air-dried and stored desiccated at room temperature. The EDTA samples were immediately centrifuged and the plasma collected and saved for future use. One hundred microlitres of pelleted cells were preserved in 500 µl of Trizol (Tri Reagent, Invitrogen). All the samples were transported to the NMIMR for analysis.
Parasite density
Thin and thick blood smears were prepared from capillary blood collected on day 0 and venous blood collected on day 7. The thin smears were used for Plasmodium species identification and thick smears used for parasite (asexual and sexual) density estimation by using 100X oil immersion light microscopy. Plasmodium falciparum parasites were counted per 200 leukocytes to estimate the parasite density per microlitre of blood. All blood smears were read by two independent microscopists.
Extraction, purification and analysis of parasite RNA
Thirty-five day 0 and day 7 paired trizol preserved samples were selected based on their antibody titres against a gametocyte specific antigen Pfs48/45-6C (19). The Pfs48/45 titres were obtained during an experiment that will be published separately and the selected samples included sero negative, low seropositive and high sero positive relative to a positive control. RNA was isolated from samples (100 μl packed blood cells) using the Quick RNA MiniPrep kit (Zymo Research) following the manufacturer’s protocol, which included an on column DNaseI treatment prior to elution with 36 μl of elution buffer. Eight microlitres of RNA extracted from the day 0 and day 7 blood samples were converted into 20 μl of cDNA using a Qiagen Omni script Reverse Transcriptase kit (Qiagen) and oligo-dT primers. To check for genomic DNA contamination after RNA extraction, conventional PCR was performed on all RNA (4 µl) samples and 2 µl of their corresponding cDNA samples diluted 1:10. Controls used in these PCR reactions were P. falciparum and Homo sapiens genomic DNA as well as a no template control (NTC) from the cDNA conversion reaction. Pfs48/45 primers (Additional file 1) were used for the day 0 samples and human blood group O genotyping primers as described by Tun et al. [27] (Additional file 1) were used for the day 7 samples that, except for one patient, were no longer positive for P. falciparum asexual parasites by microscopy. Real time (RT)-PCR was only carried out on cDNA samples after confirming that the PCR products run on 2% ethidium bromide stained agarose gels did not have amplified products in wells with the NTC and the RNA samples but there were products in the cDNA and 3D7 gDNA samples. A two-step SYBR Green 1 non-quantitative real time reverse transcription-PCR (RT-PCR) was performed on the P. falciparum cDNA isolated from the day 0 and day 7 blood samples.
Gametocyte carriage was assessed using Pfs25 transcript levels. Validation of the Pfs25 mRNA primer set (Additional file 1) was performed on cDNA converted RNA extracted from cultured NF54 stage IV and V gametocytes using the same methods describe above for the patient samples. The cDNA was diluted 1:20 with subsequent twofold serial dilution until 1: 640 and each dilution was tested in triplicate using a Pfs25 primer concentration of 300 nM and the fast SYBR® Green 2X master mix RT-PCR kit (Applied BioSystems). The reaction was run on an Applied Biosystems One-step Plus RT-PCR machine and the cycling conditions were 95 °C for 20 s, 40 cycles of 95 °C for 3 s and 60 °C for 30 s. A melt curve was performed on the final product. Applied Biosystems StepOnePlus software was used to determine the threshold cycle (CT) for each cDNA concentration and the data used to plot a standard curve (Additional file 2). The CT values of the no template control was used to determine the cut-off for the presence or absence of gametocytes. The patient samples were tested in triplicate using Fast SYBR® Green 2X master mix RT-PCR kit (Applied BioSystems). Two microlitres of cDNA diluted 20 fold (equivalent to 0.11 µl of packed blood cells) in a total reaction volume of 20 µl. The same fast SYBR® Green RT-PCR conditions described above were used and the data analysed using Applied Biosystems StepOnePlus software.
Extraction of parasite DNA
Genomic DNA was extracted from two 3 mm punches of dried blood blots using Saponin-Chelex extraction [28]. Briefly, the blood stained filter paper discs for each sample (day 0 and day 7) were incubated in 1.5 ml containing 1120 μl of 0.5% saponin/PBS solution overnight at room temperature on a shaking incubator. After the overnight incubation, the supernatant was discarded and the samples washed twice with 1 ml PBS, followed by a high-speed centrifugation at 10,000×g. Finally, 150 μl of a 6% Chelex (Sigma-Aldrich, USA) in DNase/RNase free water was added to the washed samples and incubated at 95 °C for 5 min to extract DNA from the samples. After a final high-speed centrifugation, the supernatant containing the DNA was stored at −20 °C until used for the genotyping amplification reactions.
Molecular identification and genotyping
To distinguish three major allelic families (K1, MAD 20, and RO33) block 2 of msp1 and the two allelic families (FC27 and IC3D7) central polymorphic region of msp2, nested PCR was performed using family specific primers [29] shown in Additional file 1. All amplification reactions were carried out in a final volume of 15 μl. The outer PCR reaction mix contained 200 nM dNTP, 2 mM MgCl2. 133 nM of each primer, and 0.5 unit of One Taq DNA polymerase (New England BioLab) in addition to 4 μl (about 0.25 μl of whole blood) of genomic DNA (gDNA) template. In the nested reaction, 0.5 μl of the outer PCR product was used as template in a PCR reaction mixture containing 200 nM dNTP, 1.8 mM MgCl2. 200 nM of each primer and 0.5 unit of One Taq DNA polymerase. Each amplification profile consisted of initial denaturation at 94 °C for 3 min, followed by 30 cycles at 94 °C for 1 min; 50–59 °C (depending on the primer pair annealing temperatures) for 35 s, and 68 °C for 2.5 min; with final extension at 68 °C for 3 min. The PCR reaction mixtures were run on a thermal cycler (MJ Research Tetrad PTC-225 Thermal Cycler, USA). Allelic specific positive controls 3D7, K1, HB3 and RO33 gDNA and no template negative controls were included in each set of reactions. PCR products were separated using 2% ethidium bromide-stained agarose gels respectively and visualized under UV illumination.
Multiplicity of infection
The multiplicity of infection (MOI) or number of genotypes per infection was calculated by dividing the total number of msp1 or msp2 fragments detected by the number of samples positive for the same marker. Samples with more than one genotype were considered as containing multiclonal infections while the presence of a single allele was considered as clonal infection.
G6PD genotyping
PCR based G6PD genotyping was performed on the extracted DNA using primers listed in Carter, et al. as previously reported [30]). The A376G mutation was characterized in each DNA sample using restriction fragment length polymorphism (RFLP) by digesting the 376 PCR amplicon with 1 unit of FOKI restriction enzyme at 37 °C for an hour. Only samples with the 376G genotype where further analyzed for three other sub-Saharan African cDNA mutations, G202A, G680T and T968C also using RFLP as described in our previous work [31]. The G202A PCR amplicon was digested with NlaIII restriction enzyme, the G680T amplicon was digested with BstNI restriction enzyme and the T968C amplicon digested with NciI restriction enzyme for 1 h at 37 °C. All the PCR fragments and the digested fragments were viewed under UV light after resolving on a 2% agarose gel containing 0.5 µg/ml ethidium bromide.
Data analysis
Data were entered and analysed using Excel and GraphPad Prism version 7.0. The Shapiro–Wilk normality used to determine if the data was normally distributed. The data was not normally distributed, thus the Mann–Whitney test was used to determine relationships between age and PD, MOI and Pfs25 RT-PCR CT values using GraphPad Prism v7.0. The geometric means and other column statistics were obtained using GraphPad Prism v7.0. Proportion was used to present the distribution of different allelic families. The frequency of msp1 and msp2 family alleles was calculated as the ratio of the number of PCR products obtained for each family to the total number of gene specific PCR products identified. A sample was classified as harboring a multiclonal infection when more than one amplified fragment was obtained during either or both the msp1 and msp2 genotyping. The mean MOI was calculated as a total number of P. falciparum genotypes detected per total number of positive samples. Linear regression was used to determine the relationship between MOI and day 0 and day 7 CT values obtained during the Pfs25 real time reverse transcriptase PCR. Statistical significance was defined as P ≤ 0.05.
Results
Asexual and gametocyte parasite density in the study participants were monitored by microscopy on day 0 and day 7, 4 days after a 3 day ACT regimen. All the children were PCR positive for P. falciparum after MSP genotyping, however after the thick smears prepared by the hospital laboratory staff were re-read by highly trained microscopists, only 72 of the children had microscopy confirmed parasites on day 0. The geometric mean of P. falciparum parasite density recorded by microscopy for the 96 participants that returned for the follow up visit was 20.319/µl blood (95% CI 13.568–30.428) at day 0. Three samples were positive for P. falciparum by MSP genotyping on day 7, although only one sample had microscopy confirmed asexual parasites with a density 76,080/µl.
Three samples were positive for gametocytes by microscopy on day 0 with a mean gametocyte density of 93.33 (SEM 35.28) two of these samples remained gametocytaemic on day 7, with gametocyte densities of 320 and 400/µl. Plasmodium falciparum parasite carriage measured by PCR analysis of the MSP2 and MSP1 genes revealed that all the day 0 samples were positive for P. falciparum parasites but only three day 7 samples were positive. MSP genotyping revealed one day 7 sample to be a recrudescent infection and the other two as new infections. The geometric mean participant age was 52.18 (95% CI 44.74–60.86) months, with a range from 4 months through to 156 months. No significant relationship between age and PD was identified in this study (Additional file 3). The prevalence of G6DP deficiency was estimated at 4.76% (5/105) and consisting of three hemizygous A- males and two AA- heterozygous females in the entire study population, however no data was available for seven of the children (Table 1).
Submicroscopic gametocytes
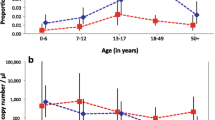
The Pfs25 RT-PCR was used to screen for submicroscopic levels of gametocytes in 35 trizol preserved samples that were selected to represent a range of titres for gametocyte specific antigen Pfs48/45-6C (Additional file 4). Although Pfs25 mRNA is not translated until uptake by a mosquito, the transcript is expressed and stored in mature female gametocytes [32] and it has been developed as a sensitive and specific marker for circulating mature gametocytes [33, 34]. Twenty-seven of the day 0 samples were classified as positive using a CT cut off of 35 (77%) (Table 2; Additional file 4). Eighteen of the 35 samples were gametocyte positive on both days (51.4%), including the sample that was gametocyte positive by microscopy on day 0 and day 7. Three samples were identified that were gametocyte negative on day 0 but gametocyte positive on day 7, making the total number of gametocyte positive samples for day 7 equal 21 (60%). Four samples which were gametocyte negative on day 0 and remained gametocyte negative on day 7 (Table 2; Additional file 4). Interestingly, significantly more children above the age of 5 years had submicroscopic gametocytes on day 7 (Additional file 3).
Genetic diversity and multiplicity of infection
Plasmodium falciparum genes, which show extensive polymorphisms, such as merozoite surface proteins 1 (msp1) and 2 (msp2) can be used as markers to study parasite genetic diversity and multiplicity of infection (MOI) [35]. Msp1 Block 2 is the most polymorphic region of the gene and is grouped into three allelic families namely K1, MAD 20, and RO33 type, while in the msp2 gene block 3 is the most polymorphic region and consists of FC27 and 3D7 families [36]. In total, PCR products were detected using at least one msp1 and msp2 family specific primer set in 86 of the 96 samples (Table 3). Only 12 samples had a single band for both msp1 and msp2, indicating that less than 14% of the subjects had monoclonal infections (as predicted by msp genotyping). The remaining samples had a range of combinations of the different allelic families. For msp1, the frequency of MAD20 and K1 family alleles were similar and higher than the R033 alleles (Fig. 2a). Of the 88 samples with PCR products for msp1, 62 had at least one K1 allele, 60 had at least one MAD20 allele and 52 had at least one R033 allele. Thirty-five percent of the subjects were multiply infected with parasites from all three msp1 families. Additional diversity was also evident within the K1 and MAD20 families, as eight subjects had two distinct MAD20 alleles and five samples had two distinct K1 alleles (Fig. 3a). Five samples had 4 distinct msp1 alleles, 30 had 3 alleles, and 24 had 2 alleles, while 29 were monoclonal for msp1 (Fig. 3a). Fifty-nine percent of the msp1 monoclonal infections belonged to the R033 family (13 of the 29). The geometric mean MOI for msp1 in the study was 1.90 (95% CI 1.71–2.11).

Representation of msp1 (a) and msp2 (b) allele families in the study population. The distribution of parasites within the major families and their combinations in patient samples are shown. The numbers in brackets represents the total number of samples that contained at least one parasite belonging to the allelic family

Prevalence and multiplicity of infection of msp1 (a) and msp2 (b) alleles. Each distinct amplicon produced by msp1 or msp2 family specific PCR represents a particular parasite clone. The number of samples that contained distinct alleles (color coded and labelled 1 through 6) for a msp1 (msp1 MOI) or each of the three msp1 families (RO33, MAD20 or K1) or b msp2 (msp2 MOI) or each of the two msp families (3D7 or FC27) are plotted
For msp2, 77 of the 94 samples with msp2 PCR products had at least one FC27 allele and 68 had at least one 3D7 allele. Again most samples were polyclonal, containing alleles from both families (51 of 94 samples) (Fig. 3b). As with msp2, some samples also contained multiple alleles within one msp2 family (Fig. 3b); with one sample containing six distinct msp2 alleles. Only thirty-three of the 94 PCR-positive msp2 samples were monoclonal for msp2. The geometric mean of msp2 MOI was 1.88 (95% CI 1.68–2.10).
Combining genotyping results, the study identified only 12 samples that carried a parasite population with a single allele for both msp1 and msp2 (Table 3), suggesting that the clonal parasite population estimated solely by msp1 genotyping is overestimated by 58.6% (17/29) and 63.6% (21/33) when msp2 genotyping is used exclusively. Two samples that were multiclonal by msp1 genotyping did not have any msp2 data and eight samples, three clonal and five multiclonal identified by msp2 genotyping did not have msp1 data. No significant correlation between the MOI for either msp1 or msp2 and submicroscopic gametocyte prevalence was found in this study (Additional file 3).
Discussion
The high prevalence (77%) of mature gametocytes detected by RT-PCR in the day 0 samples (Table 1) is consistent with the high malaria transmission rates observed in southern coastal Ghana [25] and is similar to studies in other regions of Africa using Pfs25 RT-PCR [2, 16, 37–40]. The continued persistence of gametocytes in 60% of the patients even after the clearance of asexual parasites with ACT drug treatment, including 2 of the 3 patients with microscopically detectable gametocytes on day 7, highlights the lack of efficacy of ACT against mature sexual stage parasites [31, 38, 41, 42] and the need to develop new strategies to block the spread of malaria.
Although age was not significantly associated with submicroscopic gametocytes levels on day 0 (Additional file 3), older patients had a significantly higher prevalence of submicroscopic gametocytes on day 7 compared with the younger children due to them having a lower mean CT value for Pfs25 real time reverse transcriptase PCR value (Additional file 3), suggesting an age associated decrease in gametocyte clearance following ACT. A larger study that includes a wider age range of patients would be needed to confirm this and to begin to define the contributing factors, such as the role of a maturing immune response in gametocyte clearance. The acquisition of immunity to clinical malaria is well established [43], but immune mediated clearance of gametocytes has been more difficult to demonstrate [44].
Primaquine (PQ) is currently the only WHO approved drug with potent transmission-blocking activity that targets mature P. falciparum gametocytes [45–47]. As Ghana targets to move beyond the malaria control phase into the pre elimination phase, there is a possibility of implementing PQ with first-line anti-malarial ACT to reduce gametocyte prevalence in Ghana as is on-going in some countries in the elimination and pre elimination phase [45]. The prevalence and extent of g6pd deficiency is a major concern for malaria eradication programmes, where they plan to use PQ as a gametocidal agent as although a single low dose of PQ has been suggested to be well tolerated in G6PD deficient malaria patients, PQ has been found at certain concentrations and in certain instances to cause RBC lysis in g6pd deficient individuals [48–50]. The 4.7% incidence of g6pd deficiency identified in this study was similar to another study conducted in 2015 in two different communities along the coast of Ghana where G6PD deficiency was 5.9% in hemizygous males and homozygous females [31]. The prevalence of this population needs to be taken into consideration when evaluating the use of primaquine or other 8-aminoquinolines as gametocidal agents in this region.
Multiplicity of infection within the allelic families reduced clonality of the parasite population from 37.5 to 33% in msp1 and 46 to 35% in msp2 (Fig. 2; Table 3) and it possible that even higher MOIs would be identified within the allelic families if a more sensitive sizing technique such as capillary electrophoresis was used instead of agarose gel electrophoresis. High parasite MOI (≥2) has been associated with increased parasite density, although whether this is related to parasite survival or increased production was not directly evaluated [51]. Multiclonal infections also enhance the chance of heterozygous mating after the parasites have been taken up in a blood meal of a mosquito [4–7]. Fertilization between two distinct parasites provides the opportunity for chromosome recombination during meiosis, which enhances genetic diversity in the parasite population. Geometric mean MOIs for msp1 and msp2 of 1.90 and 1.88 were slightly lower than obtained in an earlier report from the middle belt of Ghana where MOI was 2.3 in July/August, [52] but similar to another report of MOI in southern Ghana which was 1.93 [24] and 1.3 in 2013 [53]. This discrepancy may be due to differences in ecological zones [54], malaria transmission intensity [36, 55] and/or the treatment-seeking behaviour of malaria patients within the various population [56, 57].
Even at these relatively low MOIs the majority of the samples (86%) contained more than one msp1 or msp2 allele (Fig. 3) indicating the potential for cross-fertilization once gametocytes are taken up by a mosquito. Gametocytes prevalence was not associated with MOI, indicating that multiply infected individuals were just as likely to be gametocyte carriers as those with monoclonal infections. The combination of high gametocyte prevalence and multiclonal infections sets the stage for mating between two distinct parasite lines and further enhancing parasite diversity.
Limitations
Although all the volunteers were prescribed and administered received the same ACT, artemether-lumefantrine, they did so in their respective homes and as such compliance to the prescribed regimen could have been compromised. Such non-conformity in prescribed drug intake could influence the prevalence of both asexual and sexual stage parasites. This scenario reflects what happens in the community, where non-compliance can be a major obstacle for malaria control programmes.
Conclusion
This study provides critical information on factors that influence malaria transmission such as the presence of submicroscopic levels of gametocytes in 77% of the children on day 0 which persisted in 60% of the children on day 7. The high prevalence of gametocytes and multiclonal infections (86%) in the children suggests there is ample opportunity for recombination during fertilization, which would enhance genetic diversity and could contribute to the emergence of drug resistant parasites. Older children were more likely to have submicroscopic gametocytes on day 7 suggesting an age associated decrease in gametocyte clearance following ACT treatment. The high prevalence of submicroscopic gametocytes levels is consistent with other studies and it will be important to monitor drug resistance, particularly ACT clearance in future studies.
Abbreviations
- ACT:
-
artemisinin-based combination therapy
- cDNA:
-
complementary deoxyribonucleic acid
- DNA:
-
deoxyribonucleic acid
- EDTA:
-
ethylenediamine tetra acetic acid
- GLURP:
-
glutamate rich protein
- MOI:
-
multiplicity of infection
- MSP:
-
merozoite surface protein
- RNA:
-
ribonucleic acid
- RT-PCR:
-
real time reverse transcription polymerase chain reaction
References
Afudego CE. Cost effectiveness analysis of insecticide-treated mosquito nets (ITNs) and indoor residual spraying (IRS)—malaria interventions in Ghana (children under 5) [internet]. 2011. http://www.isodec.org.gh/publications/Final%20-%20HEALTH%20CEA%20Report%20June%202012.pdf.
WHO. Guidelines for the treatment of malaria. Geneva: World Health Organization; 2015.
Alano P. Plasmodium falciparum gametocytes: still many secrets of a hidden life. Mol Microbiol. 2007;66:291–302.
McConkey GA, Waters AP, McCutchan TF. The generation of genetic diversity in malaria parasites. Annu Rev Microbiol. 1990;44:479–98.
Morlais I, Nsango SE, Toussile W, Abate L, Annan Z, Tchioffo MT, et al. Plasmodium falciparum mating patterns and mosquito infectivity of natural isolates of gametocytes. PLoS ONE. 2015;10:e0123777.
Antao T, Hastings IM. Environmental, pharmacological and genetic influences on the spread of drug-resistant malaria. Proc R Soc B Biol Sci. 2011;278:1705–12.
De Roode JC, Pansini R, Cheesman SJ, Helinski MEH, Huijben S, Wargo AR, et al. Virulence and competitive ability in genetically diverse malaria infections. Proc Natl Acad Sci USA. 2005;102:7624–8.
Mardani A, Keshavarz H, Heidari A, Hajjaran H, Raeisi A, Khorramizadeh MR. Genetic diversity and natural selection at the domain I of apical membrane antigen-1 (AMA-1) of Plasmodium falciparum in isolates from Iran. Exp Parasitol. 2012;130:456–62.
Brockman A, Paul RE, Anderson TJ, Hackford I, Phaiphun L, Looareesuwan S, et al. Application of genetic markers to the identification of recrudescent Plasmodium falciparum infections on the northwestern border of Thailand. Am J Trop Med Hyg. 1999;60:14–21.
Price R, Nosten F, Simpson JA, Luxemburger C, Phaipun L, ter Kuile F, et al. Risk factors for gametocyte carriage in uncomplicated falciparum malaria. Am J Trop Med Hyg. 1999;60:1019–23.
Eksi S, Morahan BJ, Haile Y, Furuya T, Jiang H, Ali O, et al. Plasmodium falciparum gametocyte development 1 (Pfgdv1) and gametocytogenesis early gene identification and commitment to sexual development. PLoS Pathog. 2012;8:e1002964.
Lambrechts L, Halbert J, Durand P, Gouagna LC, Koella JC. Host genotype by parasite genotype interactions underlying the resistance of anopheline mosquitoes to Plasmodium falciparum. Malar J. 2005;4:3.
Nwakanma D, Kheir A, Sowa M, Dunyo S, Jawara M, Pinder M, et al. High gametocyte complexity and mosquito infectivity of Plasmodium falciparum in the Gambia. Int Parasitol. 2008;38:219–27.
Carter LM, Kafsack BFC, Llinas M, Mideo N, Pollitt LC, Reece SE. Stress and sex in malaria parasites: why does commitment vary? Evol Med Public Health. 2013;2013:135–47.
Chen PQ, Li GQ, Guo XB, He KR, Fu YX, Fu LC, et al. The infectivity of gametocytes of Plasmodium falciparum from patients treated with artemisinin. Chin Med J. 1994;107:709–11.
Bousema JT, Schneider P, Gouagna LC, Drakeley CJ, Tostmann A, Houben R, et al. Moderate effect of artemisinin-based combination therapy on transmission of Plasmodium falciparum. J Infect Dis. 2006;193:1151–9.
Saeed M, Roeffen W, Alexander N, Drakeley CJ, Targett GAT, Sutherland CJ. Plasmodium falciparum antigens on the surface of the gametocyte-infected erythrocyte. PLoS ONE. 2008;3:e2280.
Roeffen W, Lensen T, Mulder B, Teelen K, Sauerwein R, Van Druten J, et al. A comparison of transmission-blocking activity with reactivity in a Plasmodium falciparum 48/45-kD molecule-specific competition enzyme-linked immunosorbent assay. Am J Trop Med Hyg. 1995;52:60–5.
Singh SK, Roeffen W, Andersen G, Bousema T, Christiansen M, Sauerwein R, et al. A Plasmodium falciparum 48/45 single epitope R0.6C subunit protein elicits high levels of transmission blocking antibodies. Vaccine. 2015;33:1981–6.
Van Dijk MR, Janse CJ, Thompson J, Waters AP, Braks JAM, Dodemont HJ, et al. A central role for P48/45 in malaria parasite male gamete fertility. Cell. 2001;104:153–64.
Carter R. Restricted or absent immune responses in human populations to Plasmodium falciparum gamete antigens that are targets of malaria transmission-blocking antibodies. J Exp Med. 1989;169:135–47.
Drakeley CJ, Bousema JT, Akim NIJ, Teelen K, Roeffen W, Lensen AH, et al. Transmission-reducing immunity is inversely related to age in Plasmodium falciparum gametocyte carriers. Parasite Immunol. 2006;28:185–90.
Ouedraogo AL, Roeffen W, Luty AJF, de Vlas SJ, Nebie I, Ilboudo-Sanogo E, et al. Naturally acquired immune responses to Plasmodium falciparum sexual stage antigens Pfs48/45 and Pfs230 in an area of seasonal transmission. Infect Immun. 2011;79:4957–64.
Jones S, Grignard L, Nebie I, Chilongola J, Dodoo D, Sauerwein R, et al. Naturally acquired antibody responses to recombinant Pfs230 and Pfs48/45 transmission blocking vaccine candidates. J Infect. 2015;71:117–27.
Ghana Urban Malaria Study. Report of the Ghana Urban Malaria Study. JSI training and Research Institute Inc; 2013.
WHO. Methods for surveillance of antimalarial drug efficacy. Geneva: World Health Organization; 2009. http://apps.who.int/iris/handle/10665/44048. Accessed 15 Dec 2015.
Tun Z, Honda K, Nakatome M, Nakamura M, Shimada S, Ogura Y, et al. Simultaneous detection of multiple STR loci on sex chromosomes for forensic testing of sex and identity. J Forensic Sci. 1999;44:772–7.
Baidjoe A, Stone W, Ploemen I, Shagari S, Grignard L, Osoti V, et al. Combined DNA extraction and antibody elution from filter papers for the assessment of malaria transmission intensity in epidemiological studies. Malar J. 2013;12:272.
Snounou G, Zhu X, Siripoon N, Jarra W, Thaithong S, Brown KN, et al. Biased distribution of msp1 and msp2 allelic variants in Plasmodium falciparum populations in Thailand. Trans R Soc Trop Med Hyg. 1999;93:369–74.
Carter N, Pamba A, Duparc S, Waitumbi JN. Frequency of glucose-6-phosphate dehydrogenase deficiency in malaria patients from six African countries enrolled in two randomized anti-malarial clinical trials. Malar J. 2011;10:241.
Amoah LE, Opong A, Ayanful-Torgby R, Abankwa J, Acquah FK. Prevalence of G6PD deficiency and Plasmodium falciparum parasites in asymptomatic school children living in southern Ghana. Malar J. 2016;15:388.
Mair GR, Braks JAM, Garver LS, Wiegant JCAG, Hall N, Dirks RW, et al. Regulation of sexual development of Plasmodium by translational repression. Science. 2006;313:667–9.
Babiker HA, Abdel-Wahab A, Ahmed S, Suleiman S, Ranford-Cartwright L, Carter R, et al. Detection of low level Plasmodium falciparum gametocytes using reverse transcriptase polymerase chain reaction. Mol Biochem Parasitol. 1999;99:143–8.
Schneider P, Schoone G, Schallig H, Verhage D, Telgt D, Eling W, et al. Quantification of Plasmodium falciparum gametocytes in differential stages of development by quantitative nucleic acid sequence-based amplification. Mol Biochem Parasitol. 2004;137:35–41.
Medicines for Malaria Venture. Methods and techniques for clinical trials on antimalarial drug efficacy: genotyping to identify parasite populations: informal consultation organized by the Medicines for Malaria Venture and cosponsored by the World Health Organization, 29–31 May, Amsterdam, the Netherlands. Geneva: World Health Organization; 2008.
Takala SL, Escalante AA, Branch OH, Kariuki S, Biswas S, Chaiyaroj SC, et al. Genetic diversity in the Block 2 region of the merozoite surface protein 1 (MSP-1) of Plasmodium falciparum: additional complexity and selection and convergence in fragment size polymorphism. Infect Genet Evol. 2006;6:417–24.
WHO, Global Malaria Programme. World Malaria Report. Geneva: World Health Organization; 2014.
Dinko B, Oguike MC, Larbi JA, Bousema T, Sutherland CJ. Persistent detection of Plasmodium falciparum, P. malariae, P. ovale curtisi and P. ovale wallikeri after ACT treatment of asymptomatic Ghanaian school-children. Int J Parasitol Drugs Drug Resist. 2013;3:45–50.
Schneider P, Bousema JT, Gouagna LC, Otieno S, van de Vegte-Bolmer M, Omar SA, et al. Submicroscopic Plasmodium falciparum gametocyte densities frequently result in mosquito infection. Am J Trop Med Hyg. 2007;76:470–4.
Shekalaghe S, Drakeley C, Gosling R, Ndaro A, van Meegeren M, Enevold A, et al. Primaquine clears submicroscopic Plasmodium falciparum gametocytes that persist after treatment with sulphadoxine-pyrimethamine and artesunate. PLoS ONE. 2007;2:e1023.
Karl S, Laman M, Moore BR, Benjamin J, Koleala T, Ibam C, et al. Gametocyte clearance kinetics determined by quantitative magnetic fractionation in Melanesian children with uncomplicated malaria treated with artemisinin combination therapy. Antimicrob Agents Chemother. 2015;59:4489–96.
Tangpukdee N, Krudsood S, Srivilairit S, Phophak N, Chonsawat P, Yanpanich W, et al. Gametocyte clearance in uncomplicated and severe Plasmodium falciparum malaria after artesunate-mefloquine treatment in Thailand. Korean J Parasitol. 2008;46:65.
Langhorne J, Ndungu FM, Sponaas A-M, Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. 2008;9:725–32.
Stone WJR, Dantzler KW, Nilsson SK, Drakeley CJ, Marti M, Bousema T, et al. Naturally acquired immunity to sexual stage P. falciparum parasites. Parasitology. 2016;143:187–98.
WHO. Single dose primaquine as a gametocytocide in Plasmodium falciparum malaria. Updated WHO Policy Recommendation. [Internet].Geneva: World Health Organization; 2012. http://www.who.int/malaria/pq_updated_policy_recommendation_en_102012.pdf.
WHO. Evidence Review Group: The safety and effectiveness of single dose primaquine as a P. falciparum gametocytocide. Meeting Report [Internet]. Geneva: World Health Organization; 2012. http://www.who.int/malaria/mpac/sep2012/primaquine_single_dose_pf_erg_meeting_report_aug2012.pdf.
Frank JE. Diagnosis and management of G6PD deficiency. Am Fam Physician. 2005;72:1277–82.
Bancone G, Chowwiwat N, Somsakchaicharoen R, Poodpanya L, Moo PK, Gornsawun G, et al. Single low dose primaquine (0.25 mg/kg) does not cause clinically significant haemolysis in G6PD deficient subjects. PLoS ONE. 2016;11:e0151898.
Beutler E, Duparc S, G6PD Deficiency Working Group. Glucose-6-phosphate dehydrogenase deficiency and antimalarial drug development. Am J Trop Med Hyg. 2007;77:779–89.
Chen I, Poirot E, Newman M, Kandula D, Shah R, Hwang J, et al. An assessment of the supply, programmatic use, and regulatory issues of single low-dose primaquine as a Plasmodium falciparum gametocytocide for sub-Saharan Africa. Malar J. 2015;14:204.
Nassir E, Abdel-Muhsin A-MA, Suliaman S, Kenyon F, Kheir A, Geha H, et al. Impact of genetic complexity on longevity and gametocytogenesis of Plasmodium falciparum during the dry and transmission-free season of eastern Sudan. Int J Parasitol. 2005;35:49–55.
Agyeman-Budu A, Brown C, Adjei G, Adams M, Dosoo D, Dery D, et al. Trends in multiplicity of Plasmodium falciparum infections among asymptomatic residents in the middle belt of Ghana. Malar J. 2013;12:22.
Duah NO, Matrevi SA, Quashie NB, Abuaku B, Koram KA. Genetic diversity of Plasmodium falciparum isolates from uncomplicated malaria cases in Ghana over a decade. Parasit Vectors. 2016;9:416.
Thomas BN, Duru KC. Genetic diversity and allelic frequency of glutamate-rich protein (GLURP) in Plasmodium falciparum isolates from sub-Saharan Africa. Microbiol Insights. 2014;7:35–9.
Arnot D. Unstable malaria in Sudan: the influence of the dry season. Clone multiplicity of Plasmodium falciparum infections in individuals exposed to variable levels of disease transmission. Trans R Soc Trop Med Hyg. 1998;92:580–5.
Fenny AP, Asante FA, Enemark U, Hansen KS. Malaria care seeking behavior of individuals in Ghana under the NHIS: are we back to the use of informal care? BMC Public Health. 2015;15:370.
Battle KE, Bisanzio D, Gibson HS, Bhatt S, Cameron E, Weiss DJ, et al. Treatment-seeking rates in malaria endemic countries. Malar J. 2016;15:20.
Authors’ contributions
LEA and KW designed the study, RA-T, KW and LEA wrote the paper, LEA and RA-T Performed the statistical analysis, FKA, RA-T, AO and JA Performed the experiments. All authors have read and approved the final draft. All authors read and approved the final manuscript.
Acknowledgements
The authors thank the parents and children who volunteered to be a part of this study. We also thank Dr. Kwadwo A Kusi for helping with the statistical analysis and Dr. Nancy Quashie for providing access to her laboratory.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Ethics approval and consent to participate
The study was approved by the Institutional Review Board of the Noguchi Memorial Institute for Medical Research (NMIMR) and Ghana Health Services. Children were enrolled only after written parental consent had been obtained.
Funding
This work was partially supported by US-NIH Grant # 1R03AI103638 awarded to KW.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ayanful-Torgby, R., Oppong, A., Abankwa, J. et al. Plasmodium falciparum genotype and gametocyte prevalence in children with uncomplicated malaria in coastal Ghana. Malar J 15, 592 (2016). https://doi.org/10.1186/s12936-016-1640-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-016-1640-8